

Abstract
The molecular motor dynein and its associated regulatory subunit dynactin have been implicated in several neurodegenerative conditions of the basal ganglia, such as Huntington's disease (HD) and Perry syndrome, an atypical Parkinson-like disease. This pathogenic role has been largely postulated from the existence of mutations in the dynactin subunit p150Glued. However, dynactin is also able to act independently of dynein, and there is currently no direct evidence linking dynein to basal ganglia degeneration. To provide such evidence, we used here a mouse strain carrying a point mutation in the dynein heavy chain gene that impairs retrograde axonal transport. These mice exhibited motor and behavioural abnormalities including hindlimb clasping, early muscle weakness, incoordination and hyperactivity. In vivo brain imaging using magnetic resonance imaging showed striatal atrophy and lateral ventricle enlargement. In the striatum, altered dopamine signalling, decreased dopamine D1 and D2 receptor binding in positron emission tomography SCAN and prominent astrocytosis were observed, although there was no neuronal loss either in the striatum or substantia nigra. In vitro, dynein mutant striatal neurons displayed strongly impaired neuritic morphology. Altogether, these findings provide a direct genetic evidence for the requirement of dynein for the morphology and function of striatal neurons. Our study supports a role for dynein dysfunction in the pathogenesis of neurodegenerative disorders of the basal ganglia, such as Perry syndrome and HD.
INTRODUCTION
Axonal transport is a bidirectional process through which materials and signals are exchanged between the neuronal cell body and the synapse. Retrograde axonal transport is mediated by the cytoplasmic molecular motor dynein which functions in association with a multiprotein regulatory complex called dynactin (1). Loss of the dynein/dynactin function is thought to be an important factor in the pathogenesis of neurodegenerative diseases (2,3). Indeed, impairment of retrograde axonal transport appears to be one of the earliest pathogenic events during neurodegeneration (1), and transgenic inhibition of retrograde axonal transport in motor neurons is sufficient to drive the degeneration of these neurons (4,5). The involvement of dynactin in neurodegenerative diseases is supported by the discovery of mutations in the dynactin subunit p150Glued in familiar forms of motor neuron disease, including amyotrophic lateral sclerosis and distal spinal and bulbar muscular atrophy (6–8), as well as in Perry syndrome, a rare atypical form of Parkinson's disease resistant to L-DOPA (9,10). In vitro, the G59S mutation in p150Glued leads to abnormal cytoskeletal dynamics accompanied by toxic protein aggregation (11). In vivo, moderate neuron-specific overexpression of mutant dynactin causes motor neuron disease in mice (12–14).
The mechanisms underlying the toxicity of mutant p150Glued are still elusive, because dynactin is able to regulate not only the function of dynein but also that of kinesin-2 (15). Thus, dynactin disruption disables both anterograde and retrograde trafficking (16). Furthermore, dynactin may also act, independently of its role in cellular transport, as a docking protein and has been recently suggested to affect gene transcription through direct modulation of transcription factors (17,18). Despite this emerging large array of dynactin functions, it remains postulated, although largely unproven, that dynactin mutations lead to neurodegeneration through impairment of the dynein function. Several lines of evidence suggest that dynein itself is a modifier of the degeneration of striatal neurons in Huntington's disease (HD), a neurodegenerative disorder caused by an expanded CAG repeat in the huntingtin gene. First, dynein, as well as p150Glued, are binding partners of huntingtin and of huntingtin-associated protein 1 (HAP1) (19,20). Second, the activity of the dynein motor is positively regulated by wild-type huntingtin, and strongly decreased by the HD-associated expansion in the polyglutamine repeat domain of huntingtin (21,22). However, the effects of pathogenic huntingtin on axonal transport are widespread by affecting both anterograde and retrograde fast axonal transport (23). Thus, direct genetic evidence linking retrograde axonal transport, in particular dynein, and striatal degeneration is still lacking.
The aim of this study was to provide such evidence. For this, we took advantage of the existence of mouse strains bearing point mutations in the dynein heavy chain gene (dync1h1) (24). The legs at odd angles (loa) mutation represents a F580Y mutation, while the Cramping (Cra) mutation converts tyrosine 1055 to a cysteine (Y1055C). Both mutations are located in the domain involved in homodimerization of the molecular motor. Both of these mutations impair the ability of dynein motors to sustain fast retrograde transport in situations of cellular stress and lead to decreased retrograde transport in adult dynein mutant motor neurons (24,25). The phenotype of dynein mutant mice has been characterized in the peripheral nervous system, with the occurrence of a proprioceptive sensory neuropathy (26–28), but there are currently no study focusing on the central nervous system of these animals. An initial report suggested that dynein mutant mice displayed lower motor neuron degeneration, but we and others failed to reproduce these findings (26–28). In particular, we performed a longitudinal analysis of the neuromuscular phenotype of Cra/+ mice up to 24 months of age and found no evidence of motor neuron degeneration in these animals (27).
Here, we show that the Cramping dynein mutation in mice leads to distinctive signs of striatal dysfunction. Furthermore, dynein mutant striatal neurons showed profound abnormalities in neurite outgrowth in vitro. These findings strongly support a pathophysiological function for dynein in degenerative diseases of basal ganglia including HD and Perry syndrome.
RESULTS
The motor phenotype of Cra/+ mice is characterized by early muscle weakness, progressive incoordination and hyperactivity
We first performed a battery of motor and behavioural tests in mice bearing the Cramping mutation in the dynein heavy chain gene (Cra/+). In our laboratory, Cra/+ mice showed reduced total and forelimb muscle grip strength as early as 3 months of age (Fig. 1A), and suffered from an impairment in motor coordination that mildly increased with ageing, as observed using an accelerating rotarod test (Fig. 1B). Cra/+ mice displayed increased open field exploratory behaviour as revealed by increased track length and average velocity (Fig. 1C). No differences were observed in the number of rearings, indicating normal vertical behaviour (data not shown). The level of anxiety appeared similar between Cra/+ mice and their wild-type littermates as assessed using the elevated plus maze paradigm (Fig. 1D). In the Morris water maze test, Cra/+ mice tended to spend more time in reaching the platform at 12, but not 3, months of age, when compared with wild-type animals. This was likely due to impaired motor incoordination rather than to spatial memory deficits since the observed difference between genotypes was annulled when considering the distance swum by the mice (Fig. 1E). Taken together, Cra/+ mice display muscle weakness and incoordination with increased open field activity in the absence of obvious spatial working memory deficits.
Figure 1.

Locomotor and behavioural abnormalities in Cra/+ mice. (A) Grip muscle strength of forelimbs (left panel) and all limbs (right panel) in wild-type mice (+/+) and heterozygous Cra/+ mice at 3 and 12 months of age. ***P < 0.001 versus corresponding wild-type (n = 12 mice per group). (B) Latency to fall in an accelerating rotarod test in wild-type mice (+/+) and heterozygous Cra/+ mice at 3 and 12 months of age. ***P < 0.001 versus corresponding wild-type, #P < 0.05 versus 3-month-old Cra/+ mice (n = 12 mice per group). (C) Track length (left panel) and average velocity (right panel) in an open field test in wild-type mice (+/+) and heterozygous Cra/+ mice at 3 and 12 months of age. ***P < 0.001 versus corresponding wild-type (n = 12 mice per group). (D) Time spent in closed arms (upper panel) and open arms (lower panel) in an elevated plus maze test (during a 300 s session) by wild-type mice (+/+) and heterozygous Cra/+ mice at 3 and 12 months of age. Non-significant differences (n = 12 mice per group). (E) Time spent (upper panels) and distance swum (lower panel) to reach the hidden platform in a Morris water maze test by wild-type mice (+/+) and heterozygous Cra/+ mice at 3 (left panel) and 12 (right panel) months of age. The platform was in position A during the first 3 days of the test, and then moved to position C for the last 2 days. Six trials were performed per day, and each point is the mean of two consecutive trials. *P < 0.05 versus corresponding wild-type in time spent and non-significant differences in distance swum (n = 12 mice per group).
Cra/+ mice present with striatal atrophy and lateral ventricle enlargement
Our present findings suggest that the phenotype of Cra/+ mice is broader than the pure proprioceptive neuropathy previously documented and could result from damage to central areas in the brain that control movement. In fact, hyperactivity and progressive motor incoordination are also distinctive features observed in mice expressing pathological forms of huntingtin or after ablation of D1 dopamine receptor expressing cells, both paradigms affecting the striatum (29–32). Forebrain, but not hindbrain, wet weight was decreased in Cra/+ mice (Fig. 2A), suggestive of atrophy. The striatum and cerebral cortex of Cra/+ mice appeared grossly normal using haematoxylin/eosin staining (Fig. 2B), and the cortical layer organization was preserved (Fig. 2C), suggesting that the defect was not due to abnormal cortical development. In vivo brain imaging using magnetic resonance imaging (MRI) showed a significant reduction in the volume of the Cra/+ mice striata at both 5 and 10 months of age (Supplementary Material, Figs S1 and S2D–E), while, concomitantly, the volumes of the lateral ventricles were significantly increased (Fig. 2D and F). Thus, the mutation in dynein leads to striatal atrophy in mice.
Figure 2.
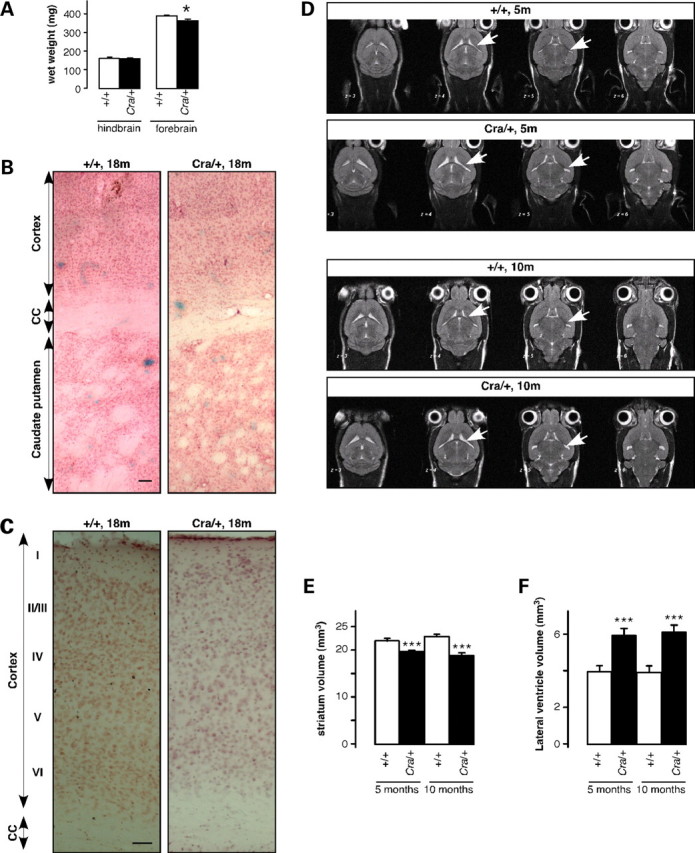
Striatal atrophy in Cra/+ mice. (A) Wet weight of hindbrain (left) and forebrain (right) of wild-type mice (+/+, empty columns) and heterozygous Cra/+ mice (black columns) at 12 months of age. *P < 0.05 versus corresponding wild-type (n = 4 mice per group). (B) Low magnification photomicrographs of haematoxylin and eosin staining of wild-type (+/+) and heterozygous Cra/+ brains at 18 months of age. CC, corpus callosum. Scale bar = 100 µm. (C) Higher magnification of B showing the aspect of the six layers of the cortex. Scale bar = 50µm. (D) Representative horizontal T2-weighted MRI slices of a wild-type (+/+) and Cra/+ mouse at 5 and 10 months of age. Note the enlargement of the lateral ventricles (white arrows) of the Cra/+ mouse. (E) Striatal volume of wild-type (+/+, empty columns) and heterozygous Cra/+ mice (black columns) at 5 (left) and 10 (right) months of age. ***P < 0.001 versus corresponding wild-type (n = 20 mice per group). (F) Lateral ventricle volume of wild-type (+/+, empty columns) and heterozygous Cra/+ mice (black columns) at 5 (left) and 10 (right) months of age. ***P < 0.001 versus corresponding wild-type (n = 20 mice per group).
Progressive astrocytosis in the absence of neurodegeneration in the striatum
Reactive astrocytosis represents a typical marker of neuronal stress and is often a sign of an underlying pathology. Interestingly, reactive astrocytosis, as revealed by glial fibrillary acidic protein (GFAP) immunoreactivity (Fig. 3A), was dramatically increased in the striatum of 8-month-old Cra/+ mice, and this increase was even higher at 18 months of age (Fig. 3B). Consistent with this observation, striatal GFAP mRNA levels as measured using RT–qPCR were higher in Cra/+ mice than in wild-type littermates at 8 months, but not at 4 months of age (Fig. 3C). To determine whether astrocytosis was associated to neurodegeneration, we determined the total number of DARPP-32 (dopamine- and cAMP-regulated phosphoprotein of a molecular weight of 32 kDa) positive medium spiny neurons (MSNs), the neuronal population comprising more than 95% of striatal neurons, using stereological analysis. The analysis of DARPP-32 positive MSNs showed a non-significant trend towards decreased number at 6 months of age (Fig. 4). These data show that the phenotype of dynein mutant mice is rather due to neuronal dysfunction than due to neurodegeneration in the striatum.
Figure 3.

Progressive striatal astrocytosis in Cra/+ mice. (A) Representative microphotographs showing haematoxylin/eosin staining (left panels) and GFAP immunoreactivity (right panels) in the striatum from wild-type mice (+/+, upper panels) and heterozygous Cra/+ mice (lower panels) at 18 months of age. Scale bar = 25 µm. (B) Quantification of the surface occupied by GFAP positive cells in the striatum from wild-type mice (+/+) and heterozygous Cra/+ mice at 8 and 18 months of age. Data are expressed as percentage of the total surface in the picture. *P < 0.05 versus indicated condition (n = 5 mice per group). (C) mRNA levels of GFAP in the striatum from wild-type mice (+/+) and heterozygous Cra/+ mice at 4 and 8 months of age. *P < 0.05 versus wild-type (n = 5–7 mice per group).
Figure 4.

No significant neuronal loss in the striatum of Cra/+ mice. (A and B) Representative photographs of striatal sections processed for DARPP-32 immunohistochemistry from wild-type mice (+/+) at 6 months of age as well as heterozygous Cra/+ mice at 6 and 18 months of age. (C) Stereological estimations of the total number of DARPP-32 positive neurons in the unilateral striatum did not reveal any statistically significant differences between the groups (n = 4–6 mice per group).
Altered dopamine signalling and D1 receptor binding in the striatum of Cra/+ mice
Our histological observations in the striatum argue in favour of neuronal dysfunction that could eventually lead to motor disturbances. To test this hypothesis, we analysed the expression of a series of genes directly involved in the striatal function. D1, but not D2, dopamine receptor mRNA levels were decreased in 8-month-old Cra/+ mice as shown using RT–qPCR (Fig. 5A). D1 receptor expressing cells synthesize substance P, whereas D2 receptor expressing cells synthesize pre-proenkephalin. Thus, substance P, but not pre-proenkephalin, mRNA levels appeared decreased in 8-month-old Cra/+ mice (Fig. 5A), which corroborates the selective down-regulation of the expression of D1 dopamine receptors in striatal neurons. It is therefore tempting to propose that Cra/+ mice suffer from D1 receptor striatal dysfunction that could underlie the motor disturbances observed in these animals. To directly determine whether this is the case, we performed positron emission tomography (PET) analysis of the binding of the D1 receptor selective ligand [11C] SCH-23390 (Fig. 5B). Quantification of [11C] SCH-23390 showed a decrease of the signal in the brains of Cra/+ mice (Fig. 5C), which further reinforces the presence of striatal dopaminergic impairment. We extended our D1-PET scans by using [18F] Fallypride, a high-affinity selective dopamine D2/3 receptor ligand with the advantage of long half-life compared with [11C] Raclopride (33). We observed a significant reduction of [18F] Fallypride uptake in the striatum of Cra/+ mice compared with wild-type animals (Fig. 5D), lending further support for the involvement of the striatal dopaminergic system in the Cra/+ pathogenesis.
Figure 5.

Altered dopamine signalling and binding in the striatum of Cra/+ mice. (A) mRNA levels of the dopamine D1 and D2 receptor, substance P and pre-proenkephalin in the striatum of wild-type mice (+/+) and heterozygous Cra/+ mice at 4 and 8 months of age. *P < 0.05 versus wild-type (n = 5–7). (B) Representative [11C] SCH-23390 images (all frames averaged together) through the striatum of a wild-type mouse brain (+/+, upper panels) and a heterozygous mouse brain (Cra/+, lower panels) are shown. (C) Binding potentials (BPND) of [11C] SCH-23390 (n = 5) and [18F] Fallypride (n = 10) calculated using SRTM. *P < 0.05 versus wild-type.
Lack of involvement of brain-derived neurotrophic factor and dopaminergic degeneration in the striatal phenotype of dynein mutant mice
The defect mediated by the dynein mutation in the striatum could be either intrinsic to the MSN or be triggered by extrinsic abnormalities in striatal afferences or efferences. Given that dynactin mutations have been associated with the Parkinson-like disease Perry syndrome (10), we hypothesized that the dynein mutation could lead to the degeneration of substantia nigra dopaminergic neurons. However, stereological assessments of tyrosine hydroxylase (TH) positive neuronal cells revealed similar numbers in the wild-type and Cra/+ substantia nigra pars compacta (Supplementary Material, Fig. S2) and TH staining of dynein mutant striatum appeared normal (data not shown), demonstrating that striatal atrophy is not a consequence of dopaminergic degeneration. Next we hypothesized that trophic factor deprivation of striatal neurons led to striatal atrophy. Brain-derived neurotrophic factor (BDNF), the major trophic factor for this neuronal population, is produced in the cortex and anterogradely transported to the striatum. Cortical BDNF ablation leads to a roughly similar striatal atrophy and behavioural impairment than the dynein mutation (34,35). At the mRNA level, BDNF expression was not decreased, but increased at 6 months of age in the cortex of Cra/+ mice (Supplementary Material, Fig. S3A). BDNF levels appeared normal in the striatum of these mice using ELISA (Supplementary Material, Fig. S3B), suggesting that BDNF was appropriately produced and targeted to the striatum. The dynein mutation could also lead to a decreased cellular response of dynein mutant neurons to BDNF exposure. Indeed, exocytosis and endocytosis, which are dynein-dependent events, appear indispensable for BDNF signal transduction (36,37). A major target of BDNF is increased DARPP-32 transcription (38) and major transcriptional responses occur 3–4 h after exposure to BDNF (39). DARPP-32 induction appeared similar between +/+, Cra/+ and Cra/Cra primary striatal neurons treated with BDNF during 4 h (Supplementary Material, Fig. S3C), suggesting that the dynein mutation did not massively impair the BDNF response of these neurons. In all, neither dopamine nor BDNF deprivation appeared to be a primary cause of dynein mutant striatal phenotype.
Cell autonomous defect in dynein mutant striatal neurons
To determine whether dynein mutant striatal neurons were intrinsically abnormal, we first studied their survival in vitro. Cultured embryonic striatal neurons of +/+, Cra/+ and Cra/Cra embryos showed similar survival as assessed by MTT assays (Fig. 6A), by cell counting (Fig. 6B) and by DNA (Fig. 6C) or RNA (Fig. 6D) yields of cultured neurons. Moreover, gene expression of neuronal markers such as neurofilament subunits NF-H and NF-M or DARPP-32 was identical between all three genotypes (Fig. 6E). Despite normal survival in culture, dynein mutant striatal neurons showed a profoundly abnormal dendritic arborization, even if bearing a single mutant allele, as revealed using β3-tubulin (neurites) or microtubule associated protein 2 (MAP2) (somatodendritic) immunostaining (Fig. 6F). Quantification revealed a reduction in the complexity of neuritic arborization of dynein mutant striatal neurons that were observed in Cra/+ neurons and even more pronounced in Cra/Cra neurons (Fig. 6G–H). The length of the longest individual MAP2 positive neurite was dramatically decreased by the presence of the dynein mutation (Fig. 6G). Thus, the dynein mutation led to dramatically impaired neuritic arborization in cultured neurons while sparing survival of these cells, suggesting that this dysfunction caused the overall pathological phenotype.
Figure 6.

Dramatic defect in dendritic morphology of cultured dynein mutant striatal neurons. (A and B) Cell survival of primary striata neuronal culture from wild-type embryo (+/+), heterozygous Cra/+ embryo and homozygous Cra/Cra embryo after 7 days in culture as assessed using MTT reduction assay (A) or direct counting of nuclei (B). (C and D) DNA (C) and RNA (D) content of primary striata neuronal culture from wild-type embryo (+/+), heterozygous Cra/+ embryo and homozygous Cra/Cra embryo after 7 days in culture. (E) mRNA levels of neurofilament heavy (NF-H) or medium (NF-M) subunits and of DARPP-32 in primary striata neuronal culture from wild-type embryo (+/+), heterozygous Cra/+ embryo and homozygous Cra/Cra embryo after 7 days in culture. (F) Representative microphotographs of MAP2 and β3-tubulin immunostaining of primary striata neuronal culture from wild-type embryo (+/+), heterozygous Cra/+ embryo and homozygous Cra/Cra embryo. (G) Length of longest neurite of individual striatal cells stained by MAP2 antibody from wild-type embryo (+/+), heterozygous Cra/+ embryo and homozygous Cra/Cra embryo. Total number of cells analysed: +/+108 cells, Cra/+162 cells and Cra/Cra 81 cells, **P < 0.01 versus wild-type (ANOVA followed by Newman–Keuls post hoc test). (H) Quantification of the number of MAP2 immunopositive primary, secondary and tertiary neurites from wild-type embryo (+/+), heterozygous Cra/+ embryo and homozygous Cra/Cra embryo. Total number of cells analysed: +/+68 cells, Cra/+126 cells and Cra/Cra 61 cells. **P < 0.01 versus wild-type, ***P < 0.001 versus wild-type (ANOVA followed by Newman–Keuls post hoc test).
DISCUSSION
This study provides evidence for the in vivo requirement of dynein in the function of the striatum and indicates that this occurs through involvement of cytoplasmic dynein in neuritic arborization. The consequences of these findings are important for neurodegenerative diseases affecting the basal ganglia.
Cra/+ mice suffer from motor and behavioural disturbances through their whole lifespan. we and others have previously shown the occurrence of an early and relatively mild proprioceptive neuropathy in Cra/+ mice (26–28), but this limited phenotype does not explain the overall behavioural and locomotor abnormalities observed here. First, muscle weakness is usually observed in patients presenting motor neuropathy, but not in patients affected by a pure sensory neuropathy (40) as in Cra/+ mice. Second, behavioural abnormalities, including limb clasping and hyperactivity, rather suggest a central involvement and do not comply with a pure sensory neuropathy. These behavioural abnormalities are suggestive of striatal involvement. Indeed, the early phase of the pathology in mouse models of HD is characterized by an increase in spontaneous motor activity (29–31,41). Similarly, transgenic mice lacking cortical BDNF or conditionally ablated for BDNF at adulthood display hindlimb and forelimb clasping (34,35), and mice deficient in PGC-1α, a transcriptional coactivator involved in the regulation of energy metabolism, also present with limb clasping and hyperactivity (42). Importantly, all these transgenic animals display a clear-cut lesion in the striatum. This is particularly interesting, because the genetic ablation of D1 dopamine receptor expressing cells (that is, within the striatum) yields a motor phenotype very similar to that observed in mutant dynein mice (32). In all, the selective down-regulation of the expression of D1 dopamine receptors, together with striatal atrophy, enlargement of lateral ventricles, decreased binding to either D1 or D2 dopamine receptors and the prominent astrocytosis that we found in the striatum of Cra/+ mice suggest that a striatal dysfunction can lead, at least in part, to the stereotyped motor disturbances in these mice.
Gene expression of D1 receptor, as well as binding potential of [11C] SCH-23390, was decreased in Cra/+ striatum. In contrast, despite the binding potential of D2 receptor was decreased, its gene expression was unchanged. There are several potential explanations to this discrepancy. First, the binding potential calculated from the PET analysis is a reflection of both the density of available receptor sides and the apparent ligand affinity (43). Since apparent ligand affinity is decreased by competition with the natural ligand, dopamine, an increase in striatal dopamine could, in principle, explain a decreased binding potential observed with PET. This explanation is, however, unlikely in our case because [18F] Fallypride is a high-affinity antagonist radioligand, and therefore less sensitive to changes in synaptic dopamine than agonist radioligands (43). Second, our stereological assessment of substantia nigra suggests that striatal dopaminergic innervation is rather normal in Cra/+ striatum. Thus, the lower D2 binding potential observed in Cra/+ mice is most likely due to a reduction of D2 receptor binding sites rather than to increased striatal dopamine. Such a decrease might be explained by defects in the various trafficking events that modulate cell surface expression of D2 receptor (44–47). Indeed, given the involvement of dynein in a number of cellular trafficking events, it cannot be excluded that the dynein mutation might directly impair D2 receptor trafficking.
Intriguingly, the behavioural phenotype of Cra/+ mice, as well as decreased D1 and D2 ligand binding and striatal atrophy, appeared between 3 and 4 months of age, while astrocytosis and transcriptional repression of D1 receptors were detectable later, after 8 months. The fact that behavioural abnormalities precede marked histopathological and biochemical changes in the striatum is not without precedent. For instance, previous studies reported that motor dysfunction in huntingtin knock-in mice occurred long before any clear signs of striatal lesions (48). We did not detect decreased DARPP-32 neuronal counts showing that the striatal phenotype was not associated with neurodegeneration, but most probably with dendritic atrophy, as suggested by our in vitro data. Our results, however, do not exclude the occurrence of a very slow and subtle process of striatal neurodegeneration that would be difficult to detect. Astrocytosis itself might be an astrocyte-autonomous event. Dynein expression and function in astrocytes has been poorly characterized and the elucidation of astrocytic dynein to the phenotype of dynein mutant mice will require the generation of conditional knock-out mice.
Our cell culture experiments show the existence of an intrinsic and massive defect in neuritic morphology of dynein mutant striatal neurons. The defect in neurite outgrowth of dynein mutant neurons is fully consistent with the recently reported critical requirement of dynein itself for dendrite outgrowth in drosophila neurons (49,50) and of the dynein-interacting protein LIS1 in dendrite morphology (51,52). Our data provide further evidence for such an involvement in mammals. The underlying molecular mechanisms remain unclear but might include perturbations of endosomal trafficking as suggested by the requirement for dynein in endosomal trafficking (53,54) and the requirement of endosomal trafficking in dendritic arborization (50,55). Interestingly, such a defect has not been noted by previous investigators that had performed dynein mutant motor neuron cultures (24,56), suggesting that motor neurons of dynein mutant mice were preserved comparatively to striatal neurons. This is in line with very recent transgenic experiments of adult BDNF deprivation that showed a specific sensitivity of MSNs to defects in dendrite morphogenesis when compared with other types of neurons (35). Furthermore, Gertler et al. (57) showed that D1 MSNs displayed more primary dendrites than D2 MSNs, suggesting a rationale for the more profound D1 involvement in the striatal pathology of Cra/+ mice. In all, our results suggest that the dynein mutation impairs dendritic arborization, and that the pathological consequences are more prominent in the striatum, likely because of a hugely developed dendritic arbour of MSNs.
Previous work in cultured cells had involved dynein and dynactin in different basal ganglia diseases, notably in HD and Perry syndrome. Indeed, huntingtin and its associated protein HAP1 bind to dynein directly, and indirectly, through dynactin subunit P150Glued. Importantly, the disease-causing protein in HD, mutant huntingtin, disrupts these interactions and decreases dynein function (21). Dynactin is also required for huntingtin-dependent transport of BDNF vesicles (22). These results suggested that dynein might be of importance for the survival and/or functioning of striatal neurons. However, most evidence to date of an in vivo role for dynein in neurons were based either on overexpression of the dynamitin subunit of dynactin or on overexpression of mutant forms of P150Glued, and mutant huntingtin affects not only retrograde but also anterograde axonal transport (23). Since dynactin and huntingtin have a number of dynein-independent functions, our results provide a direct genetic evidence for a pathophysiological role of dynein in striatal neuron health. Interestingly, crossbreeding dynein mutant mice with an animal model of HD sharply exacerbated the HD-like pathology (58). Most importantly, we observed that dynein mutant mice exhibited other HD-related peripheral phenotypes (59), including increased adiposity and impaired brown adipose thermogenesis (our unpublished data), thus strengthening the analogy between HD animal models and dynein mutant mice.
In summary, our findings provide direct evidence of the involvement of the axonal transport machinery, notably dynein, in the maintenance of striatal function and may have major implications for our understanding of basal ganglia diseases, such as HD and Perry syndrome.
MATERIALS AND METHODS
Animals
Heterozygous Cra/+ mice were obtained from Ingenium Pharmaceuticals AG, Martinsried, Germany. They were identified by tail DNA genotyping as previously described (24). Animals were maintained in a temperature- and humidity-controlled environment on a 12 h light/12 h dark cycle, and received food and water ad libitum. For the histological analysis, animals were deeply anaesthetized with 1 mg/kg body weight ketamine chlorhydrate and 0.5 mg/kg body weight xylazine, and transcardially perfused with 4% paraformaldehyde in 0.1 m pH 7.4 phosphate buffer. Tissues were then quickly dissected, post-fixed for 24 h in 4% paraformaldehyde and cryoprotected for 48 h with 30% sucrose in PBS before cryostat sectioning. For the biochemical analysis, animals were sacrificed and tissues were quickly dissected, snap frozen in liquid nitrogen and stored at −80°C until use. Animal manipulation followed current EU regulations and was performed under the supervision of authorized investigators.
Real-time RT–PCR
Total RNA was extracted using Trizol (Invitrogen, Cergy-Pontoise, France) according to the manufacturer's instructions. cDNA synthesis was performed using 1 µg of total RNA (iScript cDNA Synthesis kit; Bio-Rad, Marne La Coquette, France). PCR analysis was carried out as described (27) on a Bio-Rad CFX96 System using iQSYBR Green Supermix. A specific standard curve was performed for each gene in parallel, and each sample was quantified in duplicate. PCR conditions were 3 min at 94°C, followed by 40 cycles of 45 s at 94°C and 10 s at 60°C. Data were analysed using the iCycler software, and normalized to the reference genes encoding either the 18S ribosomal subunit and the RNA polymerase II mRNA. The primer pairs used are provided in Supplementary Material, Table S1.
Testing of motor performance and behaviour
Muscle grip strength was measured using a Bioseb gripmeter (Vitrolles, France) on forelimbs and all limbs. Each assay was performed in triplicate and measurements were averaged. For the behavioural analysis, C3H Cra/+ females were crossed with Bl6 males in order to compare with a group of mutant SOD1 (G93A) mice in mixed C3H/Bl6 background analysed in parallel (60). One week before the start of the tests, animals were brought to the behavioural analysis facility; single caged and handled every day. Only male mice were used for the tests. The rotarod test was used to assess motor coordination and balance. Mice had to keep their balance on a rotating rod at a continuous acceleration from 4 to 40 r.p.m. for 300 s (Rotarod Version 1.2.0. MED Associates Inc., St Albans, VT, USA). The time (or latency) it took the mouse to fall off the rod was measured. Each mouse had to perform three trials separated by 15 min each other, and the three trials were averaged. To identify differences in locomotor activity and exploratory behaviour, mice were tested in the open field. In this test, animals were placed at the border of a square arena (50 cm × 50 cm) and allowed to explore the arena freely for 30 min. The arena was divided into three parts, including a border zone (within 8 cm of the wall), a centre zone (inner square of 20 cm × 20 cm) and an intermediate zone. Locomotor activity was assessed by the total distance moved and the average velocity. To determine the exploratory behaviour, the number of rearings within the 30 min was measured. To assess anxiety, mice were evaluated using an elevated plus maze paradigm. The maze was elevated 92 cm above the floor, and consisted of four arms of 30 × 5 cm each, including two opposite ‘closed’ arms surrounded by dark walls and two opposite ‘open’ arms exposed without any walls. The centre of the maze was a 5 × 5 cm common area. Each mouse was placed for a single trial at the centre of the maze facing a closed arm, and allowed to explore the maze freely for a period of 5 min. The amount of time (in seconds) spent in both the open arms and closed arms was recorded. To measure reference learning (acquisition) and memory (retention), the Morris water maze was performed. The device consisted of a circular pool of 120 cm diameter, filled with water (25–27°C), which was made opaque by adding 2 l of milk. The pool was divided into four equal-sized quadrants and was surrounded by grey curtains covered with various visual cues, which helped the mice orient their location in the pool. A 10 cm platform was placed in the quadrant A, such that the platform was 1 cm below the water surface and visually indiscernible to the animals. On each trial, mice were allowed to swim for a maximum of 60 s and were released from four different defined positions. If the animal failed to discover the location of the platform in 60 s, it was guided to the platform and then allowed to stay for 30 s. After removal, mice were placed under an infrared lamp and allowed to warm up and dry off. The test was divided into two phases, an acquisition phase (18 trails, 6 per day), followed by a reversal phase during which the platform was moved to the opposite quadrant (12 trails, 6 per day). Escape latency and swum distance were analysed during both acquisition and reversal learning, and two successive trials were averaged into one block. All paths were tracked and analysed with an electronic imaging system (Viewer 2.2.0.55, BIOBSERVE GmbH, Bonn, Germany) at a frequency of 15 Hz and a spatial resolution of 720 × 576 pixels.
Assessment of the striatal and lateral ventricle volume by MRI
MRI was performed in the In-vivo-Imaging Laboratory at the research site of Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany. MRI data were acquired on a Bruker Biospec 47/40 scanner (Bruker BioSpin, Ettlingen, Germany) at 4.7 T (200 MHz proton resonance frequency). Age- and gender-matched Cra/+ and wild-type animals were used (n = 20). Acquisitions were performed at 5 and 10 months of age, respectively. Mice were anaesthetized through continuous inhalation of 1.2–1.5% isoflurane (in 70:30 N2O:O2) and fixed in a stereotactic head holder. For the anatomical analysis of the mouse brain, contiguous sets of six horizontal T2-weighted images were acquired using a RARE sequence. Imaging parameters were: TR 2500 ms, TE 12.5 ms, TEeff 50 ms, slice thickness 600 µm (no gap), FOV 28.1 × 25.6 mm, matrix size 256 × 256, RARE factor 8, six averages. Data processing was performed by the in-house developed software package Tissue Classification Software (TCS). For good visualization, the BRUKER data were transformed into a 768 × 768 grid (nearest neighbour interpolation, no filtering). TCS includes mouse-based drawing tools for tissue/voxel selection. In order to define clearly visible regions, drawing was simplified by a two-level thresholded conventional region-grow algorithm. Following the operator-defined intensity threshold, all connected voxels with respect to their intensity within the predefined range were selected. The analysis was blinded, evaluated by the same experienced investigator. The striatum was identified in four consecutive horizontal slices when compared with the Mouse Brain Library (http://www.mbl.org/mbl_main/atlas.html) between Bregma: −3.24; Interaural: 6.76 and Bregma: −5.04; Interaural: 4.96. The lateral ventricles were identified in the same horizontal sections by semi-automated region growth.
Analysis of brain astrocytosis
Brain sections comprising the anterior part of the caudate nucleus and the putamen were cut on a vibratome at a thickness of 50 µm (Leica Microsystems, Wetzlar, Germany) and were stained by indirect immunofluorescence, using an antibody directed against the specific astrocyte marker GFAP (Santa Cruz Biotechnology, Heidelberg, Germany) following the manufacturer's instructions. Quantitative analysis of immunoreactivity was performed using ImageJ.
Stereological analysis of DARPP-32 and TH-positive neurons
Coronal sections were cut in six series at a thickness of 35 µm throughout the brains using a freezing microtome. One series of free-floating brain sections were processed for immunohistochemistry either with a primary rabbit antibody against DARPP-32 (1:1000, Chemicon, AB 1656) or TH (1:1000, Pel-freez) (61). Stereological estimations of the total number of DARPP-32 positive neurons in the striatum or TH-positive neurons in the substantia nigra pars compacta were performed unilaterally on blind-coded slides with the Computer Assisted Toolbox Software (New CAST) module in VIS software (Visiopharm, Horsholm, Denmark) by applying the optical fractionator principle (62).
Micropet studies
Five wild-type and five Cra/+ mice were used for [11C] SCH-23390 PET imaging. Mice were anaesthetized with 1.5% isoflurane vaporized in 1.0 l/min oxygen gas (Vetland, Anesthesia Systems, Louisville, KY, USA). Two mice were placed head to head in the centre of field of view of a dedicated small animal PET scanner (Siemens Preclinical Solutions, Knoxville, TN, USA) yielding a spatial resolution of ∼1.4 mm in the reconstructed images. A 60 min emission scan was performed starting with bolus injection of [11C] SCH-23390 (12 ± 1 MBq) via a tail vein catheter. A heating mat was used to keep the animal warmed over the whole scan time. List-mode data were histogrammed into 64 time frames (30 × 10 s, 10 × 30 s, 10 × 60 s, 10 × 120 s, 4 × 300 s) and reconstructed using OSEM2D. To reveal quantitative images, dead-time correction, decay correction, normalization and attenuation correction were applied to all data sets. Two rectangular regions of interest (ROIs) for the left and the right striata of a size of 1 × 1 mm2 as well as for the cerebellum (3 × 1.5 mm2), which was used as reference region, were applied to all data sets. The ROIs were placed on two constitutive transversal slices of the striatum and the cerebellum. Time activity curves for the quantitative analysis were generated using PMOD software 3.0 (PMOD Technologies, Zurich, Switzerland). The simplified reference tissue model (63) was used to calculate the binding potential (BPND) of [11C] SCH-23390.
For [18F] Fallypride PET measurements, 10 animals of each group (Cra/+ and wild-type) were randomly selected at 10 months of age. Animals were anaesthetized through continuous inhalation of 1.2–1.5% isoflurane (in 70:30 N2O:O2). PET imaging was performed on a Siemens Inveon PET/CT system (Siemens Preclinical Solutions) using the D2/3 receptor ligand [18F] Fallypride (33,64). After bolus injection of radioactively labelled Fallypride (average activity 11.7 ± 2.0 MBq) through a tail vein catheter, PET data were continuously acquired for 60 min. Afterwards, list mode data were histogrammed into 21 frames (3 × 20 s, 4 × 60 s, 3 × 120 s, 3 × 180 s, 8 × 300 s) and reconstructed using a filtered back-projection algorithm without scatter correction. Quantitative analysis was assessed by SMRT implemented on the Inveon Research Workplace (IRW) software package (Siemens Molecular Imaging, Erlangen, Germany). ROIs were defined on co-registered PET/CT images using IRW.
Striatal neuron culture
Primary striatal neurons were cultured from E15 embryos issued from a cross between two heterozygous Cra/+ mice. Neurons from each embryo were cultured separately and genotype was determined post hoc by DNA genotyping of remaining embryos tissues. After enzymatic and mechanical dissociation, primary striatal neurons were plated at a density of 2.105 cells/ml on 0.1 mg/ml polyornithine pre-coated culture dishes and grown at 37°C in a humidified atmosphere (5% CO2/95% air). Culture media were composed of Neurobasal defined medium supplemented with B27, glutamax (2 mm), KCl (25 mm) and gentamycin (50 µg/ml). Primary neurons were cultured for 7 days, and the culture media were changed every 2 days. BDNF treatment (Tocris Bioscience, Ellisville, MO, USA) was performed the seventh day at the indicated concentration for 4 h.
For immunocytochemistry, primary striatal neurons were post-fixed with 4% paraformaldehyde in PBS pH 7.4 and were permeabilized for 30 min in 1% Triton X-100/PBS. Primary antibodies anti-MAP-2 and β3 Tubulin (Millipore, Molsheim, France) were applied overnight, and Rhodamine-conjugated secondary antibody (Jackson Immunoresearch Laboratories, West Grove, PA, USA) was applied for 1 h. Cover slips were mounted in Mowiol solution for observation under a conventional epifluorescence microscope (Nikon, Tokyo, Japan) and pictures were analysed with ImageJ software.
MTT assays
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich, Lyon, France) tests were done with primary striatal neurons cultured in 96-well culture dishes (Costar, NY, USA). Briefly, MTT was dissolved in PBS (pH 7.2) to obtain a concentration of 5 mg/ml, then diluted 10 times in freshly prepared culture medium and the plates were incubated for 1 h at 37°C. Medium was then removed and dark blue crystals formed during reaction were dissolved by adding 100 µl/well of 0.04 m HCl in isopropyl alcohol. Plates were read on a Bio-Rad 680 micro-ELISA plate-reader, using a wavelength of 490 nm. Results are given in percentage of the +/+ values.
Cell counting
Cultured embryonic striatal neurons' survival was evaluated after nuclei staining with Hoechst 33342 (Sigma-Aldrich). Hoescht solution was dissolved in 100% methanol to obtain a final concentration of 0.5 mg/ml. After paraformaldehyde fixation, cultured embryonic striatal neurons were incubated 30 min with 0.5 µg/ml Hoechst 33342 in 0.1% Triton X-100/PBS and then washed three times in 0.1% Triton X-100/PBS. A total of five embryos of each genotype were analysed and six fields per embryo were randomly photographed on a Nikon fluorescence microscope. The total number of nuclei per field was count. Results represented the average number of nuclei per field.
BDNF ELISA
BDNF ELISA (Promega, Charbonnières-les-Bains, France) was performed as described by the provider.
Statistical analysis
Data are expressed as the mean ± SEM. Statistical analysis was accomplished using non-parametric Student's t-test or ANOVA followed by Newman–Keuls multiple comparisons test (PRISM Version 4.0b; GraphPad, San Diego, CA, USA). Differences at P < 0.05 were considered significant.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants from Fondation pour la Recherche Médicale, Association pour la Recherche sur la Sclérose Latérale Amyotrophique (ARS) and Amyotrophic Lateral Sclerosis Association (grant 1698) to L.D.; and Association Française contre les Myopathies, ARS and Association pour la Recherche et le Développement de Moyens de Lutte contre les Maladies Neurodégénératives to J.-P.L. and by Deutsche Forschungsgemeinschaft (KFO142) to A.C.L. J.L.G.D.A. is the recipient of chaire INSERM/Université de Strasbourg; K.R.-V. held an EFNS Scientific Fellowship at the University of Ulm. Å.P. is supported by grants from the Swedish Research Council.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge the skilful technical assistance of Marie-José Ruivo, Annie Picchinenna, Lise Nuss and Caroline Mursch. Pr Ehret helped for behavioural studies. David Kind and Michael Neumaier provided help with MRI and PET imaging. We thank the radiopharmacy group of the University Hospital Tübingen for synthesizing the 11C tracer.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Morfini G.A., Burns M., Binder L.I., Kanaan N.M., LaPointe N., Bosco D.A., Brown R.H., Jr, Brown H., Tiwari A., Hayward L., et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. doi:10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chevalier-Larsen E., Holzbaur E.L. Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta. 2006;1762:1094–1108. doi: 10.1016/j.bbadis.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Levy J.R., Holzbaur E.L. Cytoplasmic dynein/dynactin function and dysfunction in motor neurons. Int. J. Dev. Neurosci. 2006;24:103–111. doi: 10.1016/j.ijdevneu.2005.11.013. doi:10.1016/j.ijdevneu.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 4.LaMonte B.H., Wallace K.E., Holloway B.A., Shelly S.S., Ascano J., Tokito M., Van Winkle T., Howland D.S., Holzbaur E.L. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. doi:10.1016/S0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 5.Teuling E., van Dis V., Wulf P.S., Haasdijk E.D., Akhmanova A., Hoogenraad C.C., Jaarsma D. A novel mouse model with impaired dynein/dynactin function develops amyotrophic lateral sclerosis (ALS)-like features in motor neurons and improves lifespan in SOD1-ALS mice. Hum. Mol. Genet. 2008;17:2849–2862. doi: 10.1093/hmg/ddn182. doi:10.1093/hmg/ddn182. [DOI] [PubMed] [Google Scholar]
- 6.Puls I., Jonnakuty C., LaMonte B.H., Holzbaur E.L., Tokito M., Mann E., Floeter M.K., Bidus K., Drayna D., Oh S.J., et al. Mutant dynactin in motor neuron disease. Nat. Genet. 2003;33:455–456. doi: 10.1038/ng1123. doi:10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 7.Munch C., Sedlmeier R., Meyer T., Homberg V., Sperfeld A.D., Kurt A., Prudlo J., Peraus G., Hanemann C.O., Stumm G., et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63:724–726. doi: 10.1212/01.wnl.0000134608.83927.b1. [DOI] [PubMed] [Google Scholar]
- 8.Puls I., Oh S.J., Sumner C.J., Wallace K.E., Floeter M.K., Mann E.A., Kennedy W.R., Wendelschafer-Crabb G., Vortmeyer A., Powers R., et al. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann. Neurol. 2005;57:687–694. doi: 10.1002/ana.20468. doi:10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wider C., Dachsel J.C., Farrer M.J., Dickson D.W., Tsuboi Y., Wszolek Z.K. Elucidating the genetics and pathology of Perry syndrome. J. Neurol. Sci. 289:149–154. doi: 10.1016/j.jns.2009.08.044. doi:10.1016/j.jns.2009.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farrer M.J., Hulihan M.M., Kachergus J.M., Dachsel J.C., Stoessl A.J., Grantier L.L., Calne S., Calne D.B., Lechevalier B., Chapon F., et al. DCTN1 mutations in Perry syndrome. Nat. Genet. 2009;41:163–165. doi: 10.1038/ng.293. doi:10.1038/ng.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levy J.R., Sumner C.J., Caviston J.P., Tokito M.K., Ranganathan S., Ligon L.A., Wallace K.E., LaMonte B.H., Harmison G.G., Puls I., et al. A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J. Cell Biol. 2006;172:733–745. doi: 10.1083/jcb.200511068. doi:10.1083/jcb.200511068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laird F.M., Farah M.H., Ackerley S., Hoke A., Maragakis N., Rothstein J.D., Griffin J., Price D.L., Martin L.J., Wong P.C. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J. Neurosci. 2008;28:1997–2005. doi: 10.1523/JNEUROSCI.4231-07.2008. doi:10.1523/JNEUROSCI.4231-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chevalier-Larsen E.S., Wallace K.E., Pennise C.R., Holzbaur E.L. Lysosomal proliferation and distal degeneration in motor neurons expressing the G59S mutation in the p150Glued subunit of dynactin. Hum. Mol. Genet. 2008;17:1946–1955. doi: 10.1093/hmg/ddn092. doi:10.1093/hmg/ddn092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai C., Lin X., Chandran J., Shim H., Yang W.J., Cai H. The G59S mutation in p150(glued) causes dysfunction of dynactin in mice. J. Neurosci. 2007;27:13982–13990. doi: 10.1523/JNEUROSCI.4226-07.2007. doi:10.1523/JNEUROSCI.4226-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berezuk M.A., Schroer T.A. Dynactin enhances the processivity of kinesin-2. Traffic. 2007;8:124–129. doi: 10.1111/j.1600-0854.2006.00517.x. doi:10.1111/j.1600-0854.2006.00517.x. [DOI] [PubMed] [Google Scholar]
- 16.King S.J., Schroer T.A. Dynactin increases the processivity of the cytoplasmic dynein motor. Nat. Cell Biol. 2000;2:20–24. doi: 10.1038/71338. [DOI] [PubMed] [Google Scholar]
- 17.Lee S.J., Chae C., Wang M.M. p150/glued modifies nuclear estrogen receptor function. Mol. Endocrinol. 2009;23:620–629. doi: 10.1210/me.2007-0477. doi:10.1210/me.2007-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shrum C.K., Defrancisco D., Meffert M.K. Stimulated nuclear translocation of NF-kappaB and shuttling differentially depend on dynein and the dynactin complex. Proc. Natl Acad. Sci. USA. 2009;106:2647–2652. doi: 10.1073/pnas.0806677106. doi:10.1073/pnas.0806677106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li S.H., Gutekunst C.A., Hersch S.M., Li X.J. Interaction of huntingtin-associated protein with dynactin P150Glued. J. Neurosci. 1998;18:1261–1269. doi: 10.1523/JNEUROSCI.18-04-01261.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X.J., Li S.H., Sharp A.H., Nucifora F.C., Jr, Schilling G., Lanahan A., Worley P., Snyder S.H., Ross C.A. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995;378:398–402. doi: 10.1038/378398a0. doi:10.1038/378398a0. [DOI] [PubMed] [Google Scholar]
- 21.Caviston J.P., Ross J.L., Antony S.M., Tokito M., Holzbaur E.L. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl Acad. Sci. USA. 2007;104:10045–10050. doi: 10.1073/pnas.0610628104. doi:10.1073/pnas.0610628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gauthier L.R., Charrin B.C., Borrell-Pages M., Dompierre J.P., Rangone H., Cordelieres F.P., De Mey J., MacDonald M.E., Lessmann V., Humbert S., et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. doi:10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 23.Morfini G.A., You Y.M., Pollema S.L., Kaminska A., Liu K., Yoshioka K., Bjorkblom B., Coffey E.T., Bagnato C., Han D., et al. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 2009;12:864–871. doi: 10.1038/nn.2346. doi:10.1038/nn.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hafezparast M., Klocke R., Ruhrberg C., Marquardt A., Ahmad-Annuar A., Bowen S., Lalli G., Witherden A.S., Hummerich H., Nicholson S., et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. doi:10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- 25.Perlson E., Jeong G.B., Ross J.L., Dixit R., Wallace K.E., Kalb R.G., Holzbaur E.L. A switch in retrograde signaling from survival to stress in rapid-onset neurodegeneration. J. Neurosci. 2009;29:9903–9917. doi: 10.1523/JNEUROSCI.0813-09.2009. doi:10.1523/JNEUROSCI.0813-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ilieva H.S., Yamanaka K., Malkmus S., Kakinohana O., Yaksh T., Marsala M., Cleveland D.W. Mutant dynein (Loa) triggers proprioceptive axon loss that extends survival only in the SOD1 ALS model with highest motor neuron death. Proc. Natl Acad. Sci. USA. 2008;105:12599–12604. doi: 10.1073/pnas.0805422105. doi:10.1073/pnas.0805422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dupuis L., Fergani A., Braunstein K.E., Eschbach J., Holl N., Rene F., Gonzalez De Aguilar J.L., Zoerner B., Schwalenstocker B., Ludolph A.C., et al. Mice with a mutation in the dynein heavy chain 1 gene display sensory neuropathy but lack motor neuron disease. Exp. Neurol. 2009;215:146–152. doi: 10.1016/j.expneurol.2008.09.019. doi:10.1016/j.expneurol.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 28.Chen X.J., Levedakou E.N., Millen K.J., Wollmann R.L., Soliven B., Popko B. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic Dynein heavy chain 1 gene. J. Neurosci. 2007;27:14515–14524. doi: 10.1523/JNEUROSCI.4338-07.2007. doi:10.1523/JNEUROSCI.4338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luesse H.G., Schiefer J., Spruenken A., Puls C., Block F., Kosinski C.M. Evaluation of R6/2 HD transgenic mice for therapeutic studies in Huntington's disease: behavioral testing and impact of diabetes mellitus. Behav. Brain Res. 2001;126:185–195. doi: 10.1016/s0166-4328(01)00261-3. doi:10.1016/S0166-4328(01)00261-3. [DOI] [PubMed] [Google Scholar]
- 30.Schiefer J., Landwehrmeyer G.B., Luesse H.G., Sprunken A., Puls C., Milkereit A., Milkereit E., Kosinski C.M. Riluzole prolongs survival time and alters nuclear inclusion formation in a transgenic mouse model of Huntington's disease. Mov. Disord. 2002;17:748–757. doi: 10.1002/mds.10229. doi:10.1002/mds.10229. [DOI] [PubMed] [Google Scholar]
- 31.Bolivar V.J., Manley K., Messer A. Early exploratory behavior abnormalities in R6/1 Huntington's disease transgenic mice. Brain Res. 2004;1005:29–35. doi: 10.1016/j.brainres.2004.01.021. doi:10.1016/j.brainres.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 32.Gantois I., Fang K., Jiang L., Babovic D., Lawrence A.J., Ferreri V., Teper Y., Jupp B., Ziebell J., Morganti-Kossmann C.M., et al. Ablation of D1 dopamine receptor-expressing cells generates mice with seizures, dystonia, hyperactivity, and impaired oral behavior. Proc. Natl Acad. Sci. USA. 2007;104:4182–4187. doi: 10.1073/pnas.0611625104. doi:10.1073/pnas.0611625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siessmeier T., Zhou Y., Buchholz H.G., Landvogt C., Vernaleken I., Piel M., Schirrmacher R., Rosch F., Schreckenberger M., Wong D.F., et al. Parametric mapping of binding in human brain of D2 receptor ligands of different affinities. J. Nucl. Med. 2005;46:964–972. [PubMed] [Google Scholar]
- 34.Baquet Z.C., Gorski J.A., Jones K.R. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J. Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. doi:10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rauskolb S., Zagrebelsky M., Dreznjak A., Deogracias R., Matsumoto T., Wiese S., Erne B., Sendtner M., Schaeren-Wiemers N., Korte M., et al. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J. Neurosci. 2010;30:1739–1749. doi: 10.1523/JNEUROSCI.5100-09.2010. doi:10.1523/JNEUROSCI.5100-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santi S., Cappello S., Riccio M., Bergami M., Aicardi G., Schenk U., Matteoli M., Canossa M. Hippocampal neurons recycle BDNF for activity-dependent secretion and LTP maintenance. EMBO J. 2006;25:4372–4380. doi: 10.1038/sj.emboj.7601303. doi:10.1038/sj.emboj.7601303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou P., Porcionatto M., Pilapil M., Chen Y., Choi Y., Tolias K.F., Bikoff J.B., Hong E.J., Greenberg M.E., Segal R.A. Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron. 2007;55:53–68. doi: 10.1016/j.neuron.2007.05.030. doi:10.1016/j.neuron.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bogush A., Pedrini S., Pelta-Heller J., Chan T., Yang Q., Mao Z., Sluzas E., Gieringer T., Ehrlich M.E. AKT and CDK5/p35 mediate brain-derived neurotrophic factor induction of DARPP-32 in medium size spiny neurons in vitro. J. Biol. Chem. 2007;282:7352–7359. doi: 10.1074/jbc.M606508200. doi:10.1074/jbc.M606508200. [DOI] [PubMed] [Google Scholar]
- 39.Gokce O., Runne H., Kuhn A., Luthi-Carter R. Short-term striatal gene expression responses to brain-derived neurotrophic factor are dependent on MEK and ERK activation. PLoS ONE. 2009;4:e5292. doi: 10.1371/journal.pone.0005292. doi:10.1371/journal.pone.0005292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Auer-Grumbach M. Hereditary sensory neuropathy type I. Orphanet J. Rare Dis. 2008;3:7. doi: 10.1186/1750-1172-3-7. doi:10.1186/1750-1172-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reddy P.H., Williams M., Charles V., Garrett L., Pike-Buchanan L., Whetsell W.O., Jr, Miller G., Tagle D.A. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat. Genet. 1998;20:198–202. doi: 10.1038/2510. doi:10.1038/2510. [DOI] [PubMed] [Google Scholar]
- 42.Lin J., Wu P.H., Tarr P.T., Lindenberg K.S., St-Pierre J., Zhang C.Y., Mootha V.K., Jager S., Vianna C.R., Reznick R.M., et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. doi:10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 43.Laruelle M. Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J. Cereb. Blood Flow Metab. 2000;20:423–451. doi: 10.1097/00004647-200003000-00001. [DOI] [PubMed] [Google Scholar]
- 44.Xiao M.F., Xu J.C., Tereshchenko Y., Novak D., Schachner M., Kleene R. Neural cell adhesion molecule modulates dopaminergic signaling and behavior by regulating dopamine D2 receptor internalization. J. Neurosci. 2009;29:14752–14763. doi: 10.1523/JNEUROSCI.4860-09.2009. doi:10.1523/JNEUROSCI.4860-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Namkung Y., Dipace C., Urizar E., Javitch J.A., Sibley D.R. G protein-coupled receptor kinase-2 constitutively regulates D2 dopamine receptor expression and signaling independently of receptor phosphorylation. J. Biol. Chem. 2009;284:34103–34115. doi: 10.1074/jbc.M109.055707. doi:10.1074/jbc.M109.055707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tirotta E., Fontaine V., Picetti R., Lombardi M., Samad T.A., Oulad-Abdelghani M., Edwards R., Borrelli E. Signaling by dopamine regulates D2 receptors trafficking at the membrane. Cell Cycle. 2008;7:2241–2248. doi: 10.4161/cc.7.14.6307. [DOI] [PubMed] [Google Scholar]
- 47.Kim O.J., Ariano M.A., Namkung Y., Marinec P., Kim E., Han J., Sibley D.R. D2 dopamine receptor expression and trafficking is regulated through direct interactions with ZIP. J. Neurochem. 2008;106:83–95. doi: 10.1111/j.1471-4159.2008.05348.x. doi:10.1111/j.1471-4159.2008.05348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Menalled L.B., Sison J.D., Wu Y., Olivieri M., Li X.J., Li H., Zeitlin S., Chesselet M.F. Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington's disease knock-in mice. J. Neurosci. 2002;22:8266–8276. doi: 10.1523/JNEUROSCI.22-18-08266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng Y., Wildonger J., Ye B., Zhang Y., Kita A., Younger S.H., Zimmerman S., Jan L.Y., Jan Y.N. Dynein is required for polarized dendritic transport and uniform microtubule orientation in axons. Nat. Cell Biol. 2008;10:1172–1180. doi: 10.1038/ncb1777. doi:10.1038/ncb1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Satoh D., Sato D., Tsuyama T., Saito M., Ohkura H., Rolls M.M., Ishikawa F., Uemura T. Spatial control of branching within dendritic arbors by dynein-dependent transport of Rab5-endosomes. Nat. Cell Biol. 2008;10:1164–1171. doi: 10.1038/ncb1776. doi:10.1038/ncb1776. [DOI] [PubMed] [Google Scholar]
- 51.Fleck M.W., Hirotsune S., Gambello M.J., Phillips-Tansey E., Suares G., Mervis R.F., Wynshaw-Boris A., McBain C.J. Hippocampal abnormalities and enhanced excitability in a murine model of human lissencephaly. J. Neurosci. 2000;20:2439–2450. doi: 10.1523/JNEUROSCI.20-07-02439.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Z., Steward R., Luo L. Drosophila Lis1 is required for neuroblast proliferation, dendritic elaboration and axonal transport. Nat. Cell Biol. 2000;2:776–783. doi: 10.1038/35041011. [DOI] [PubMed] [Google Scholar]
- 53.Murray J.W., Wolkoff A.W. Roles of the cytoskeleton and motor proteins in endocytic sorting. Adv. Drug Deliv. Rev. 2003;55:1385–1403. doi: 10.1016/j.addr.2003.07.008. doi:10.1016/j.addr.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 54.Aniento F., Emans N., Griffiths G., Gruenberg J. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J. Cell Biol. 1993;123:1373–1387. doi: 10.1083/jcb.123.6.1373. doi:10.1083/jcb.123.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jan Y.N., Jan L.Y. Branching out: mechanisms of dendritic arborization. Nat. Rev. Neurosci. 2010;11:316–328. doi: 10.1038/nrn2836. doi:10.1038/nrn2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kieran D., Hafezparast M., Bohnert S., Dick J.R., Martin J., Schiavo G., Fisher E.M., Greensmith L. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J. Cell Biol. 2005;169:561–567. doi: 10.1083/jcb.200501085. doi:10.1083/jcb.200501085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gertler T.S., Chan C.S., Surmeier D.J. Dichotomous anatomical properties of adult striatal medium spiny neurons. J. Neurosci. 2008;28:10814–10824. doi: 10.1523/JNEUROSCI.2660-08.2008. doi:10.1523/JNEUROSCI.2660-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ravikumar B., Acevedo-Arozena A., Imarisio S., Berger Z., Vacher C., O'Kane C.J., Brown S.D., Rubinsztein D.C. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet. 2005;37:771–776. doi: 10.1038/ng1591. doi:10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- 59.Weydt P., Pineda V.V., Torrence A.E., Libby R.T., Satterfield T.F., Lazarowski E.R., Gilbert M.L., Morton G.J., Bammler T.K., Strand A.D., et al. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. doi:10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 60.Teuchert M., Fischer D., Schwalenstoecker B., Habisch H.J., Bockers T.M., Ludolph A.C. A dynein mutation attenuates motor neuron degeneration in SOD1(G93A) mice. Exp. Neurol. 2006;198:271–274. doi: 10.1016/j.expneurol.2005.12.005. doi:10.1016/j.expneurol.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 61.Bode F.J., Stephan M., Suhling H., Pabst R., Straub R.H., Raber K.A., Bonin M., Nguyen H.P., Riess O., Bauer A., et al. Sex differences in a transgenic rat model of Huntington's disease: decreased 17beta-estradiol levels correlate with reduced numbers of DARPP32+ neurons in males. Hum. Mol. Genet. 2008;17:2595–2609. doi: 10.1093/hmg/ddn159. doi:10.1093/hmg/ddn159. [DOI] [PubMed] [Google Scholar]
- 62.West M.J., Slomianka L., Gundersen H.J. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat. Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. doi:10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 63.Lammertsma A.A., Hume S.P. Simplified reference tissue model for PET receptor studies. Neuroimage. 1996;4:153–158. doi: 10.1006/nimg.1996.0066. doi:10.1006/nimg.1996.0066. [DOI] [PubMed] [Google Scholar]
- 64.Yakushev I., Hammers A., Fellgiebel A., Schmidtmann I., Scheurich A., Buchholz H.G., Peters J., Bartenstein P., Lieb K., Schreckenberger M. SPM-based count normalization provides excellent discrimination of mild Alzheimer's disease and amnestic mild cognitive impairment from healthy aging. Neuroimage. 2009;44:43–50. doi: 10.1016/j.neuroimage.2008.07.015. doi:10.1016/j.neuroimage.2008.07.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.