

Abstract
Neuronal ceroid lipofuscinoses (NCLs) constitute a group of progressive neurodegenerative disorders resulting from mutations in at least eight different genes. Mutations in the most recently identified NCL gene, MFSD8/CLN7, underlie a variant of late-infantile NCL (vLINCL). The MFSD8/CLN7 gene encodes a polytopic protein with unknown function, which shares homology with ion-coupled membrane transporters. In this study, we confirmed the lysosomal localization of the native CLN7 protein. This localization of CLN7 is not impaired by the presence of pathogenic missense mutations or after genetic ablation of the N-glycans. Expression of chimeric and full-length constructs showed that lysosomal targeting of CLN7 is mainly determined by an N-terminal dileucine motif, which specifically binds to the heterotetrameric adaptor AP-1 in vitro. We also show that CLN7 mRNA is more abundant in neurons than astrocytes and microglia, and that it is expressed throughout rat brain, with increased levels in the granular layer of cerebellum and hippocampal pyramidal cells. Interestingly, this cellular and regional distribution is in good agreement with the autofluorescent lysosomal storage and cell loss patterns found in brains from CLN7-defective patients. Overall, these data highlight lysosomes as the primary site of action for CLN7, and suggest that the pathophysiology underpinning CLN7-associated vLINCL is a cell-autonomous process.
INTRODUCTION
Neuronal ceroid lipofuscinoses (NCLs) comprise a group of genetically heterogeneous neurodegenerative disorders, with many similarities in clinical manifestation and disease pathology. The patients present with deterioration of cognitive and motor skills, epileptic seizures, progressive loss of vision, and premature death. To date, mutations in eight different genes have been shown to result in NCLs (1). Mutations in CLN7/MFSD8 (MIM 611124) underlie a variant form of late-infantile neuronal ceroid lipofuscinosis (vLINCL), originally identified in a subset of mainly Turkish patients (2). Since the identification of CLN7/MFSD8, a further 22 disease-causing mutations have been reported in patients of various ethnic origins, suggesting that the geographical distribution of defects in this gene is more widespread than originally anticipated (2–6). Electron microscopy has revealed an intralysosomal accumulation of membranous material with various profiles, including fingerprints, in the lysosomes of CLN7-defective patients (2,7).
CLN7/MFSD8 encodes the polytopic protein CLN7 with a suggested topology of 12 transmembrane domains, and both the N- and C-terminal tails are thought to be facing the cytosol. Based on sequence homology, CLN7 belongs to the major facilitator superfamily (MFS) of secondary active transporters. MFS transporters translocate small solutes across membranes, generally using chemiosmotic ion gradients (8). To date, 63 MFS families have been described (9), which are often characterized by similar types of substrates or a similar ion-coupling mechanism within a given family. The substrates transported include sugars, drugs, metabolites, amino acids, nucleosides and vitamins but not macromolecules (8). Both the function and the substrate specificity of CLN7 remain unknown.
NCL proteins are classified as soluble [CLN1/PPT1 (10), CLN2/TPP1 (11), CLN10/CTSD (12,13), CLN5 (14,15)] or transmembrane proteins [CLN3 (16), CLN6 (17,18), CLN8 (19)], localizing to the endoplasmic reticulum (ER) or to lysosomes. Although NCL proteins are ubiquitously expressed, the endogenous expression levels of many of them are relatively low, making determination of their intracellular localization difficult. This has resulted in controversial data and debate concerning the precise site of action of different NCL proteins (20). Like the majority of NCL proteins (excluding CLN6 and CLN8), overexpressed CLN7 has been shown to co-localize with lysosomal markers, and is therefore suggested to perform its transport function across the lysosomal membrane (2).
Targeting of lysosomal membrane proteins is typically mediated by short stretches of amino acid residues situated in their cytosolic domains (21). These sorting motifs belong to two major classes, the tyrosine-based and the dileucine-based motifs. Tyrosine-based signals conform to the consensus motif YXXΦ, in which Y is a tyrosine, X any amino acid and Φ a bulky hydrophobic amino acid. Dileucine-based motifs can be divided into two distinct classes, [DE]XXXL[IL] and DXXLL, which differ by the position of the acidic residues preceding the pair of leucines (or leucine–isoleucine) and are recognized by a distinct class of membrane coat adaptor proteins (21). In addition to typical tyrosine- and dileucine-based motifs, unconventional targeting motifs have also been reported to facilitate targeting of some lysosomal membrane proteins (22,23), including the lysosomal NCL protein CLN3 (24). Tyrosine-based and [DE]XXXL[IL]-type motifs are recognized by heterotetrameric adaptor protein (AP) complexes (AP-1, AP-2, AP-3, and AP-4), whereas DXXLL-type motifs are recognized by monomeric GGAs (Golgi-localized, gamma-ear containing, ARF-binding proteins) (21,25). As these adaptor proteins function at distinct sites of the exocytic and endocytic pathways, adaptor/motif binding events determine which trafficking route delivers membrane proteins to the lysosome. Lysosomal membrane proteins can be targeted from the trans-Golgi network either directly to early or late endosomes (direct route) or indirectly via the plasma membrane (indirect route).
In the present study, we investigated the expression and trafficking of CLN7. We show that the regional and cellular expression of CLN7 corresponds well with the pathological findings characteristic of the brains of vLINCL patients. Furthermore, we show that lysosomal sorting of CLN7 is mainly determined by an N-terminal dileucine motif. Mutation of this targeting motif leads to a significant misrouting of CLN7 to the plasma membrane, thus providing a tool to identify its transport function in future studies.
RESULTS
Native CLN7 localizes to lysosomes and late endosomes
To confirm that tagging of CLN7 does not interfere with its intracellular localization, an antibody was raised against a peptide derived from the mouse sequence and the distributions of EGFP-tagged and -untagged mouse CLN7 (mCLN7) were compared in transiently transfected HeLa cells. To facilitate subsequent experiments, human CLN7 (hCLN7) was also tagged with EGFP. Both EGFP-tagged mouse and human CLN7 extensively co-localized with the late endosomal/lysosomal marker lysosomal-associated membrane protein 1 (LAMP1) (Fig. 1A and B), in agreement with previous findings (2). When HeLa cells transiently expressing EGFP-mCLN7 were analyzed by immunofluorescence with our immunopurified anti-mCLN7 antibody (UBP119ip), extensive overlap was found between the EGFP and immunoreactivity signals (Fig. 1C), thus validating the selectivity of our antibody in this technique. Expression of the untagged mCLN7 construct in HeLa cells, followed by staining with our novel antibody also showed co-localization with LAMP1 (Fig. 1D). The lysosomal nature of mCLN7-immunopositive puncta was also demonstrated using the fluorogenic proteolysis probe DQ-Red-BSA, a highly conjugated protein substrate which undergoes dequenching upon digestion in endocytic compartments after fluid-phase endocytosis. When live transfected HeLa cells were incubated with this probe prior to immunofluorescence analysis, DQ-Red-BSA fluorescence appeared as intracellular puncta which all tested positive for untagged mCLN7 immunoreactivity (Fig. 1E).
Figure 1.

CLN7 localizes to lysosomes and late endosomes in transfected HeLa cells. EGFP-tagged human (A) and mouse CLN7 (B and C) or the untagged mouse protein (D and E) were transiently expressed in HeLa cells and analyzed by deconvolution fluorescence microscopy. Detection of EGFP tags (A–C) and of mCLN7 stained with an antibody termed UBP119ip (D and E) are shown on the left. Lysosomes are stained with LAMP1 (A, B, D) in the second column. In (C), the EGFP signal is compared with UBP119p staining. In (E), the proteolytic activity in the endocytic pathway was determined using the fluorogenic probe DQ-Red-BSA prior to cell fixation. Merged images and enlarged views of the boxed areas are shown in the third and fourth columns, respectively. Arrows indicate co-localization. In (E), some mCLN7-positive puncta were not stained with the DQ-RED-BSA probe (arrowheads). Scale bar, 20 µm.
We next examined whether endogenous CLN7 can also be detected with our antibody. As the anti-mCLN7 antibody showed poor sensitivity by immunoblotting (Supplementary Material, Fig. S1), subcellular fractionation was precluded to address this issue. Since CLN7 deficiency causes a neurological disease, we first asked which neural cell types express CLN7 using purified cell-type cultures from rat brain and real-time reverse transcriptase (RT)–PCR. For comparison, the transcript levels of four other NCL-related genes, CTSD, PPT1, TPP1 and CLN3, were also quantified. Interestingly, the mRNA level of CLN7 was 6- and 12-fold higher in rat neurons than in astrocytes and microglial cells, respectively, whereas neuronal expression did not predominate for CLN3 (neuron = microglia > astrocyte), PPT1 and TPP1 (neuron ≈ astrocyte > microglia) or CTSD (microglia > astrocyte ≈ neuron) (Fig. 2A). Comparison of transcript levels from distinct brain regions indicated that CLN7 mRNA is 5-fold more abundant in hippocampus than midbrain, cerebellum or striatum, while cerebral cortices showed an intermediate level (Fig. 2B).
Figure 2.
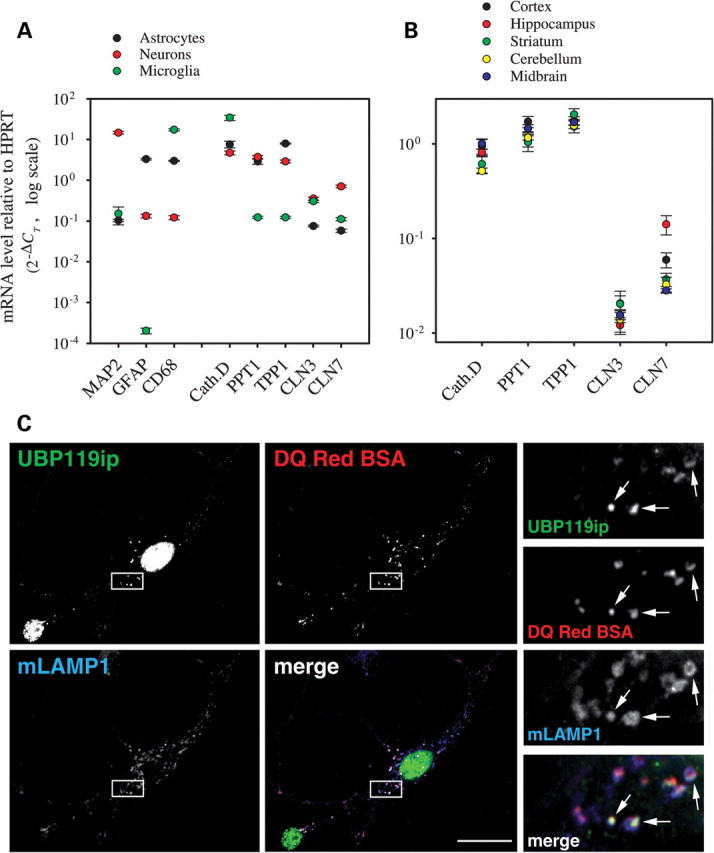
Expression and intracellular localization of native CLN7 in rodent neural cells. (A) mRNAs from five NCL-associated genes—Cathepsin D (Cath. D), Palmitoyl-Protein Thioesterase 1 (PPT1), Tripeptidyl Peptidase I (TPP1), CLN3 and CLN7—were quantified in rat neuronal, astroglial and microglial cultures using real-time RT–PCR. Microtubule Associated Protein 2 (MAP2), Glial Fibrillary Acidic Protein (GFAP) and CD68 transcripts were analyzed to assess the cell-type purity of the cultures (astrocytes were partially contaminated with microglial cells). Expression levels are expressed as the relative number of copies compared with the HPRT transcript using the comparative threshold cycle (CT) method. CLN7 mRNA was 12 and 6.4 times more abundant in neurons than in astrocytes and microglial cells, respectively. (B) NCL-associated transcripts were quantified in several rat brain regions using the same technique. CLN7 mRNA was 2.4- and 5-fold more abundant in hippocampus than in cortex and midbrain, respectively. (C) Mouse hippocampal neurons were incubated with the endolysosomal proteolysis probe DQ-Red-BSA, fixed, stained with mLAMP1 and UBP119ip (detecting mCLN7) and analyzed by deconvolution fluorescence microscopy. Two triple-stained neurons are shown. Enlarged views of the boxed area are shown on the right. Puncta stained with UBP119ip co-localize with mLAMP1 and DQ-Red-BSA-positive puncta (arrows) in the soma and proximal dendrites. The nuclear staining obtained with the UBP119ip antibody is due to a cross-reaction artifact.
We thus focused on primary cultures of mouse hippocampal neurons to determine the intracellular localization of endogenous CLN7. Unexpectedly, the UBP119ip antibody strongly labeled the nuclei of mouse hippocampal neurons (Fig. 2C) in an antigenic peptide-dependent manner (data not shown). This nuclear staining resulted from a cross-reaction artifact, also observed in human cell lines (see nuclear staining in Fig. 1C), although the UBP119ip antibody does not recognize the human protein (Supplementary Material, Fig. S1). However, the UBP119ip staining also exhibited a punctuate profile which co-localized with DQ-Red-BSA staining and with mLAMP1 in the soma and proximal dendrites of hippocampal neurons (Fig. 2C). We thus concluded that native mCLN7 predominantly localizes to lysosomes and late endosomes in neurons.
Putative lysosomal targeting motifs of CLN7
The intracellular localization of native CLN7 suggests the existence of lysosomal targeting signals in its sequence. The amino acid sequence and topological prediction of hCLN7 were evaluated for the identification of putative dileucine- or tyrosine-based sorting motifs (21), which are typically located at the cytosolic tails of lysosomal membrane proteins. The N-terminus of hCLN7 contains a canonical [DE]XXXL[IL]-type putative dileucine motif, 9-EQEPLL-14 (Fig. 3), which is conserved among vertebrates (Supplementary Material, Fig. S2). We also considered 22-EWDIL-26 as a putative dileucine-type motif, although it is neither canonical nor conserved among mammals. The C-terminus contains two putative tyrosine-based motifs, 503-YKRL-506 and 513-YGRI-516, which are fully and partially conserved among vertebrates, respectively. Finally, the fifth cytosolic loop of CLN7 also carries a putative tyrosine-based motif, 441-YSKIL-445 (Fig. 3).
Figure 3.

Schematic representation of human CLN7 topology, N-glycosylation sites and position of potential tyrosine- and dileucine-based motifs. Based on topological predictions, CLN7 is a type III transmembrane protein with 12 transmembrane domains. The N-terminus of hCLN7 contains a classical [DE]XXXL[IL] dileucine-type motif (9-EQEPLL-14) and a non-canonical dileucine-type motif 22-EWDIL-26. The C-terminus contains two putative tyrosine-based motifs, the conserved 503-YKRL-506 and the partially conserved 513-YGRI-516. Additionally, a putative tyrosine-based motif, 441-YSKIL-445, is located in the fifth cytosolic loop of CLN7. Two N-glycosylation consensus sites are present in the luminal loop connecting transmembrane domains 9 and 10.
Expression of chimeric proteins suggests the presence of sorting motifs in both N- and C-terminal tails
Chimeric proteins carrying the N- and C-terminal tails of hCLN7 (residues 1–40, CD8-CLN7Nterm chimera, and residues 505–518, CD8-CLN7Cterm) were produced and analyzed by immunofluorescence microscopy to examine whether they contain targeting information. The human T cell surface marker CD8 was chosen for the first set of chimeras because it has widely been used in previous trafficking studies (24,26,27). After transient expression in HeLa cells, wild-type CD8 localized to the plasma membrane, whereas CD8-CLN7Cterm accumulated in the Golgi, probably due to chimera misfolding, and in lysosomes, as shown by its co-localization with the lysosomal enzyme aspartylglucosaminidase (AGA). Mutation of the putative tyrosine motif present in CD8-CLN7Cterm (Y513A; full-length hCLN7 numbering) abolished the co-localization with AGA and significantly misrouted the chimera to the plasma membrane, thus suggesting the presence of a targeting motif in the C-terminal tail (Supplementary Material, Fig. S3).
Expression of the CD8-CLN7Nterm resulted in large aggregates, which did not co-localize with lysosomes (data not shown) and most probably reflected folding problems due to presenting the N-terminus of CLN7 as a C-terminal tail of a type I monotopic CD8 protein. To resolve this, a sorting mutant of the NCL-associated lysosomal membrane protein CLN3 (CLN3dm) was used as a reporter to place the N-terminal tail of CLN7 in the right orientation. CLN3dm carries mutations in two targeting motifs (LI/AA + MX9G/AX9A mutant) and localizes to the plasma membrane and early endosomes, but not to the lysosomes [Fig. 4A (24)]. Replacement of the N-terminus of CLN3dm by that of CLN7 (CLN7-CLN3dm-Δ1-38 chimera) induced a significant co-localization with LAMP1 (Fig. 4B), although plasma membrane staining was partially preserved. To identify the underlying sorting signal, we introduced a mutation (L13A) into the putative dileucine-type motif 9-EQEPLL-14 (numbering according to the full-length hCLN7). This abolished the lysosomal localization of the chimera (Fig. 4C), indicating that EQEPLL is also a potential targeting motif of hCLN7.
Figure 4.
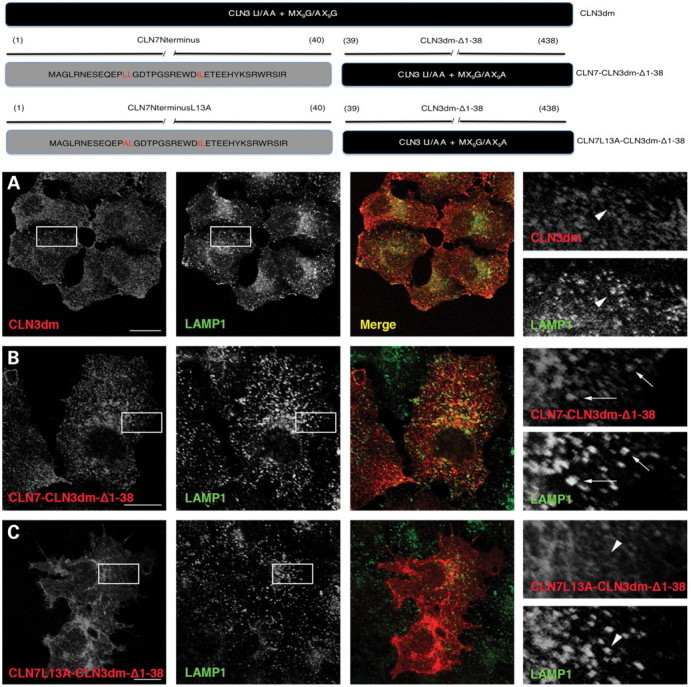
Expression of CLN7-CLN3dm-Δ1-38 chimeras in HeLa cells. Generation of CLN7-CLN3dm-Δ1-38 constructs is shown in the schematic representation. Localization of the CLN3dm and CLN7-CLN3dm-Δ1-38 chimeric proteins in HeLa cells was evaluated by confocal microscopy. The CLN3dm chimeric proteins were detected with an antibody against CLN3 (left panel) and lysosomes were visualized with LAMP1 staining (middle panel). The CLN3dm sorting mutant localizes to the plasma membrane and early endosomes, but shows no co-localization with lysosomes (A). The CLN7-CLN3dm-Δ1-38 chimera shows partial co-localization with lysosomes (B, inserts). Mutation in a putative dileucine motif of the CLN7 N-terminus (L13A) abolishes the lysosomal localization of the chimera (C, inserts). Co-localization is indicated with arrows, while absence of co-localization is shown with arrowheads. Scale bars, 20 µm.
The N-terminal dileucine motif of CLN7 is a major determinant for lysosomal sorting
To determine whether these two potential motifs actually contribute to the lysosomal localization of CLN7, mutations were introduced in critical residues of full-length CLN7 and the resulting constructs were expressed in HeLa cells. Strikingly, mutation of the N-terminal motif (LL13/14AA) in EGFP-tagged hCLN7 or in the untagged mouse protein dramatically redistributed CLN7 to the cell surface, as shown by epifluorescence microscopy (Fig. 5A and C). However, optical sections within the cells using confocal (data not shown) or deconvolution fluorescence microscopy and comparison to LAMP1 revealed a remaining lysosomal pool of CLN7-LL13/14AA (Fig. 5B and D), implying the existence of additional sorting signals. We thus mutated the tyrosine motif (Y513A) revealed by the CD8 chimera experiments. However, this mutation alone did not alter the lysosomal distribution of EGFP-hCLN7 (Supplementary Material, Fig. S4). Despite its unfavorable proximity to the last predicted transmembrane domain of CLN7, we also mutated an upstream putative sorting motif (Y503), but found again no effect. Combined mutations of the dileucine-based motif (LL13/14AA) with either Y503A or Y513A did not notably increase the plasma membrane localization over that seen with EGFP-hCLN7 carrying LL13/14AA alone, thus suggesting that C-terminal tyrosine-based motifs play a minor role in CLN7 targeting (Supplementary Material, Fig. S4).
Figure 5.
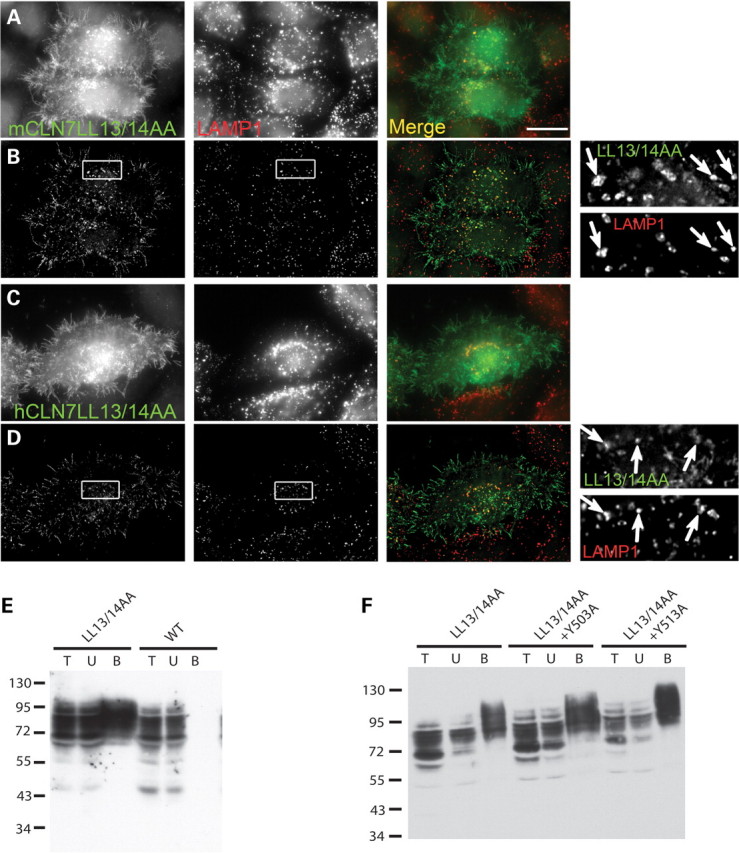
Mutation of the N-terminal dileucine motif misroutes full-length CLN7 to the plasma membrane. Untagged mouse CLN7 (A and B) or EGFP-tagged human CLN7 (C and D) constructs carrying a LL13/14AA mutation were transiently expressed in HeLa cells and analyzed by epifluorescence (A and C) or deconvolution fluorescence microscopy (B and D) as in Figure 1. (A and B) Two transfected cells double stained with UBP119ip and LAMP1 antibodies are shown. In (A), the epifluorescence image highlights the prominent distribution of mCLN7-LL13/14AA at the plasma membrane, including microvilli. In (B), an optical slice obtained by deconvolution of a z-stack of images shows that most LAMP1-positive lysosomes also contain residual mCLN7-LL13/14AA protein. (C and D) Similar observations were made for EGFP-hCLN7-LL13/14AA. (E and F) Surface biotinylation analysis of the EGFP-hCLN7 sorting mutants. HEK293 cells transiently expressing EGFP-hCLN7 constructs were treated with an impermeant biotinylation reagent, solubilized and fractionated using streptavidin-agarose beads. Total cell lysate, unbound and streptavidin-bound materials (lanes T, U, B, respectively) were analyzed by immunoblotting using anti-GFP antibodies. Bound material (surface pool) was concentrated 37-fold relative to the cell lysate prior to SDS–PAGE analysis. Two independent experiments are shown in (E) and (F). Whereas wild-type EGFP-hCLN7 is not detected at the cell surface, the LL13/14AA mutation dramatically increased the biotinylated (surface) pool of protein (E, lanes B). Additional mutation of a C-terminal tyrosine residue did not alter (Y503A) or slightly increased (Y513A) the amount of biotinylated EGFP-CLN7 (F).
To confirm these findings, we evaluated the redistribution of CLN7 sorting mutants to the plasma membrane using cell surface biotinylation. Whereas the wild-type protein was not detected at the cell surface, mutation of the N-terminal dileucine-based motif substantially misrouted EGFP-hCLN7 to the plasma membrane (Fig. 5E), in agreement with immunofluorescence data. On the other hand, combining the LL13/14AA mutation with either Y503A or Y513A did not alter, or only slightly increased (Y513A), the surface level of EGFP-hCLN7 (Fig. 5F).
Since simultaneous disruption of the two targeting motifs suggested by the chimera approach did not result in a complete loss of lysosomal localization, additional mutations (Fig. 3) were introduced into full-length CLN7. The unconventional dileucine-type motif (IL25-26) in the N-terminus was disrupted by introduction of the I25A mutation. Residue I444 was replaced by an alanine to disrupt simultaneously a potential YXXΦ and a non-canonical dileucine-type motif (lacking the acidic patch) in the fifth cytoplasmic loop of CLN7. However, these mutations had no effect on the subcellular localization of full-length CLN7 when introduced individually (data not shown) or in combination with L13A and Y513A mutations (Supplementary Material, Fig. S5). We also examined whether the pathogenic vLINCL mutations impair the lysosomal localization of hCLN7 and would thereby unveil unanticipated sorting signals. Previous studies showed that mutations R139H, T294K, G310D, G429D and R465W do not alter the intracellular distribution of hCLN7 (2,3). We introduced mutations G52R, Y121C, A157P, P412L and P447L, recently identified in vLINCL patients (3–6), into EGFP-hCLN7. However, none of them interfered with the lysosomal localization of hCLN7 (Supplementary Material, Fig. S6).
These findings show that the N-terminal dileucine-based motif plays a major, but non-exclusive, role in the lysosomal targeting of CLN7. Additional motifs, possibly the C-terminal 513-YGRI-516 motif but also unidentified, presumably unconventional signals, can partially direct CLN7 to the lysosome when the dileucine-based motif is impaired.
The N-terminal dileucine motif binds the heterotetrameric AP-1 adaptor
To analyze whether CLN7 follows the direct or indirect route to the lysosome, HeLa cells were transiently transfected with wild-type CLN7 together with an AP180 mutant, known to block clathrin-mediated endocytosis (28). Inhibition of endocytosis in transfected cells was confirmed by their inability to take up fluorescent transferrin. Cells with impaired endocytosis showed only a small amount of CLN7 at the plasma membrane under immunofluorescence microscopy, while most of the protein remained in lysosomes (Fig. 6A), suggesting that CLN7 reaches lysosomes mainly via the direct trafficking route. To identify adaptor proteins mediating lysosomal targeting of CLN7, an epitope-tagged hCLN7 (HA-CLN7pAHC) was transiently expressed in AP-1 (Δµ1A) and in AP-3 deficient (pearl) mouse fibroblasts. Wild-type CLN7 strictly localized to lysosomes in both cell lines, suggesting that targeting of CLN7 may not depend on the action of AP-1 or AP-3 adaptor proteins alone (Fig. 6B). Expression of CLN7 Y513A in these cell lines also showed solely lysosomal localization (data not shown), suggesting that the strong dileucine motif may also be recognized by more than one adaptor. Expression of the L13A/Y513A double mutant in either of the defective cell lines showed strong plasma membrane localization, with a remaining lysosomal pool (Supplementary Material, Fig. S7). These findings support the idea that several motifs and, consequently, several adaptor proteins are involved in lysosomal trafficking of CLN7.
Figure 6.
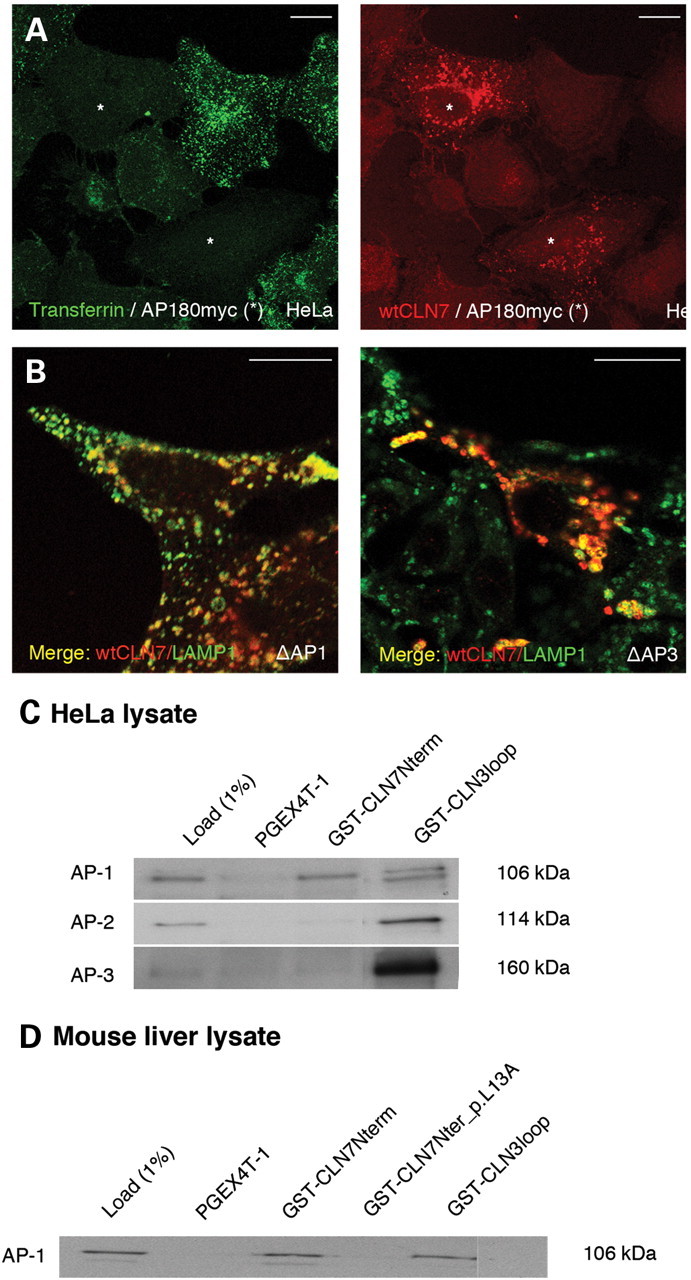
Determination of the heterotetrameric adaptor proteins recognizing the N-terminal dileucine motif. HeLa cells were transiently co-transfected with wild-type CLN7 (red) and mutated AP180. Inhibition of endocytosis was confirmed by feeding the cells with FITC-transferrin (green). The transfected cells showing defective endocytosis are indicated by an asterisk (A). Wild-type CLN7 bearing a HA epitope was transiently expressed in AP-1 (Δµ1A) and in AP-3 deficient (pearl) mouse fibroblasts (Δ AP-1 and Δ AP-3, respectively). Co-localization of CLN7 (HA immunoreactivity, red) and mLAMP1 (green) in AP-1 and AP-3 deficient cells is indicated in yellow (B). Scale bars, 20 µm. (C) The N-terminus (amino acids 1–40) of CLN7 was expressed as a GST fusion protein, purified and used to pull-down adaptor proteins from HeLa cell lysate. GST-CLN3loop was used as a positive control for all the pull-downs (28). SDS–PAGE separated proteins were immunoblotted and stained with specific antibodies against AP-1, AP-2 and AP-3 (C). (D) The specificity of AP-1 binding to the N-terminal dileucine motif was verified by repeating the pull-down experiments from the mouse liver extract and including the GST-CLN7Nterm construct carrying an alanine substitution at L13 (GST-CLN7Nterm_p.L13A). Molecular masses of protein standards, in kDa (C and D). One percent of the cell lysate with which the affinity-purified GST proteins were incubated was used as load (left lanes, C and D).
To determine which adaptor proteins interact with the major N-terminal dileucine motif, GST pull-down experiments were performed. The N-terminal domain of CLN7 was expressed as a GST fusion protein and used to pull-down adaptors from a HeLa cell lysate. Binding of adaptors was detected by immunoblotting with antibodies recognizing specific subunits of AP-1, AP-2 and AP-3. GST-CLN3loop was used as a positive control in the experiments, since it contains a dileucine-type motif which is known to bind all the tested adaptors (29). The N-terminal domain of CLN7 was found to bind only to AP-1 (Fig. 6C). Further pull-down experiments with mouse liver extract showed that the interaction of AP-1 with the N-terminus was completely abolished by the L13A mutation, thus indicating that it specifically occurs via the dileucine motif (Fig. 6D).
N-glycosylation of CLN7 and intracellular distribution of unglycosylated mutants
Binding to lectins, such as galectins, can regulate the trafficking of membrane glycoproteins by decreasing their availability for endocytosis (30,31) or by recruiting them into sorting microdomains (32). Although such mechanisms have not been described for lysosomal proteins, we investigated the glycosylation status of CLN7 and its possible contribution to the intracellular distribution of the protein.
Treatment of cell lysates of EGFP-hCLN7 and EGFP-mCLN7 with endoglycosidase PNGase F resulted in increased electrophoretic mobility of both proteins, indicating the presence of N-glycosylation. hCLN7 possesses four N-glycosylation consensus sites at asparagine residues 6, 171, 371 and 376, of which only the last two are predicted to locate in the luminal compartment (Fig. 3). We thus mutated N371 and N376 to glutamine, individually or simultaneously, and analyzed the mutants by immunoblotting. Single mutations increased the electrophoretic mobility of EGFP-hCLN7, but preserved its sensitivity to PNGase F. In contrast, the double mutant migrated as a sharp, 70 kDa band which was insensitive to PNGase F treatment (Fig. 7A), thus showing that N-glycans are carried at both N371 and N376 residues in human CLN7. We also mutated homologous positions in mouse CLN7 (N372, N377), but the protein electrophoretic mobility remained heterogeneous and sensitive to PNGase F (Fig. 7B). However, additional mutation at a third N-x-S/T consensus site (N389), which is absent from the human sequence, converted EGFP-mCLN7 into a sharp ∼67 kDa band insensitive to the endoglycosidase, thus showing that mouse CLN7 is glycosylated at all the putative asparagine residues in the fifth luminal loop. These mutated constructs were then expressed in HeLa cells and their intracellular distribution was analyzed by deconvolution fluorescence microscopy. EGFP-tagged hCLN7 N371Q/N376Q and mCLN7 N372Q/N377Q/N389Q as well as the untagged mouse mutant showed extensive overlap with LAMP1 (Fig. 7C–E), thus excluding a role of N-glycans in the intracellular targeting of CLN7.
Figure 7.
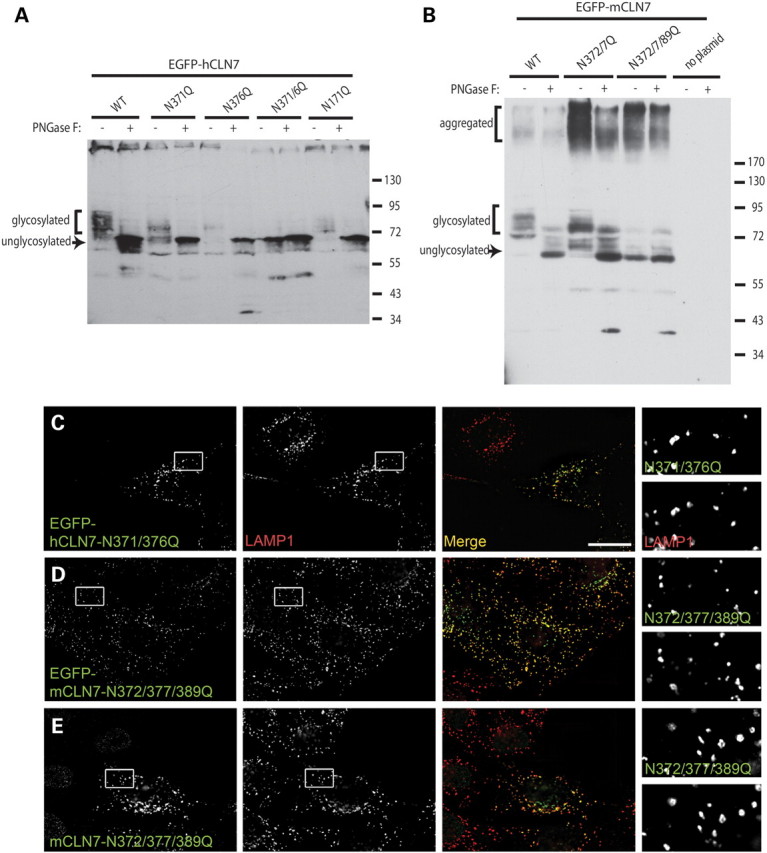
Lysosomal localization of human and mouse CLN7 does not depend on N-glycans. (A and B) EGFP-tagged human and mouse CLN7 constructs carrying N-glycosylation site mutations were transiently expressed in HEK293 and analyzed by immunoblotting with or without prior treatment with endoglycosidase PNGase F. Molecular masses of protein standards, in kDa, are shown on the right. (A) PNGase F converted all EGFP-hCLN7 constructs to a sharp (unglycosylated) ∼70 kDa band. Whereas single N371Q or N376Q mutations increased the electrophoretic mobility of EGFP-hCLN7 but did not abolish its heterogeneity (smear appearance), only the double mutation N371Q/N376Q fully mimicked the effect of PNGase F. Mutation N171Q had no effect. (B) For the mouse protein, complete disruption of the electrophoretic heterogeneity and PNGase F sensitivity was achieved only after mutating three asparagine residues: N372, N377 and N389. (C, D, E) Genetic ablation of the N-glycans does not interfere with the intracellular distribution of human and mouse CLN7. Multiple asparagine mutants were analyzed for EGFP fluorescence (C and D) or UBP119ip (E) and LAMP1 immunoreactivity as in Figure 1. Scale bar, 20 µm.
CLN7 expression profile in rodent brain matches vLINCL neuropathology
In the last part of this study, we investigated how the expression profile of CLN7 correlates with the pattern of cell vulnerability observed in vLINCL patient brains. Since CLN7 was predominantly found to be expressed in mouse neurons (Fig. 2A), we examined whether the expression concentrates within a specific neuron type, and thus performed in situ hybridization in rat brain. These analyses showed that the CLN7 mRNA is present throughout the brain, with increased levels in the granular layer of the cerebellar cortex, the pyramidal layer of the hippocampus and, to some extent, in the deeper layers of the neocortex (Fig. 8).
Figure 8.
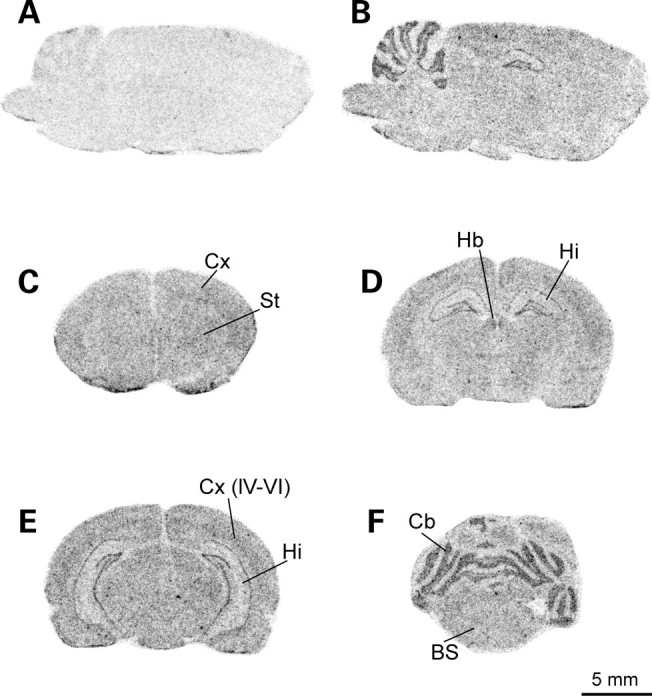
Distribution of CLN7 transcript in rat brain. Autoradiograms shown represent parasagittal (A and B) and coronal sections (C–F) of rat brain taken at the level of the forebrain (C), midbrain (D and E) and hindbrain (F). Sections were incubated with a mix of sense (A) or antisensense (B–F) radiolabeled oligonucleotides derived from the rat CLN7 mRNA sequence. Bs, brain stem; Cx, cerebral cortex; Cb, cerebellum; Hb, habenula; Hi, hippocampus; St, striatum. IV–VI, layers IV to VI of the cerebral cortex. Scale bar, 5 mm.
We also performed a histological analysis of the postmortem brains and retinas from CLN7-defective vLINCL patients with ages ranging from 7 to 18 years old. Neurons generally displayed maximum lysosomal storage, demonstrated by autofluorescence of accumulated ceroid (Fig. 9) and immunoreactivity for Cathepsin D (data not shown). In agreement with the dominant expression of CLN7 mRNA in mouse neurons (Fig. 2A), storage was found to be less abundant in other cell types. There was discrete, just detectable storage in oligodendrocytes, in agreement with the absence of myelin alteration (data not shown). The amount of storage in astrocytes was low, but easily detectable and did not interfere with reactive astrocytosis, which is a feature of vLINCL neuropathology (Fig. 9C). Microglial phagocytes detected by CD68 staining were small and numerous, and showed barely detectable autofluorescence (Fig. 9E and G). In the areas of neuronal degeneration they were enlarged, often clustered in the form of neuronophagic granulomas and loaded with autofluorescent material (Fig. 10).
Figure 9.
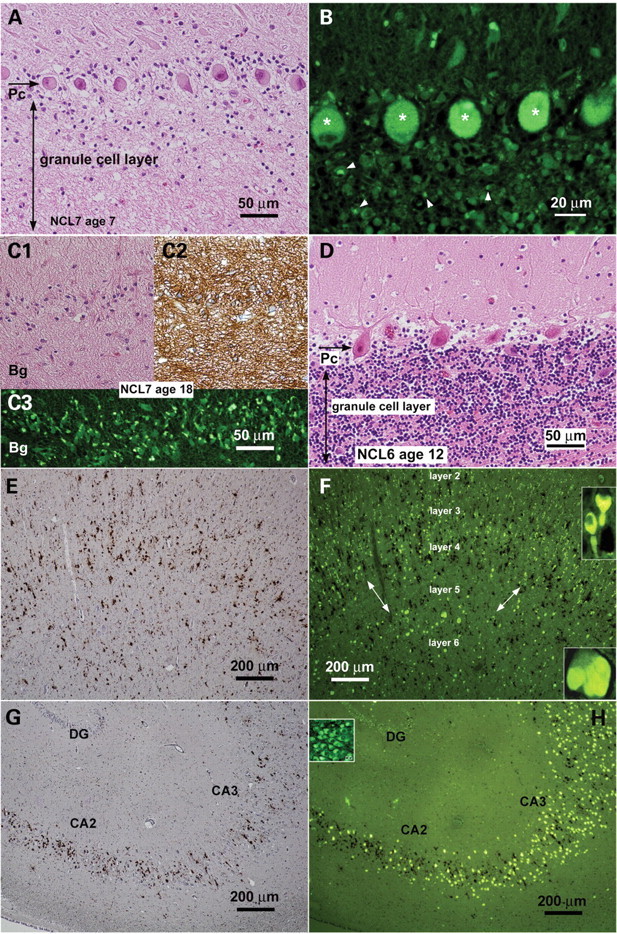
Histological abnormalities in brain from CLN7-defective vLINCL patients. (A–D) Cerebellar cortex sections showing progression of neuronal depletion between ages 7 (A and B) and 18 years (C). Sections were stained with haematoxylin and eosin (A, C1, D) or analyzed for autofluorescence under UV light (B, C3). Granule cell layer neurons are dramatically reduced in the cerebellum of the younger patient (A) and completely disappeared in the older patient (C1). Purkinje cells (‘Pc’ arrow) disappear much later (compare A with C1). For comparison, the cerebellar cortex of a CLN6-defective LINCL patient (D; age 12 years) is practically intact. Autofluorescence is mostly expressed in Purkinje cells (asterisks in B) and is discrete in the remaining granule neurons (arrowheads). Only Bergmann reactive astroglia (Bg in C) persist in the older patient, expressing strong signal for GFAP (C2) and a low but discernible degree of storage (C3). (E and F) Frontal cortex section (7-year patient): CD68 immunoreactivity (E) and autofluorescence (F) are present in all neuronal layers except layer V (marked by double arrows). The discrete residual storage in layer V represents reactive astrocytes. Neuronal storage occurs even in axon hillocks (F, upper insert). Some neurons in layer VI (lower insert) show massive perikaryal storage. Macrophages, demonstrated by CD68 staining, are dispersed throughout the cortex, thus suggesting they came from the scavenged layer V. (G and H) Hippocampus section (7-year patient) analyzed for CD68 immunoreactivity (G) and autofluorescence (H). Pyramidal neurons of the CA sector show strong autofluorescence; their number is significantly reduced in the CA2 sector. Macrophages are concentrated in the CA sector, thus indicating active scavenging of damaged neurons. Insert in (H) shows discrete uniform storage in dentate gyrus (DG) neurons, whose number is not apparently reduced.
Figure 10.
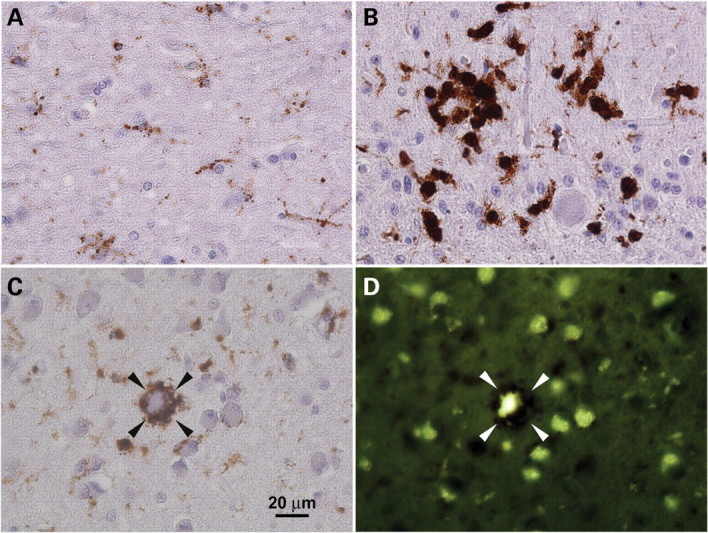
Microglial scavenging activity in CLN7-associated vLINCL. Brain sections (patients aged 9 and 12) were analyzed for CD68 immunoreactivity (A–C) and autofluorescence (D). (A and B) Morphological changes associated with microglial activation. Only discrete microglial cells are present in white matter and grey matter (A), whereas cerebellar cortex (B) shows both discrete microglia and enlarged microglia active in scavenging and forming granulomas. (C and D) Phagocytic microglial cell engulfing a degenerated storage neuron in basal ganglia (arrowheads). The scavenged neuron autofluorescence (D) is clearly visible within the immunostained phagocyte surrounding the neuronal body (compare with CD68 staining in C). Note the remaining storage neurons which are free of any scavenging attack.
Interestingly, the loss of human neurons correlates well with the level of CLN7 expression observed in rodent brain. In the cerebellar cortex, Purkinje cells persisted much longer than granule layer neurons. Whereas granule neurons were highly reduced in number in patients who died at ages 7 and 8, beyond that age the granule cell layer was totally absent (Fig. 9A–C; compare with Fig. 9D). Purkinje cells survived significantly longer (Supplementary Material, Fig. S8). Ceroid storage was higher in Purkinje cells, in both perikarya and dendrites, the latter often displaying torpedo-like distensions. Storage in remaining granule neurons was discrete and perinuclear (Fig. 9B), thus suggesting that neurodegeneration may not directly result from autofluorescent material storage. Highest storage was detected in neurons of the neocortical area where the autofluorescence was present in both perikarya and axon hillocks. Maximal neuron loss was found in the cortical layer V (Fig. 9E and F). Hippocampal pyramidal neurons showed intense storage and maximum vulnerability in the CA2 sector. Storage was discrete in perikarya of the dentate gyrus neurons, which did not display significant reduction in number (Fig. 9G and H). Neurons in other regions (basal ganglia, thalamus, oblongata, brain stem, spinal medulla) showed storage without significant large-scale cell loss. Some local variability in the storage intensity was detected (data not shown).
Retinal neurons in all layers showed high level of degeneration irrespective of the degree of storage. Neurodegeneration was maximal in neurons of the ganglionic layer. Other (remaining) neurons in the nuclear layers displayed discrete signs of storage (Supplementary Material, Fig. S9).
DISCUSSION
In this study, we sought to characterize the distribution of CLN7, the polytopic membrane protein underlying a variant form of late-infantile NCL, to identify the determinants responsible for its intracellular distribution and to compare its cellular and regional distribution with human neuropathological features. We show that CLN7 mRNA is preferentially expressed in neurons and that the native protein localizes to lysosomes in hippocampal neurons. Furthermore, we identified the major determinant responsible for the lysosomal targeting of CLN7. Finally, we show that the distribution of ceroid storage and of neuron loss in patient brains correlates with the cellular and regional expression of CLN7 in rat brain, suggesting that vLINCL is a cell-autonomous disorder.
Intracellular distribution of CLN7
Previous data had shown that recombinant, epitope-tagged CLN7 overexpressed in cell lines localizes to lysosomes and late endosomes at steady state (2). However, it was important to confirm the localization of CLN7 in a more natural context, given the non-lysosomal localization of CLN6 and CLN8 (33–35) and the controversial data published in relation to CLN3 (20). We thus raised an antibody against the mouse CLN7 and analyzed the distribution of the untagged protein in transfected cells and that of the native protein in cultured neurons. The lysosomal and late endosomal localization of CLN7 was confirmed in both cell types, thus establishing lysosomes as the primary site of action of CLN7 and the intracellular target in CLN7-associated vLINCL. The fact that CLN7 sequences have been detected in mass-spectrometry analyses of rat liver lysosomes (36) and of highly purified placental lysosomes (37) confirms the intracellular distribution of native CLN7 in other tissues.
Determinants of the lysosomal localization
In a second line of investigation, we examined the determinants responsible for the lysosomal and late endosomal localization of CLN7. We found that a classical dileucine-type motif, present in the cytosolic N-terminal tail, is a major determinant of this localization. In the absence of this motif, a large fraction of the protein is misrouted to the cell surface, although another pool of CLN7 molecules is still delivered to the lysosomes. Similar N-terminal dileucine-based sorting motifs have been previously reported for other transporters, such as the lysosomal sialic acid transporter sialin (38,39), the glucose transporter GLUT8 (40) and the melanosomal, transporter-like protein OCA2 (41). During revision of this manuscript, the role of the N-terminal dileucine motif of CLN7, and the lack of contribution of N-glycosylation, was independently confirmed by another study (42).
However, these authors also reported that transport of CLN7 to the lysosomes is mediated via the plasma membrane (42). This is in contradiction with the absence of detectable wild-type CLN7 at the cell surface in our biotinylation experiments and with the fact that most wild-type CLN7 remained in the lysosomes after impairment of clathrin-mediated endocytosis. The direct lysosomal trafficking route of CLN7 is also supported by our in vitro pull-down experiments, which revealed a strong and specific binding of the N-terminal dileucine motif to the heterotetrameric adaptor AP-1. However, the steady-state lysosomal distribution of wild-type CLN7 was fully preserved in AP-1-deficient cells, in contrast with the strong effect of the dileucine motif mutation. Such an apparent discrepancy between motif and cognate adaptor inactivation suggests that the dileucine motif may also be recognized by other adaptors within the cell.
Detection of a small fraction of CLN7 at the plasma membrane when endocytosis is blocked, and of a significant lysosomal pool of CLN7 in the absence of the N-terminal dileucine motif, indicates the existence of additional targeting information and the obvious complexity of targeting of CLN7. Our chimera experiments revealed that the C-terminal tail of CLN7 carries such information and that it depends on a tyrosine-based putative motif. However, mutation of the Y513 residue in the full-length protein did not alter its steady-state localization, suggesting that it plays a minor targeting role. Due to the strong trafficking defect associated with the mutation of the dileucine motif alone, the hidden targeting signal(s) are suggested to play only a minor, or maybe a cell-specific role in the targeting of CLN7. It is noteworthy that none of the missense mutations associated with vLINCL analyzed previously (2,3) or, in this study, seem to affect the lysosomal localization of CLN7, indicating that vLINCL is primarily caused by loss or impaired function of CLN7 rather than disturbed trafficking of the protein as has been detected in many other NCLs. For example, most of the pathogenic mutations in the lysosomal membrane protein CLN3, underlying the juvenile NCL, result in retention of the protein in the ER (24,43). Similarly, lysosomal trafficking of the soluble NCL proteins, CLN1/PPT1 and CLN5, is reported to be very sensitive to disease-causing mutations (15,44).
The identification of the dileucine motif as the major determinant of CLN7 lysosomal targeting also has practical interest for future functional studies, since the possibility to re-direct lysosomal transporters to the cell surface allows monitoring their activity as a whole-cell uptake, which is more accessible than, and topologically equivalent to, lysosomal efflux (38,45,46). Candidate substrates can thus be screened on whole cells expressing the dileucine mutant of CLN7 to elucidate its putative transport activity.
Cellular and regional expression of CLN7 predicts human neuropathology
In a third line of investigation, we asked which regions in the brain and which cell types predominantly express CLN7 and compared this expression, on one hand, to that of other lysosomal NCL proteins in rodent brain and, on the other hand, to the neuropathological features of CLN7-associated vLINCL.
At the cellular level, CLN7 was the only one, among the analyzed NCL mRNAs, which is more prominently expressed in neurons than in microglial cells and astrocytes, suggesting a prominent function of CLN7 in neurons. Quantification of regional mRNA expression in rat brain showed that the highest CLN7 mRNA levels were detected in hippocampus and, overall, the levels displayed greater variation between the different brain regions when compared with those of the other analyzed NCL genes. However, the mRNA levels of CLN7 in different brain regions were generally 10- to 50-fold less abundant than those detected for the soluble enzymes CTSD, PPT1 and TPP1. Only the mRNA levels of CLN3, another transmembrane protein of the NCL group, were lower than those of CLN7. Similar differences were observed in neural cell cultures. Altogether, these findings draw a line between the mRNA levels of genes encoding soluble and membrane-bound NCL proteins.
In situ hybridization analyses of the rat brain confirmed that the expression level of CLN7 is overall low since a long exposure time was required to produce autoradiograms. Our data show that CLN7 is expressed throughout the brain, but show higher mRNA levels in the pyramidal cell layers of the hippocampus and in the granular layer of the cerebellum. Most importantly, these findings are in a good agreement with the early loss of cerebellar granule neurons and, more generally, the prominent storage of ceroid in neurons over other cell types in the post-mortem brains of CLN7-defective vLINCL patients analyzed in this study. In contrast to soluble lysosomal enzymes which can be transferred from one cell to another by mannose-6-phoshate-dependent endocytosis, lysosomal membrane proteins act in the cells which synthesized them (47). Their deficiency should thus generally induce cell-autonomous defects. Accordingly, it has been observed that disruption of the Niemann-Pick type C1 or the chloride transporter ClC-7 genes in mice results in cell-autonomous defects (48–50)—but see ref. (51) for a non-cell-autonomous defect of glial cells in a Drosophila model of mucolipidosis type IV. The correlation observed in this study between CLN7 mRNA distribution and neuropathological findings suggests that, similarly, cell-autonomous defects may underpin the pathophysiology of CLN7-associated vLINCL. Animal models should help test this proposal in the future.
MATERIALS AND METHODS
cDNA constructs
The construct for full-length hCLN7 (amino acids 1–518) carrying an aminoterminal hemagglutinin (HA) tag (HA-CLN7pAHC) has been described earlier (2). The untagged mCLN7 construct was generated by PCR from Mus musculus IMAGE cDNA no. 40061889 (primers in Supplementary Material, Table S1) and cloned at the HindIII and XbaI sites of pcDNA3 (Invitrogen). Mouse and human CLN7 constructs tagged at their N-terminus by EGFP were generated by PCR (Supplementary Material, Table S1) and cloned into pEGFP-C2 (Clontech). To generate CD8 chimeric proteins, the N-terminus (amino acids 1–40) and C-terminus (amino acids 505–518) of hCLN7 were PCR amplified from a Marathon Ready Human Brain cDNA (Clontech) using CLN7 specific primers, and cloned into CD8-pBluescript in replacement of the CD8 cytosolic tail (Supplementary Material, Table S1). The CD8-CLN7Nterm and CD8-CLN7Cterm inserts were then subcloned into pcDNA3.1(+). To generate the CLN7/CLN3 chimera, a CLN3 cDNA (CLN3dm) carrying mutations in two lysosomal targeting motifs [LI/AA+ MX9G/AX9A (24)] was used as a PCR template to produce an N-terminally truncated construct lacking the first 38 amino acids (CLN3dm-Δ1-38) and cloned into pcDNA3.1. A PCR product coding for the N-terminus of hCLN7 (amino acids 1–40) was then ligated in frame with CLN3dm-Δ1-38 in pcDNA3.1 (Supplementary Material, Table S1). The myc-tagged AP180pCMV construct used in the endocytosis blocking experiments was a gift from Dr H. McMahon, Cambridge, UK. For the production of GST fusion protein, the N-terminal fragment (amino acids 1–40) of hCLN7 was cloned in frame with GST in the pGEX4T-1 vector (Amersham Biosciences) (Supplementary Material, Table S1). All amino acid substitutions introduced into the above constructs were generated using the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) or an equivalent approach using Phusion polymerase (Finnzymes) and two complementary mutated primers (Supplementary Material, Table S2). All constructs were verified by sequence analysis.
Antibodies
An antibody against mouse CLN7 was produced by immunizing New Zealand White rabbits against a 22-mer peptide (CKSVNFQEENTDEPQIPEGSID) coupled at its N-terminus to Keyhole Limpet Haemocyanin according to standard procedures (Agro-Bio, France). Immunoglobulins were affinity-purified using the antigenic peptide and stored frozen at −20°C in phosphate buffer saline (PBS) supplemented with 0.09% sodium azide.
Mouse monoclonal anti-CD8 was used for the detection of CD8 chimeras (mCD8; 1:200; kindly provided by Dr Matthew Seaman, Cambridge Institute for Medical Research, Cambridge, UK). CLN7-CLN3dm-Δ1-38 chimeras were detected with a rabbit peptide antibody raised against the amino acids 242–258 of mouse CLN3 [m385; 1:300 (52)]. Full-length hCLN7 constructs carrying an N-terminal HA tag or EGFP tag were detected with rabbit polyclonal anti-HA (Y-11; 1:500; Santa Cruz Biotechnology) and with a mixture of two monoclonal anti-GFP antibodies (1:1000, Roche Molecular Biochemicals), respectively. A mouse monoclonal antibody (H4A3; 1:100; Developmental Studies Hybridoma Bank, University of Iowa, IA, USA) against LAMP1, or a rabbit polyclonal anti-AGA [1:400 (53)], was used to visualize lysosomes in HeLa cells. In Δµ1A (AP-1 deficient cells, kindly provided by Peter Schu, Göttingen, Germany) and AP-3βA pearl cells (AP-3 deficient cells, kindly provided by Scottie Robinson, Cambridge Institute for Medical Research, Cambridge, UK), lysosomes were detected using a monoclonal rat antibody 1D4B against mouse LAMP1 (1:100 from Developmental Studies Hybridoma Bank, or 1:500 from BD Biosciences). Monoclonal antibodies against AP-1γ and AP-2α were from BD Biosciences (Pharmingen). AP-3 was detected with a purified antibody against δ-adaptin kindly provided by Dr A. Peden (54). Conjugated secondary antibodies used in immunofluorescence stainings were from Jackson ImmunoResearch (West Grove, PA, USA) or Molecular Probes (Invitrogen) and HRP-conjugated antibodies used for western blot detection were from DAKO or Sigma.
Cell culture and transfections
HeLa and HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal calf serum (FCS), 50 mg/ml streptomycin and 100 IU/ml penicillin in a humidified atmosphere containing 5% CO2 at 37°C. Δµ1A and AP-3βA pearl cells were cultured in DMEM supplemented with 20% FCS and antibiotics. Cells were transfected by electroporation (HeLa cells) as previously described (38) or by lipofection on 6-well plates using FuGENE-HD (HeLa cells; Roche Molecular Biochemicals) or Lipofectamine 2000 (Invitrogen) transfection reagents according to the guidelines provided by the manufacturers.
For immunofluorescence analysis, hippocampal neuron cultures were performed as previously described (55). Hippocampi of mouse embryos were dissected at days 17–18. After trypsinization, tissue dissociation was achieved with a Pasteur pipette. Cells were counted and plated on poly-l-lysine-coated 14 mm diameter cover slips, at a density of 60 000–75 000 cells per 15 mm dish (300–375 cells per square millimeter), in complete Neurobasal medium (Invitrogen) supplemented with 2% B-27 (Invitrogen), 0.5 mm l-glutamine, 10 U/ml penicillin G and 10 mg/ml streptomycin. Four to 5h after plating, cover slips were transferred to a 90 mm dish containing conditioned medium obtained by incubating rat glial cultures (70–80% confluency) for 24 h in the complete medium described above.
For RT–PCR analysis, cortical neurons were prepared from E16 Sprague-Dawley rat embryos according to Huettner and Baughman (56). Cells dissociated with 1 mm papain (Sigma) were plated in Neurobasal medium supplemented with B-27 at a density of 1.5×105 cells/cm2 in poly-d-lysine coated dishes. Medium was changed after 3 days and neurons harvested after 7 days.
Microglial cultures were obtained from 9- to 12-day mixed primary glial cultures prepared from cerebral cortices of 1-day-old rats (57). Microglial cells were harvested by mild shaking, resuspended in Basal Eagle's Medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mm glutamine and 100 µg/ml gentamicin, and plated on uncoated plastic wells at a density of 1.25×105 cells/cm2. Cells were allowed to adhere for 20 min and washed to remove non-adhering cells.
Astrocytes were prepared from 7- to 10-day mixed primary glial cultures derived from 1-day-old rat cerebral cortices. Cells were cultured in MEM/FCS and re-plated twice, at confluence, to eliminate microglial cells (58).
Experiments were performed in agreement with the institutional guidelines for use of animals and their care, in compliance with national and international laws and policies (European Community Council Directive no. 86-609/87-848/EEC and agreement no. 75-974 from the Ministère de l'Agriculture et de la Forêt, Service Vétérinaire de la Santé et de la Protection Animale to M.D.).
Real-time RT-PCR
RNA was extracted from primary cultures using the Tri-Reagent isolation system (Sigma) according to the manufacturer's instructions. Yield and integrity of RNA were determined by 260 nm light absorbance and agarose gel electrophoresis. Total RNA was treated with the DNA-free kit (Ambion) to eliminate possible DNA contaminations. Four micrograms of RNA were reverse transcribed with M-MLV Reverse Transcriptase (Invitrogen) using random hexamers in a final volume of 20 µl. Real-time PCR was performed on the RT products with the Power SYBR Green PCR Master Mix (Applied Biosystem) and primers listed in Supplementary Material, Table S3, in a 7900HT Fast Real-Time PCR system apparatus (Applied Biosystem), following the manufacturer's instructions. Thermal cycling conditions comprised initial steps at 50°C for 2 min and 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. All samples were run in triplicate. Amplification efficiency of each primer pair was verified by performing RT–PCR using different template dilutions.
Gene expression levels were quantified from real-time PCR data by the comparative threshold cycle (CT) method (59) using hypoxanthine–guanine phosphoribosyltransferase (HPRT) as an internal control gene. The fractional number of PCR cycles CT required to obtain a given amount of RT–PCR product in the exponential phase of amplification was determined for the gene of interest and for HPRT in each RNA sample. The relative expression level of the genes of interest was then expressed as 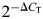 where ΔCT = CT gene of interest − CT HPRT.
where ΔCT = CT gene of interest − CT HPRT.
Immunofluorescence analysis
For immunofluorescence analysis in HeLa and AP-deficient cells, transfected cells were grown on cover slips for 48 h and fixed with 4% paraformaldehyde (PFA) for 15 min. To visualize proteolytic activity of late endosomes and lysosomes, live HeLa cells were incubated overnight in complete medium supplemented with 50 µg/ml DQ™ Red BSA (Molecular Probes) and washed twice with PBS prior to fixation. In the endocytosis blocking experiments, HeLa cells transfected 24 h earlier were starved in serum-free DMEM for 2 h and then incubated for 20 min in serum-free DMEM containing 25 µg/ml FITC-conjugated human serum transferrin (Molecular Probes). The cells were then washed and subsequently fixed with 4% PFA. Cells were permeabilized with 0.05–0.2% saponin in PBS supplemented with 0.5% bovine serum albumin (BSA) for 30 min at room temperature and stained with the specific antibodies described above.
Immunofluorescence analysis in neurons was performed 9–10 days after plating. Cells were washed with PBS fixed 10 min with 4% PFA in PBS and washed three times with PBS before permeabilization for 5 min with 0.2% Triton X-100. Primary and secondary antibodies were applied overnight at 4°C and for 1 h at room temperature, respectively, in PBS supplemented with 3% BSA, 2% normal goat serum and 2% normal donkey serum. The cover slips were mounted with Gel Mount (Sigma) or FluoromountG (Southern Biotechnology Associates) on object glasses and viewed using a Leica DMR confocal microscope or a Nikon Eclipse TE-2000 microscope equipped with a CCD camera (Coolsnap). When indicated, out of focus signal in the selected focal plane was corrected by deconvolution microscopy: five z-sections (0.2 µm spacing) were collected on both sides of the selected plane and the stacked images were deconvoluted using the PSF-based Iterative 3D Deconvolution module of Metamorph software (Universal Imaging Corporation). Image processing was done using Adobe Photoshop software.
Biotinylation assay
Two days after transfection, 107 HEK293 cells were washed twice with ice-cold PBS/Ca/Mg and biotinylated for 2 × 20 min at 4°C using 2 mg/ml of the cell-impermeant, cleavable reagent sulfo-NHS-SS-biotin (Pierce) in PBS/Ca/Mg. Unbound biotin was quenched for 20 min at 4°C with 100 mm glycine in PBS/Ca/Mg. After two washes, cells were lysed for 1 h in 1 ml/well lysis buffer (150 mm NaCl, 5 mm EDTA, 50 mm Tris–HCl pH 7.5, 0.1% SDS, 1% Triton X-100, 1 mm PMSF, 0.1 mm leupeptin, 0.1 µm pepstatin A). The cell lysate (1 mg protein in 3 ml) was clarified by sedimentation at 20 000g for 10 min and the supernatant was incubated for 2 h at 4°C with 750 µl streptavidin-agarose bead suspension (Fluka) under gentle agitation. Beads were sedimented at 100g for 30 s. Supernatants (unbound material) were recovered and beads were washed three times successively with 5 ml of lysis buffer, then 500 mm NaCl containing 50 mm Tris–HCl pH 7.5 and finally 10 mm Tris–HCl pH 7.5. Streptavidin-bound material was eluted by adding 50 µl of 2× Laemmli's sample buffer to the streptavidin-agarose beads (pellet volume: 150 µl). Fifteen microliters of cell lysate and unbound proteins and 30 µl of the bead eluate were resolved by 10% SDS–PAGE and analyzed by immunoblotting with anti-GFP antibodies.
GST pull-down analyses
The wild-type and mutated GST-CLN7 fusion proteins were produced in E. coli BL21 strain at 30°C. Bacterial pellets were solubilized in the STE buffer (100 mm NaCl, 10 mm Tris–HCl, pH 8.0, 1 mm EDTA) supplemented with 10 ng/ml lysozyme, 5 mm DTT, 1.5% sarcosyl and 1 × protease inhibitor cocktail. The soluble fusion proteins were then affinity-purified by binding to glutathione-Sepharose 4B beads (Amersham Biosciences) over night at 4°C. The cytosolic extract of HeLa cells was prepared by lysing cells in 25 mm Hepes, pH 7.4, 0.5 mm MgCl2, 150 mm NaCl, 1 mm EDTA and 0.5% Triton X-100, and removing cell debris by centrifugation. The mouse liver extract was prepared by homogenizing one mouse liver in 8 ml of extraction buffer (50 mm Hepes-KOH, pH 7.4, 100 mm NaCl, 5 mm MgCl2 and 0.5% Triton X-100) followed by centrifugation at 100 000g for 30 min in a Beckman TLA 100.3 rotor. GST pull-down experiments were performed by incubating equalized GST fusion protein samples either with 2 ml of cytosolic extract of HeLa cells (∼0.7 mg protein/ml) overnight or with 0.5 ml (10 mg protein/ml) of mouse liver extract for 4 h at 4°C. Samples were separated by 10% SDS–PAGE and analyzed by western blotting with the above-mentioned antibodies against AP complexes.
Deglycosylation assay
Transfected HEK-293 were briefly washed with PBS and scrapped in ice-cold PBS supplemented with protease inhibitors (Halt Protease Inhibitor Cocktail, Pierce Biotechnology). Cell extracts (20 µg proteins) were incubated overnight at room temperature with 2.5 µl (1250U) PNGaseF (New England Biolabs) in 50 mm sodium phosphate pH 7.5 containing 40 mm DTT, 1% NP-40 and 0.5% SDS before SDS–PAGE and immunoblotting analysis with anti-GFP antibodies.
In situ hybridization
Rats were rapidly sacrificed; brains were removed and frozen in isopentane at −30°C. Sagittal and coronal sections were taken at −20°C, thaw-mounted on glass slides and stored at −80°C before use. CLN7 mRNA expression was determined by in situ hybridization with radiolabeled oligonucleotides, as previously described (60,61). Four antisense oligonucleotide probes were designed from the Rattus norvegicus sequence (accession number NM_001047910.1) using the following sequences (from 5′ to 3′): GCT GAC TGC CCA TCG TGG GCC CAA GTA AGT GTA C; GCT GTA GGA ACC TGG ACC CAG AAT AAA GCC CAG GGC; GGC ACC CGT TCT TTC CTG AAG GGA AGT AGC ACC G; and CCG TGC TGC ACT TCC AGA AGT GGT TAA CCA GCC C. Sense probes were synthesized as negative controls.
Oligonucleotides were labeled with [35S]dATP (Perkin Elmer, Ontario, Canada) to 5×108 cpm/µg using terminal transferase (Promega, Fisher Scientific Ltd, Ontario, Canada). On the day of the experiment, slides were fixed with 3.7% formaldehyde in PBS, washed with PBS, rinsed with water, dehydrated with 70% ethanol and air-dried. Sections were then covered with 100 µl of hybridization medium containing 4 × 106 cpm of a mix of the four antisense, or sense, labeled oligonucleotides in the HELIOS buffer (Helios Biosciences; http://www.heliosbioscience.com). Slides were incubated overnight at 42°C, washed and dried. After exposure for 2 weeks to BAS-TR Fuji Imaging screens (Fuji Film Photo Co., Tokyo, Japan), screens were scanned with a Fuji Bioimaging Analyzer BAS-5000.
Human brain neuropathology
Neuropathology was studied in samples from various regions of the central nervous system of formalin-fixed brains from autopsies of patients with vLINCL defined by pathogenic mutation in the CLN7 gene. All the mutations were homozygous: c.881 C > A was proved in four of them, aged 7, 7, 8 and 16; in two of them, aged 8 and 9, it was c.754+2 T > A. The samples were embedded in paraffin and studied by the following list of methods. The presence of lysosomal storage was detected by the intensity and distribution of autofluorescence under UV light excitation (400–440 nm; Nikon BV-2A filter block) and by immunohistochemical detection of Cathepsin D as a luminal lysosomal marker (polyclonal antibody, DAKO, Copenhagen, Denmark). Vulnerability of the affected cell populations was analyzed by histological staining directly revealing the changes in regional cell populations (hematoxylin and eosin, Klüver-Barrera) and by indirect indicators of neuronal degeneration: microglial phagocyte response using CD68 antibodies (clone PGM1, DAKO, Copenhagen, Denmark) and astrocytosis using mouse monoclonal antibody against GFAP (DAKO, Copenhagen, Denmark; dilution 1:200). Sections stained for CD68 were also used for simultaneous evaluation of autofluorescence in non-phagocytic cells. This strongly decreased autofluorescence in phagocytes, but preserved autofluorescence in other cell types, thereby allowing simultaneous evaluation of primary storage (neurons, astrocytes) and of the scavenging phagocyte response of microglia. Scavenged autofluorescent material in glial phagocytes could be evaluated better in sections with reduced exposure to the CD68 antibody (data not shown). Primary antibody reactivity was detected using the appropriate DAKO Envision® kits with DAB chromogen (DAKO, Glostrup, Denmark).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Centre National de la Recherche Scientifique (B.G.); the Agence Nationale de la Recherche (ANR-05-MRAR-007 to B.G.); the Fédération pour la Recherche sur le Cerveau (B.G.); the Fondation Louis D. (B.G.); the Consiglio Nazionale delle Ricerche (G.C.B.); the Fondo per gli Investimenti della Ricerca di Base (RBIN062YH4 to G.C.B.); the Folkhälsan Research Foundation (M.K. and A.-E.L.); the Centre of Excellence in Complex Disease Genetics of the Academy of Finland (Grant no. 129680 to A.-E.L. and A.J.) and the Ministry of Education of the Czech Republic (MSM 0021620806 to M.E. and H.H.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Seija Puomilahti for excellent technical assistance. DNA analysis of the human cases was done by Dr L. Dvorakova and her group in the Prague Institute of Inherited Metabolic Disorders. A.S. is a PhD fellow from the charity Vaincre les Maladies Lysosomales. C.S. is a scientist from the Institut National de la Santé et de la Recherche Médicale. M.K. is a fellow of the Helsinki Biomedical Graduate School.
Conflict of Interest statement. None declared.
References
- 1.Jalanko A., Braulke T. Neuronal ceroid lipofuscinoses. Biochim. Biophys. Acta. 2009;1793:697–709. doi: 10.1016/j.bbamcr.2008.11.004. doi:10.1016/j.bbamcr.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Siintola E., Topcu M., Aula N., Lohi H., Minassian B.A., Paterson A.D., Liu X.Q., Wilson C., Lahtinen U., Anttonen A.K., et al. The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am. J. Hum. Genet. 2007;81:136–146. doi: 10.1086/518902. doi:10.1086/518902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kousi M., Siintola E., Dvorakova L., Vlaskova H., Turnbull J., Topcu M., Yuksel D., Gokben S., Minassian B.A., Elleder M., et al. Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain. 2009;132:810–819. doi: 10.1093/brain/awn366. doi:10.1093/brain/awn366. [DOI] [PubMed] [Google Scholar]
- 4.Aiello C., Terracciano A., Simonati A., Discepoli G., Cannelli N., Claps D., Crow Y.J., Bianchi M., Kitzmuller C., Longo, D., et al. Mutations in MFSD8/CLN7 are a frequent cause of variant-late infantile neuronal ceroid lipofuscinosis. Hum. Mutat. 2009;30:E530–E540. doi: 10.1002/humu.20975. doi:10.1002/humu.20975. [DOI] [PubMed] [Google Scholar]
- 5.Stogmann E., El Tawil S., Wagenstaller J., Gaber A., Edris S., Abdelhady A., Assem-Hilger E., Leutmezer F., Bonelli S., Baumgartner, C., et al. A novel mutation in the MFSD8 gene in late infantile neuronal ceroid lipofuscinosis. Neurogenetics. 2009;10:73–77. doi: 10.1007/s10048-008-0153-1. doi:10.1007/s10048-008-0153-1. [DOI] [PubMed] [Google Scholar]
- 6.Aldahmesh M.A., Al-Hassnan Z.N., Aldosari, M., Alkuraya F.S. Neuronal ceroid lipofuscinosis caused by MFSD8 mutations: a common theme emerging. Neurogenetics. 2009;10:307–311. doi: 10.1007/s10048-009-0185-1. doi:10.1007/s10048-009-0185-1. [DOI] [PubMed] [Google Scholar]
- 7.Topcu M., Tan H., Yalnizoglu D., Usubutun A., Saatci I., Aynaci M., Anlar B., Topaloglu H., Turanli G., Kose, G., et al. Evaluation of 36 patients from turkey with neuronal ceroid lipofuscinosis: clinical, neurophysiological, neuroradiological and histopathologic studies. Turk. J. Pediatr. 2004;46:1–10. [PubMed] [Google Scholar]
- 8.Pao S.S., Paulsen I.T., Saier M.H., Jr Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998;62:1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Transport Classification Database. http://www.tcdb.org/tcdb/index.php?tc=2.A.1 . [Google Scholar]
- 10.Vesa J., Hellsten E., Verkruyse L.A., Camp L.A., Rapola J., Santavuori P., Hofmann S.L., Peltonen L. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature. 1995;376:584–587. doi: 10.1038/376584a0. doi:10.1038/376584a0. [DOI] [PubMed] [Google Scholar]
- 11.Sleat D.E., Donnelly R.J., Lackland H., Liu C.G., Sohar I., Pullarkat R.K., Lobel, P. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 1997;277:1802–1805. doi: 10.1126/science.277.5333.1802. doi:10.1126/science.277.5333.1802. [DOI] [PubMed] [Google Scholar]
- 12.Siintola E., Partanen S., Stromme P., Haapanen A., Haltia M., Maehlen J., Lehesjoki A.E., Tyynela, J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain. 2006;129:1438–1445. doi: 10.1093/brain/awl107. doi:10.1093/brain/awl107. [DOI] [PubMed] [Google Scholar]
- 13.Steinfeld R., Reinhardt K., Schreiber K., Hillebrand M., Kraetzner R., Bruck W., Saftig P., Gartner J. Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am. J. Hum. Genet. 2006;78:988–998. doi: 10.1086/504159. doi:10.1086/504159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savukoski M., Klockars T., Holmberg V., Santavuori P., Lander E.S., Peltonen, L. CLN5, a novel gene encoding a putative transmembrane protein mutated in finnish variant late infantile neuronal ceroid lipofuscinosis. Nat. Genet. 1998;19:286–288. doi: 10.1038/975. doi:10.1038/975. [DOI] [PubMed] [Google Scholar]
- 15.Schmiedt M.L., Bessa C., Heine C., Gil Ribeiro M., Jalanko A., Kyttala A. The neuronal ceroid lipofuscinosis protein CLN5: new insights into cellular maturation, transport and consequences of mutations. Hum. Mutat. 2010;31:356–365. doi: 10.1002/humu.21195. doi:10.1002/humu.21195. [DOI] [PubMed] [Google Scholar]
- 16.International Batten Disease Consortium. Isolation of a novel gene underlying batten disease, CLN3. Cell. 1995;82:949–957. doi: 10.1016/0092-8674(95)90274-0. doi:10.1016/0092-8674(95)90274-0. [DOI] [PubMed] [Google Scholar]
- 17.Gao H., Boustany R.M., Espinola J.A., Cotman S.L., Srinidhi L., Antonellis K.A., Gillis T., Qin X., Liu S., Donahue L.R., et al. Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. Am. J. Hum. Genet. 2002;70:324–335. doi: 10.1086/338190. doi:10.1086/338190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wheeler R.B., Sharp J.D., Schultz R.A., Joslin J.M., Williams R.E., Mole S.E. The gene mutated in variant late-infantile neuronal ceroid lipofuscinosis (CLN6) and in nclf mutant mice encodes a novel predicted transmembrane protein. Am. J. Hum. Genet. 2002;70:537–542. doi: 10.1086/338708. doi:10.1086/338708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ranta S., Zhang Y., Ross B., Lonka L., Takkunen E., Messer A., Sharp J., Wheeler R., Kusumi K., Mole, S., et al. The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant mice are associated with mutations in CLN8. Nat. Genet. 1999;23:233–236. doi: 10.1038/13868. doi:10.1038/13868. [DOI] [PubMed] [Google Scholar]
- 20.Kyttala A., Lahtinen U., Braulke T., Hofmann S.L. Functional biology of the neuronal ceroid lipofuscinoses (NCL) proteins. Biochim. Biophys. Acta. 2006;1762:920–933. doi: 10.1016/j.bbadis.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 21.Bonifacino J.S., Traub L.M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. doi:10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- 22.Blagoveshchenskaya A.D., Norcott J.P., Cutler D.F. Lysosomal targeting of P-selectin is mediated by a novel sequence within its cytoplasmic tail. J. Biol. Chem. 1998;273:2729–2737. doi: 10.1074/jbc.273.5.2729. doi:10.1074/jbc.273.5.2729. [DOI] [PubMed] [Google Scholar]
- 23.Cherqui S., Kalatzis V., Trugnan G., Antignac C. The targeting of cystinosin to the lysosomal membrane requires a tyrosine-based signal and a novel sorting motif. J. Biol. Chem. 2001;276:13314–13321. doi: 10.1074/jbc.M010562200. doi:10.1074/jbc.M010562200. [DOI] [PubMed] [Google Scholar]
- 24.Kyttala A., Ihrke G., Vesa J., Schell M.J., Luzio J.P. Two motifs target batten disease protein CLN3 to lysosomes in transfected nonneuronal and neuronal cells. Mol. Biol. Cell. 2004;15:1313–1323. doi: 10.1091/mbc.E03-02-0120. doi:10.1091/mbc.E03-02-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Braulke T., Bonifacino J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta. 2009;1793:605–614. doi: 10.1016/j.bbamcr.2008.10.016. doi:10.1016/j.bbamcr.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 26.Ihrke G., Gray S.R., Luzio J.P. Endolyn is a mucin-like type I membrane protein targeted to lysosomes by its cytoplasmic tail. Biochem. J. 2000;345:287–296. doi:10.1042/0264-6021:3450287. [PMC free article] [PubMed] [Google Scholar]
- 27.Metcalf D.J., Calvi A.A., Seaman M.N., Mitchison H.M., Cutler D.F. Loss of the batten disease gene CLN3 prevents exit from the TGN of the mannose 6-phosphate receptor. Traffic. 2008;9:1905–1914. doi: 10.1111/j.1600-0854.2008.00807.x. doi:10.1111/j.1600-0854.2008.00807.x. [DOI] [PubMed] [Google Scholar]
- 28.Ford M.G., Pearse B.M., Higgins M.K., Vallis Y., Owen D.J., Gibson A., Hopkins C.R., Evans P.R., McMahon H.T. Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science. 2001;291:1051–1055. doi: 10.1126/science.291.5506.1051. doi:10.1126/science.291.5506.1051. [DOI] [PubMed] [Google Scholar]
- 29.Kyttala A., Yliannala K., Schu P., Jalanko A., Luzio J.P. AP-1 and AP-3 facilitate lysosomal targeting of batten disease protein CLN3 via its dileucine motif. J. Biol. Chem. 2005;280:10277–10283. doi: 10.1074/jbc.M411862200. doi:10.1074/jbc.M411862200. [DOI] [PubMed] [Google Scholar]
- 30.Partridge E.A., Le Roy C., Di Guglielmo G.M., Pawling J., Cheung P., Granovsky M., Nabi I.R., Wrana J.L., Dennis J.W. Regulation of cytokine receptors by golgi N-glycan processing and endocytosis. Science. 2004;306:120–124. doi: 10.1126/science.1102109. doi:10.1126/science.1102109. [DOI] [PubMed] [Google Scholar]
- 31.Cha S.K., Ortega B., Kurosu H., Rosenblatt K.P., Kuro, O M., Huang C.L. Removal of sialic acid involving klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc. Natl Acad. Sci. USA. 2008;105:9805–9810. doi: 10.1073/pnas.0803223105. doi:10.1073/pnas.0803223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delacour D., Cramm-Behrens C.I., Drobecq H., Le Bivic A., Naim H.Y., Jacob R. Requirement for galectin-3 in apical protein sorting. Curr. Biol. 2006;16:408–414. doi: 10.1016/j.cub.2005.12.046. doi:10.1016/j.cub.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 33.Heine C., Koch B., Storch S., Kohlschutter A., Palmer D.N., Braulke T. Defective endoplasmic reticulum-resident membrane protein CLN6 affects lysosomal degradation of endocytosed arylsulfatase A. J. Biol. Chem. 2004;279:22347–22352. doi: 10.1074/jbc.M400643200. doi:10.1074/jbc.M400643200. [DOI] [PubMed] [Google Scholar]
- 34.Lonka L., Kyttala A., Ranta S., Jalanko A., Lehesjoki A.E. The neuronal ceroid lipofuscinosis CLN8 membrane protein is a resident of the endoplasmic reticulum. Hum. Mol. Genet. 2000;9:1691–1697. doi: 10.1093/hmg/9.11.1691. doi:10.1093/hmg/9.11.1691. [DOI] [PubMed] [Google Scholar]
- 35.Mole S.E., Michaux G., Codlin S., Wheeler R.B., Sharp J.D., Cutler D.F. CLN6, which is associated with a lysosomal storage disease, is an endoplasmic reticulum protein. Exp. Cell Res. 2004;298:399–406. doi: 10.1016/j.yexcr.2004.04.042. doi:10.1016/j.yexcr.2004.04.042. [DOI] [PubMed] [Google Scholar]
- 36.Bagshaw R.D., Mahuran D.J., Callahan J.W. A proteomic analysis of lysosomal integral membrane proteins reveals the diverse composition of the organelle. Mol. Cell. Proteomics. 2005;4:133–143. doi: 10.1074/mcp.M400128-MCP200. [DOI] [PubMed] [Google Scholar]
- 37.Schroder B., Wrocklage C., Pan C., Jager R., Kosters B., Schafer H., Elsasser H.P., Mann, M., Hasilik, A. Integral and associated lysosomal membrane proteins. Traffic. 2007;8:1676–1686. doi: 10.1111/j.1600-0854.2007.00643.x. doi:10.1111/j.1600-0854.2007.00643.x. [DOI] [PubMed] [Google Scholar]
- 38.Morin P., Sagne, C., Gasnier, B. Functional characterization of wild-type and mutant human sialin. EMBO J. 2004;23:4560–4570. doi: 10.1038/sj.emboj.7600464. doi:10.1038/sj.emboj.7600464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wreden C.C., Wlizla, M., Reimer R.J. Varied mechanisms underlie the free sialic acid storage disorders. J. Biol. Chem. 2005;280:1408–1416. doi: 10.1074/jbc.M411295200. doi:10.1074/jbc.M411295200. [DOI] [PubMed] [Google Scholar]
- 40.Augustin R., Riley, J., Moley K.H. GLUT8 contains a [DE]XXXL[LI] sorting motif and localizes to a late endosomal/lysosomal compartment. Traffic. 2005;6:1196–1212. doi: 10.1111/j.1600-0854.2005.00354.x. doi:10.1111/j.1600-0854.2005.00354.x. [DOI] [PubMed] [Google Scholar]
- 41.Sitaram A., Piccirillo R., Palmisano I., Harper D.C., Dell'Angelica E.C., Schiaffino M.V., Marks M.S. Localization to mature melanosomes by virtue of cytoplasmic dileucine motifs is required for human OCA2 function. Mol. Biol. Cell. 2009;20:1464–1477. doi: 10.1091/mbc.E08-07-0710. doi:10.1091/mbc.E08-07-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steenhuis P., Herder S., Gelis S., Braulke T., Storch S. Lysosomal targeting of the CLN7 membrane glycoprotein and transport via the plasma membrane require a dileucine motif. Traffic. 2010;11:987–1000. doi: 10.1111/j.1600-0854.2010.01073.x. [DOI] [PubMed] [Google Scholar]
- 43.Jarvela I., Lehtovirta M., Tikkanen R., Kyttala A., Jalanko A. Defective intracellular transport of CLN3 is the molecular basis of Batten disease (JNCL) Hum. Mol. Genet. 1999;8:1091–1098. doi: 10.1093/hmg/8.6.1091. doi:10.1093/hmg/8.6.1091. [DOI] [PubMed] [Google Scholar]
- 44.Lyly A., von Schantz C., Heine C., Schmiedt M.L., Sipila T., Jalanko A., Kyttala A. Novel interactions of CLN5 support molecular networking between neuronal ceroid lipofuscinosis proteins. BMC Cell Biol. 2009;10:83. doi: 10.1186/1471-2121-10-83. doi:10.1186/1471-2121-10-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalatzis V., Cherqui S., Antignac, C., Gasnier, B. Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. EMBO J. 2001;20:5940–5949. doi: 10.1093/emboj/20.21.5940. doi:10.1093/emboj/20.21.5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sagne, C., Gasnier, B. Molecular physiology and pathophysiology of lysosomal membrane transporters. J. Inherit. Metab. Dis. 2008;31:258–266. doi: 10.1007/s10545-008-0879-9. doi:10.1007/s10545-008-0879-9. [DOI] [PubMed] [Google Scholar]
- 47.Ruivo R., Anne C., Sagne, C., Gasnier, B. Molecular and cellular basis of lysosomal transmembrane protein dysfunction. Biochim. Biophys. Acta. 2009;1793:636–649. doi: 10.1016/j.bbamcr.2008.12.008. doi:10.1016/j.bbamcr.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 48.Elrick M.J., Pacheco C.D., Yu T., Dadgar N., Shakkottai V.G., Ware C., Paulson H.L., Lieberman A.P. Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum. Mol. Genet. 2010;19:837–847. doi: 10.1093/hmg/ddp552. doi:10.1093/hmg/ddp552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ko D.C., Milenkovic L., Beier S.M., Manuel H., Buchanan, J., Scott M.P. Cell-autonomous death of cerebellar purkinje neurons with autophagy in niemann-pick type C disease. PLoS Genet. 2005;1:81–95. doi: 10.1371/journal.pgen.0010007. doi:10.1371/journal.pgen.0010081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wartosch L., Fuhrmann J.C., Schweizer M., Stauber, T., Jentsch T.J. Lysosomal degradation of endocytosed proteins depends on the chloride transport protein ClC-7. FASEB J. 2009;23:4056–4068. doi: 10.1096/fj.09-130880. doi:10.1096/fj.09-130880. [DOI] [PubMed] [Google Scholar]
- 51.Venkatachalam K., Long A.A., Elsaesser R., Nikolaeva D., Broadie K., Montell C. Motor deficit in a Drosophila model of mucolipidosis type IV due to defective clearance of apoptotic cells. Cell. 2008;135:838–851. doi: 10.1016/j.cell.2008.09.041. doi:10.1016/j.cell.2008.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luiro K., Kopra O., Lehtovirta, M., Jalanko A. CLN3 protein is targeted to neuronal synapses but excluded from synaptic vesicles: new clues to batten disease. Hum. Mol. Genet. 2001;10:2123–2131. doi: 10.1093/hmg/10.19.2123. doi:10.1093/hmg/10.19.2123. [DOI] [PubMed] [Google Scholar]
- 53.Halila R., Baumann M., Ikonen E., Enomaa N., Peltonen L. Human leucocyte aspartylglucosaminidase. Evidence for two different subunits in a more complex native structure. Biochem. J. 1991;276:251–256. doi: 10.1042/bj2760251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peden A.A., Park G.Y., Scheller R.H. The di-leucine motif of vesicle-associated membrane protein 4 is required for its localization and AP-1 binding. J. Biol. Chem. 2001;276:49183–49187. doi: 10.1074/jbc.M106646200. doi:10.1074/jbc.M106646200. [DOI] [PubMed] [Google Scholar]
- 55.Carrel D., Masson J., Al Awabdh S., Capra C.B., Lenkei Z., Hamon M., Emerit M.B., Darmon M. Targeting of the 5-HT1A serotonin receptor to neuronal dendrites is mediated by Yif1B. J. Neurosci. 2008;28:8063–8073. doi: 10.1523/JNEUROSCI.4487-07.2008. doi:10.1523/JNEUROSCI.4487-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huettner J.E., Baughman R.W. Primary culture of identified neurons from the visual cortex of postnatal rats. J. Neurosci. 1986;6:3044–3060. doi: 10.1523/JNEUROSCI.06-10-03044.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levi G., Patrizio M., Bernardo A., Petrucci T.C., Agresti C. Human immunodeficiency virus coat protein gp120 inhibits the beta-adrenergic regulation of astroglial and microglial functions. Proc. Natl Acad. Sci. USA. 1993;90:1541–1545. doi: 10.1073/pnas.90.4.1541. doi:10.1073/pnas.90.4.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Agresti C., Aloisi F., Levi G. Heterotypic and homotypic cellular interactions influencing the growth and differentiation of bipotential oligodendrocyte-type-2 astrocyte progenitors in culture. Dev. Biol. 1991;144:16–29. doi: 10.1016/0012-1606(91)90474-h. doi:10.1016/0012-1606(91)90474-H. [DOI] [PubMed] [Google Scholar]
- 59.Schmittgen T.D., Livak K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. doi:10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 60.Gras C., Amilhon B., Lepicard E.M., Poirel O., Vinatier J., Herbin M., Dumas S., Tzavara E.T., Wade M.R., Nomikos G.G., et al. The vesicular glutamate transporter VGLUT3 synergizes striatal acetylcholine tone. Nat. Neurosci. 2008;11:292–300. doi: 10.1038/nn2052. doi:10.1038/nn2052. [DOI] [PubMed] [Google Scholar]
- 61.Amilhon B., Lepicard E., Renoir T., Mongeau R., Popa D., Poirel O., Miot S., Gras C., Gardier A.M., Gallego J., et al. VGLUT3 (vesicular glutamate transporter type 3) contribution to the regulation of serotonergic transmission and anxiety. J. Neurosci. 2010;30:2198–2210. doi: 10.1523/JNEUROSCI.5196-09.2010. doi:10.1523/JNEUROSCI.5196-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.