

Abstract
Mitochondrial ferritin (FtMt) is a nuclear-encoded iron-sequestering protein that specifically localizes in mitochondria. In mice it is highly expressed in cells characterized by high-energy consumption, while is undetectable in iron storage tissues like liver and spleen. FtMt expression in mammalian cells was shown to cause a shift of iron from cytosol to mitochondria, and in yeast it rescued the defects associated with frataxin deficiency. To study the role of FtMt in oxidative damage, we analyzed the effect of its expression in HeLa cells after incubation with H2O2 and Antimycin A, and after a long-term growth in glucose-free media that enhances mitochondrial respiratory activity. FtMt reduced the level of reactive oxygen species (ROS), increased the level of adenosine 5'triphosphate and the activity of mitochondrial Fe-S enzymes, and had a positive effect on cell viability. Furthermore, FtMt expression reduces the size of cytosolic and mitochondrial labile iron pools. In cells grown in glucose-free media, FtMt level was reduced owing to faster degradation rate, however it still protected the activity of mitochondrial Fe-S enzymes without affecting the cytosolic iron status. In addition, FtMt expression in fibroblasts from Friedreich ataxia (FRDA) patients prevented the formation of ROS and partially rescued the impaired activity of mitochondrial Fe-S enzymes, caused by frataxin deficiency. These results indicate that the primary function of FtMt involves the control of ROS formation through the regulation of mitochondrial iron availability. They are consistent with the expression pattern of FtMt observed in mouse tissues, suggesting a FtMt protective role in cells characterized by defective iron homeostasis and respiration, such as in FRDA.
INTRODUCTION
Since living organisms developed oxidative metabolism, limitation of the highly damaging by-products of respiration, the reactive oxygen species (ROS), has become fundamental. Oxidation of ferrous iron generates superoxide anions and toxic hydroxyl radicals, thus the control of cellular iron homeostasis represents an important mechanism for ROS limitation (1). Ferritins are proteins dedicated for a safe cellular iron storage; they assemble in 24-subunit shells that oxidize free ferrous iron and sequester it inside the cavity in a non-reactive ferric form (2). Mammalian ferritins are ubiquitous cytoplasmic proteins made by the combination of two different subunits, ferritin Heavy chain (FtH) with ferroxidase activity and ferritin Light chain (FtL) that promotes iron nucleation (3). Recently, a ferritin encoded by a nuclear gene and with specific mitochondrial localization has been characterized in human, mouse, Drosophila and plants (4–7). Mitochondrial ferritin (FtMt) is a 24-mer homopolymer (8) that has ferroxidase activity (9). It is different from cytosolic ferritins; FtMt is not ubiquitous and not found in cultured cells such as HeLa or H1299 (10). Over-expression of FtMt influences cellular iron homeostasis, driving the metal to mitochondria, resulting in cytosolic iron starvation (11,12). This effect explained the reduced growth in nude mice of a H1299 clone expressing FtMt (13). In humans, the protein is highly expressed in testis (4) and in the sideroblasts of patients affected by sideroblastic anemia (14,15). This is a heterogeneous group of disorders that have in common the presence of ringed erythroblasts with iron-loaded mitochondria (16). In mouse, FtMt is highly expressed in testis, particularly in spermatocytes and Leydig cells, and was also found in other organs, including heart, nervous system, pancreas (islet of Langerhans) and kidney, but only in cells characterized by high metabolic activity. FtMt was not found in liver and spleen, which store iron and express high levels of cytosolic ferritins (17).
Mitochondria require iron for heme and iron–sulfur cluster (ISC) synthesis, but also generate large amounts of hydrogen peroxide as by-products of respiratory activity (18). FtMt might be important to reduce the harmful interaction between Fe and H2O2, which may be particularly dangerous in cells with sustained respiratory activity. FtMt may also be important in pathological conditions where both mitochondrial iron homeostasis and respiration are defective, such as in Friedreich's ataxia (FRDA) (19). FRDA is caused by decreased expression of frataxin, a mitochondrial protein that acts as an iron chaperone for the synthesis of heme and ISC (20,21). Heart tissues of FRDA patients showed defective activity of aconitase and mitochondrial respiratory complexes, and increased iron deposits (22–24). The cultured skin fibroblasts and lymphoblasts from FRDA patients were more sensitive to oxidative insults than those from healthy donors (25). Also frataxin-free pancreas and liver of conditional knock-out mice (26,27) and tissues of FRDA mouse models with GAA repeat expansion showed oxidative damage and mitochondrial iron accumulation (28). However, the frataxin-free hearts of conditional knock-out mice did not show signs of oxidative damage, and mitochondrial iron overload was a late event in disease progression (29). Various studies showed that frataxin-deficient yeasts have respiratory deficiency, a 10-fold increase in mitochondrial iron content, increased sensitivity to oxidants, defective Fe-S enzymes activities and mitochondrial DNA damage (30–32). The expression of FtMt rescued the respiratory deficiency, reduced the mitochondrial iron accumulation and improved Fe-S enzyme activities; moreover, it increased the resistance to oxidants and protected from mitochondrial DNA damage (33). The finding of FtMt in cardiomyocytes and in mitochondrial fraction of fibroblasts from two FRDA patients (22,34) suggests that its expression may be a response to the disorder. A protective role of FtMt was recently demonstrated in frataxin-silenced HeLa cells (35). Thus, we considered interesting to verify if FtMt expression could protect from oxidative damage and compensate for frataxin deficiency in cells from FRDA patients.
In this work, we expressed FtMt in HeLa cells and we studied their properties in response to stress conditions induced by H2O2 and Antimycin A, with changes of growth media and after prolonged growth in galactose-supplemented glucose-free medium, a condition that increases respiratory activity (36). We also expressed FtMt in FRDA fibroblasts that have defective respiratory activity caused by frataxin deficiency. We found that, under all tested conditions, FtMt protected the cells from oxidative damage and maintained the balance between mitochondria iron availability and ROS formation.
RESULTS
Mitochondrial ferritin and oxidative stress
We studied a HeLa-tTA-FtMt clone in which FtMt expression was modulated by the addition (control cells) or the removal (FtMt-cells) of doxycycline to culture medium (11). After 7 days of growth in the absence of doxycycline, FtMt reached a maximum concentration of 85 ± 15 ng for milligrams of total protein. To determine the effect of FtMt on ROS generation, the cells were incubated with the ROS-sensitive mitochondrial probe Dihydrorhodamine-123 (DHR-123) (37) and then exposed to 200 µm H2O2 for 30 min. In the FtMt cells, the relative increase of fluorescence was <50% than that of the control cells (Fig. 1A). In other experiments, the 200 µm H2O2 treatment was extended up to 2 h; in FtMt cells, the adenosine 5'triphosphate (ATP) concentration remained >60% of the initial, while in control cells it decreased to <40% (Fig. 1B). A 2 h treatment with 600 µm H2O2 reduced the number of viable cells to <30% in the control cells, and to >40% in the FtMt cells (Fig. 1C). We also analyzed the HeLa-tTA-FtMt222 clone that expresses an FtMt variant with an inactive ferroxidase activity (11), the cells viability after the H2O2 treatments was the same as that of the control cells (not shown), indicating that the ferroxidase activity is essential for increasing resistance of the cell to oxidative damages.
Figure 1.

HeLa-tTA-FtMt clone subjected to H2O2 treatment. Effects of H2O2 treatment was analyzed on HeLa-tTA-FtMt clone, not induced (-FtMt) or induced (+FtMt) for 7 days. (A) Reactive oxygen species (ROS) generation was measured by Rhodamine-123 fluorescence development after incubation of cells with 200 µm H2O2 for 30 min. Values are presented as relative increase. (B) Adenosine 5'triphosphate (ATP) production was evaluated after 2 h cellular treatment with 200 µM H2O2 by ATP luciferase assay. (C) Cell viability was assayed after 2 h of treatment with 600 µm H2O2, by thiazolyl blue tetrazolium bromide assay. ATP and cell viability are presented as percentage relative to untreated cells. A representative of three sets of results is shown and error bars represent standard deviations referred to six independent determinations. Horizontal bars point at significant differences. p: result of Student's t-test.
For a different and more localized oxidative insult, we used Antimycin A, an uncoupler reagent that generates oxidative stress specifically at the mitochondrial level (38). Treatment of control cells with 5 µm Antimycin A for 18 h reduced the activity of the succinate dehydrogenase (SDH) and mitochondrial aconitase (mtACO) Fe-S enzymes to 30% and 18%, respectively, while it did not affect the activity of the non-Fe-S enzyme malate dehydrogenase (MDH) (Fig. 2A). The same treatment had lower effects on FtMt cells, where the activities of the SDH and mtACO were reduced to 50% and 30%, respectively (Fig. 2A). A high concentration of Antimycin A (10 µm for 18 h) induced a 60% increase of ROS above the basal level in control cells (Fig. 2B, left panel), but a non-significant increase in FtMt cells, which also showed a lower basal level (Fig. 2B, left panel). This treatment caused a reduction of ATP to approximately 20% of the initial level in the control cells, and to 50% in FtMt cells (Fig. 2B, right panel). Moreover, it induced the partial release of cytochrome c from the mitochondria of the control cells (not shown). For more evident effects we used harsh conditions by treating cells with 100 µm Antimycin A for 5 h, and then we analyzed the mitochondrial (MF) and the post-mitochondrial fractions (PMF) by western blotting. The release of cytochrome c was detected in the PMF fraction of control cells, as expected, while it was barely visible in the PMF of FtMt cells (Fig. 2C).
Figure 2.

HeLa-tTA-FtMt clone subjected to Antimycin A treatment. Effects of Antimicyn A treatment was analyzed on HeLa-tTA-FtMt clone, not induced (-FtMt) or induced (+FtMt) for 7 days. (A) Succinate dehydrogenase, aconitase and malate dehydrogenase activities were measured spectrophotometrically in mitochondrial fractions of cells treated or not with 5 µm Antimycin A for 18 h. The experiment was repeated three times and one representative experiment is shown. Error bars represent standard deviations referred to activity measurements. (B) Reactive oxygen species (ROS) and adenosine 5'triphosphate (ATP) production were measured after cellular treatments with 10 µm Antimycin A for 18 h. ATP production is presented as percentage relative to untreated cells. A representative of three sets of results is shown and error bars represent standard deviations referred to six independent determinations. Results were normalized on protein content. Horizontal bars point at significant differences. p: result of Student's t-test. (C) Cells were treated or not with 100 µm Antimycin A for 6 h. Protein extracts (30 µg) of mitochondrial (MF) and post-mitochondrial (PMF) fractions were analyzed for cytochrome c content by western blot; β-actin was assayed to confirm equal loading of PMFs. The experiment was repeated two times.
Mitochondrial ferritin and replacement of glucose with galactose in culture medium
In the experiments shown above HeLa-tTA-FtMt clone was grown in a medium containing 25 mm glucose, condition in which cells are thought to produce most ATP from glucose through glycolysis and from l-glutamine through tricarboxylic acid cycle. To force oxidative phosphorylation we grew the cells in glucose-free medium supplemented with 5 mm galactose (39). After 7 days of FtMt induction in glucose medium, cells were transferred to a galactose medium for another 72 h and ROS level was analyzed. It increased about two-fold in control cells, while it remained unchanged in FtMt cells (Fig. 3A). Interestingly, ROS level was only marginally influenced by FtMt expression in the cells grown in glucose (Figs 3A and 2B, left panel). To understand the link between FtMt and ROS level, we analyzed the labile iron pool (40) in the mitochondrial (mtLIP) and cytosolic (cytLIP) compartments. We used the iron-sensitive fluorescent probes Rhodamine B 4-[(1,10-Phenanthrolin-5-yl) Aminocarbonyl] benzyl ester (RPA) for mitochondria (41) and Calcein-AM for cytosol (42). In control cells, the switch to galactose caused a two-fold increase of mtLIP level and a decrease of the cytLIP level (Fig. 3B). In the FtMt cells, the effect was similar, but the mtLIP and the cytLIP levels were 25–30% lower. The finding suggested that the change from glucose to galactose causes an oxidative stress to the cells, and that FtMt protects in this condition also.
Figure 3.

HeLa-tTA-FtMt clone grown 3 days in glucose-free medium supplemented with galactose. Reactive oxygen species (ROS), mitochondrial (mtLIP) and cytosolic (cytLIP) labile iron pools were measured in HeLa-tTA-FtMt clone, not induced (−FtMt) or induced (+FtMt) for 7 days in glucose medium and then transferred to galactose medium for 3 days. (A) Left panel: representative pictures of Rhodamine-123 fluorescence showing differences in the two growth conditions. Right panel: ROS quantification. A representative of three sets of results is shown and error bars represent standard deviations referred to six independent determinations. (B) Estimation of mtLIP and cytLIP. Error bars indicate standard deviations referred to three independent experiments. Data are presented as percentage relative to the value obtained in cells without FtMt and grown in glucose medium. Results were normalized on protein content. Horizontal bars point at significant differences. p: result of Student's t-test.
Mitochondrial ferritin during extended growth in glucose and galactose media
HeLa-tTA-FtMt cells, whether or not induced for FtMt expression, were plated in glucose or in galactose media and maintained in exponential growth rate for 21 days. In glucose medium, FtMt concentration, monitored by enzyme-linked immunosorbent assay (ELISA), reached the plateau after 7 days of induction, while in galactose FtMt kept increasing for 21 days and reached a much lower level (Fig. 4A, left panel). It was unlikely that glucose or galactose affected FtMt expression by acting on cytomegalovirus promoter, while probably they modified FtMt degradation. To verify this, FtMt cells were metabolically labeled for 18 h, and FtMt immunoprecipitated with the α-FtMt antibody at 0-, 16- and 24 h after labeling. The specificity of the assay was demonstrated by the absence of immunoprecipitates from cells not expressing FtMt (Fig. 4A, central panel). During growth in glucose medium, 75% and 70% of the labeled FtMt were recovered after 16- and 24 h, respectively; while in galactose it was only 47% and 40%, suggesting an increased protein degradation (Fig. 4A, right panel). After 21 days in glucose medium, the cytLIP and mtLIP in FtMt cells were reduced by 25–30% with respect to those measured in control cells (Fig. 4B, left and central panels). The 21 days of growth in galactose caused a reduction of mtLIP in control cells, which was even further reduced in FtMt cells, reaching 20% (Fig. 4B, central panel). No reduction occurred to the cytLIP in the FtMt-cells grown in galactose medium (Fig. 4B, left panel) and the variation of cytosolic aconitase (cytACO) activities measured in the two conditions paralleled that of the cytLIP values, 25% reduction in glucose and no reduction in galactose (not shown). The expression of FtMt reduced ROS levels by approximately 20% in glucose medium and prevented the ROS increase in galactose medium (Fig. 4B, right panel). In glucose medium, the continuous expression of FtMt also affected SDH activity which was reduced by 30% (Fig. 4C), differently from what was observed during the first 7 days of induction where it was unchanged (Fig. 2A). The activities of mtACO and MDH were unaffected (Fig. 4C). In FtMt-cells grown in galactose, the SDH, mtACO and MDH activities were significantly increased with respect to those measured in FtMt-cells grown in glucose (Fig. 4C). In contrast, in control cells, we observed only a slight increase in enzyme activities, which was not statistically significant (Fig. 4C). In particular, the deficit of SDH activity, observed in the FtMt-cells grown in glucose medium, was rescued in galactose medium and mtACO activity was approximately 20% higher with respect to that of control cells (Fig. 4C). To establish whether the variation of Fe-S enzyme activities were related to altered protein levels, we performed SDH and mtACO immunoblotting analysis. The results indicated that both the enzyme levels were not changed by FtMt expression in both growth conditions (not shown). Finally, population doubling time (PDT) measured in glucose medium was 23.2 ± 2.3 h for FtMt-cells and 19.8 ± 2.1 h for control cells (P < 0,025), while in galactose medium FtMt- and control cells did not show differences in PDT (31.3 ± 2.1 h).
Figure 4.

HeLa-tTA-FtMt clone during long-term-induced mitochondrial ferritin expression in glucose and galactose media. Data were collected in HeLa-tTA-FtMt clone, not induced (−FtMt) or induced (+FtMt) for 21 days, grown in glucose or galactose media. (A) Left panel: mitochondrial ferritin content, measured by enxyle-linked immunosorbent assay, during a representative induction. The experiment was repeated twice. Central and right panels: autoradiography after sodium dodecyl sulfate–polyacrylamide gel electrophoresis of samples collected at indicated times after 35S-met and 35S-cys labeling and immunoprecipitated by α-FtMt antibody. Central panel: assay for α-FtMt antibody specificity. Right panel: FtMt pulse-chase experiments were repeated three times and one representative is shown. Arrows indicated FtMt. (B) Left and central panels: cytosolic (cytLIP) and mitochondrial (mtLIP) labile iron pools determined in three independent experiments. Data are presented as percentage relative to the values obtained in cells without FtMt and grown in glucose medium. Right panel: reactive oxygen species quantification. A representative of three sets of results is shown and error bars represent standard deviations referred to six independent determinations. (C) Enzymatic activities were measured in mitochondrial fractions and determined in four independent experiments. Data are presented as percentage relative to the values obtained in cells without FtMt and grown in glucose medium. Results were normalized to protein content. Error bars represent standard deviations. Horizontal bars indicate significant differences. p: result of Student's t-test.
Mitochondrial ferritin in Friedreich ataxia cells
Reduced mitochondrial activity, susceptibility to oxidative stress and defective mitochondrial iron utilization occur in FRDA cells (43). We investigated the effect of FtMt expression in skin fibroblasts from two FRDA patients [cell lines 1037 (25) and 117] and from two healthy controls. They were transiently transfected with the pcDNA3-huFtMt plasmid-encoding FtMt (11), reaching an average transfection efficiency of approximately 25%. Immunocytochemical analysis confirmed that FtMt was correctly localized (Fig. 5A, left panel), and electrophoretic data showed that it assembled into a ferritin shell (not shown). First, we evaluated the sensitivity to H2O2 insults of FtMt-transfected FRDA fibroblasts (FRDA–FtMt) and of FRDA fibroblasts transfected with an empty plasmid (FRDA-mock). After 3 h treatments with 200 µm and 300 µm H2O2 cell viability was 20% higher in FRDA–FtMt cells than in FRDA-mock cells (Fig. 5A, central panel). However, if the cells were first treated with 100 µm Desferrioxamine for 15 h, the incubation with 800 µm H2O2 for 3 h did not reduce cell viability (Fig. 5A, right panel). It is thought that increased sensitivity to H2O2 depends on iron misregulation that occurs in FRDA at mitochondrial level (25). ROS level were three times higher in FRDA than in control fibroblasts, even in the absence of any oxidizing insult (Fig. 5B, left and central panels). FtMt transfection caused a small, but significant, decrease of ROS formation (Fig. 5B, right panel), even with a low transfection efficiency of these primary cells. Co-staining showed that cells expressing FtMt had low ROS level (not shown). Finally, we evaluated if increased ROS contributed to affect mitochondrial Fe-S enzyme activities in FRDA cells. In 1037 and 117 cells, the activities of mtACO and SDH were reduced by 20% and approximately 50%, respectively, compared with controls, while the one that of MDH was unaffected (Fig. 6A). After FtMt transfection, the mtACO activity was increased by 15% in FRDA fibroblasts, while MDH activity was unaffected (Fig. 6B). A direct evaluation of enzymatic functionality on live cells showed that FtMt expressing fibroblasts had higher SDH activity than the non-expressing ones (Fig. 6C).
Figure 5.

Mitochondrial ferritin expression in Friedreich ataxia (FRDA) fibroblasts. (A) Left panel: representative picture showing localization of mitochondrial ferritin (FtMt) in transfected FRDA fibroblasts assayed by immunocytochemistry using α-FtMt antibody. Central panel: cells viability was revealed by trypan blue assay on FRDA-mock and FRDA–FtMt fibroblasts subjected to increasing amounts of H2O2 for 3 h. Data were collected on four independent transfections for each of the two FRDA patients. Significant differences are pointed by P-values. Right panel: representative experiment in which transfected cells were pretreated with 100 µm iron chelator DFO prior to incubation with 800 µm H2O2. (B) Reactive oxygen species (ROS) were measured following the fluorescence of Rhodamine-123 in controls and FRDA fibroblasts. Left panel: representative pictures of fluorescence intensity. Central panel: ROS quantification on eight independent determinations. Right panel: ROS quantification on eight independent transfections with FtMt encoding or empty plasmids. Results were normalized on protein content. Error bars indicate standard deviations. Horizontal bars point significant differences. DFO: desferoxamine. p: result of Student's t-test.
Figure 6.
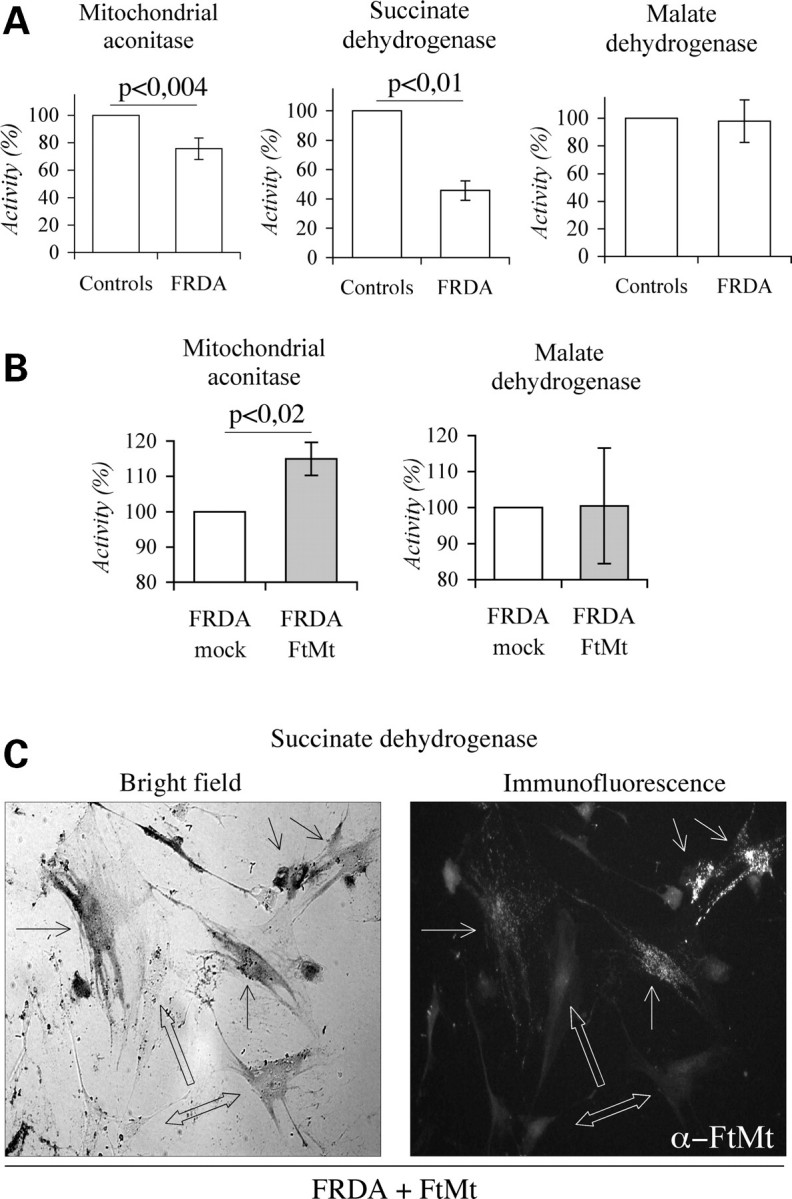
Mitochondrial ferritin expression in Friedreich ataxia (FRDA) fibroblasts, iron–sulfur cluster enzymatic activities. Mitochondrial enzymatic activities were measured in mitochondrial fractions of controls and FRDA fibroblasts. (A) Basal enzymatic activities determined in two independent experiments. Results are presented as percentage relative to control fibroblasts. (B) Activities measured after three independent transfections with mitochondrial ferritin (FtMt) encoding or empty plasmids on FRDA cells. Values are presented as percentage relative to activities measured in mock cells. Error bars represent standard deviations. Horizontal bars point at significant differences. p: result of Student's t-test. (C) Representative pictures illustrating co-determination of succinate dehydrogenase (SDH) activity and FtMt-expressing cells in transfected FRDA fibroblasts. Bright field: simple arrows point cells with high SDH activity. Empty arrows indicate low or negative fibroblasts. Immunofluorence: simple arrows indicate FtMt-positive cells. Empty arrows indicate FtMt-negative cells.
DISCUSSION
Mitochondrial ferritin protects HeLa cells from oxidative damage
H-ferritin has an antioxidant activity, not only because it oxidizes the potentially toxic ferrous iron, but also because it consumes ROS, such as H2O2 (44,45). This partially explains why the expression of cytosolic H-ferritin protected HeLa cells from H2O2 toxicity, while its silencing yield the opposite effect (46,47). FtMt is thought to have a similar role, although in vitro data suggested that it has lower capacity to inhibit free radical formation than cytosolic H-ferritin (9). This work shows that FtMt expression protected HeLa cells from oxidative damage that caused a decrease of cell viability, of ATP production and the inhibition of mitochondrial Fe-S enzymes, and that the protection was linked to FtMt ferroxidase activity. In fact, expression of the FtMt222 variant with inactivated ferroxidase activity (11) did not show any protection. Protective role of FtMt was observed in the HeLa-tTA-FtMt clone subjected to various types of oxidative insults, including H2O2, Antimicyn A, that acts more specifically on the mitochondria, and the switch from glucose to galactose media, a pro-oxidant condition that caused an evident increase in ROS production. Also FRDA fibroblasts are characterized by chronic oxidative stress secondary to frataxin deficiency (19), and also in these cells FtMt showed a protective effect.
We previously demonstrated that FtMt positively competes with cytosolic ferritin for iron incorporation (11). Here, we add that FtMt expression reduces LIP level both in the cytosolic and in the mitochondrial compartments of cells grown in normal glucose medium. Interestingly, we observed that, under all conditions, the variation of the mtLIP size was accompanied by a parallel variation of cellular ROS, suggesting a causal relationship. This was particularly evident in control cells grown in galactose medium, where both mtLIP and ROS levels increased in the initial growth phase and significantly decreased under long-term growth condition. The reduction of mtLIP promoted by FtMt might also affect superoxide dismutase 2 (SOD2) activity. This enzyme has Mn2+ as cofactor, and this can be competed out by iron resulting in enzyme inactivation (48). This has been described in yeast with alteration of mitochondrial iron metabolism and in the condition of low manganese concentration (49). Thus, it is conceivable that FtMt may contribute to cellular antioxidant systems by preventing the inactivation of SOD2 even under conditions of major mitochondrial iron requirement, as recently shown in frataxin-silenced HeLa cells (35).
Mitochondrial ferritin and oxidative phosphorylation
The analysis of the FtMt clone induced for about 3 weeks showed some interesting relationships between FtMt and oxidative phosphorylation. When in glucose, the FtMt level reached a plateau after one week and then it leveled-off. In contrast, SDH activity strongly decreased on day 21 but not on day 7, a finding that, together with the reduction of the cytosolic and mitochondrial LIP, suggested that during the two weeks of high expression, FtMt reduced iron availability even for mitochondrial Fe-S enzymes. Moreover, the levels of SDH and mtACO enzymes did not change, confirming that in glucose growth condition, FtMt may interfere with ISC synthesis by iron sequestration. Surprisingly, the cells grown in galactose showed a much lower level of FtMt, which we found to be caused by a faster protein degradation rate. This effect, probably related to the increased respiratory activity, seems to be specific for FtMt, with a mechanism that remains to be clarified. Possibly, the Lon mitochondrial protease was activated in response to oxidative stress; as it occurs for the yeast homologue Pim1, in frataxin-deficient yeast strains (50). The increased degradation of ferritin would release iron, thus explaining why in the cells grown in galactose the cytLIP, cytACO and SDH activity did not decrease, and mtACO actually increased. Nevertheless, the residual FtMt was sufficient to reduce mtLIP, and, more importantly, also ROS level.
Taken together, these data suggest an interesting interplay between respiratory activity and FtMt. FtMt regulates mtLIP and ROS production to protect against oxidative damage. On the other side, rate of respiratory activity regulates FtMt degradation and consequently iron availability for the synthesis of Fe-S enzymes and possibly for heme. This mechanism implies that mainly cells with high respiratory activity may take advantages from FtMt expression. This situation was exemplified by the HeLa cells grown in galactose for 3 weeks, in which FtMt expression showed protective activity while preserving the functionality of mitochondrial enzymes. This model is in accordance with the FtMt expression profile in mouse; it was identified in cells with high respiratory activity like neurons, cardiomyocytes, muscle cells, ciliated epithelial cells, Leydig cells and spermatozoa (17), where it may play an important protective role. On the other hand, endogenous FtMt was not observed in cells with low respiratory activity, as the tumor cell lines HeLa or H1299. This would partially explain the slow in vivo growth of the tumor cells H1299 expressing FtMt (13); they may behave similar to the HeLa cells grown in glucose that have reduced proliferation (PDT: FtMt cells 23.2 h and control cells 19.8 h) probably due to inhibition of Fe-S enzymes.
Mitochondrial ferritin and Friedreich ataxia
In FRDA cells the pathological condition causes a chronic oxidative stress, and they represent an example in which the expression of FtMt is advantageous. A protective role of FtMt in artificial frataxin-deficient cells has been already demonstrated (35). To verify if it acts also on more physiological cellular models, we expressed FtMt in FRDA fibroblasts and confirmed that it reduced ROS formation and protected Fe-S enzyme activities. The low transfection efficiency of these primary cells (25%) explains why the overall protection was less evident than that observed in the HeLa cells. In fact, immunocytochemical examination of single cells showed that the fibroblasts expressing FtMt were characterized by: (i) preferential survival after H2O2 treatments; (ii) accumulation of lower levels of ROS (not shown) and (iii) increased SDH activity (Fig. 4C).
We confirm that the expression of FtMt partially compensates for frataxin deficiency, as previously shown in yeast (33) and HeLa cells (35), suggesting that FtMt and frataxin share some similar properties. Frataxin has been defined as a mitochondrial iron protein with iron detoxification and chaperone functions directly involved in ISC synthesis (21). In accordance with what demonstrated on HeLa cells, we assume that FtMt reduces ROS level by limiting the mtLIP through iron sequestration also in FRDA. This could be sufficient to improve the FRDA phenotype, although we cannot exclude that the protein might have the property to make iron available for mitochondria functions. FtMt binds and eventually releases iron, thus it may act as an iron donor, particularly in cells with high respiratory activity where it is rapidly degraded. This mechanism of iron regulation may be done directly or mediated by its interaction with other mitochondrial iron chaperons. To support this hypothesis, recently, it was reported that ferritin A (FtnA), a major iron-storage protein in Escherichia coli, was able to sequester the iron released from the disrupted iron–sulfur clusters under oxidative stress; this iron stored in FtnA can be made available to the iron chaperon IscA and utilized for the re-assembly of the iron–sulfur cluster (51). Unfortunately, the available data on FtMt expression in tissues of FRDA patients are limited. The presence of FtMt was documented in the heart of one FRDA patient (22) and it was found to be upregulated in two FRDA fribroblast cell lines (34) suggesting that cells could respond to frataxin deficiency promoting FtMt expression. Further in vitro and in vivo experiments will be needed to verify if FtMt is able to fully complement frataxin functionality and if this role depends on high FtMt concentration, a situation typical of transiently transfected cells such as FRDA–FtMt fibroblasts.
Altogether, these results show that FtMt has a role in controlling ROS production via the regulation of mitochondrial iron availability, and that this function may be more important than its iron storage property. FtMt expression affects many indices of total cellular iron, supporting the hypothesis that mitochondria are central to iron homeostasis. The mechanism of the regulation of FtMt expression remains unclear: our evidence shows that it may also act at a post-transcriptional level involving protein degradation. The data on the FRDA fibroblasts stimulates further studies to evaluate if FtMt expression corrects the defects of FRDA and to asses the efficacy of therapies aimed at moderating cell-labile iron levels.
MATERIALS AND METHODS
Cell culture
HeLa-tTA-FtMt and HeLa-tTA-FtMt222 clones were generated as previously described (11) and grown in high glucose Dulbecco's modified Eagle's medium (DMEM, Invitrogen) or in glucose-free DMEM (GIBCO) complemented with 25 mm glucose (glucose medium) or 5 mm galactose (galactose medium). All media were further supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 mg/ml streptomycin, 2 mm L-glutamine, 100 mg/ml G418 (Geneticin, Sigma), 150 mg/ml hygromycin B (Clontech). In the presence of doxycycline (2 ng/ml, Sigma), FtMt synthesis was repressed, whereas in its absence, the synthesis was induced. PDT studies were carried out during log-phase by plating 1 × 105 cells/well, on six-well plates containing glucose or galactose media. Three wells were plated for each condition. After 24 h (time 0) and then at daily intervals, one well for each condition was harvested by trypsinization and cells counted. The experiment was repeated three times.
We analyzed primary skin fibroblasts from two unaffected subjects (Control 1570 and Control 1270) and two FRDA patients (1037SS and 117DC). The size of the GAA repeat expansion was analyzed by polymerase chain reaction on genomic DNA, and the following allele sizes were determined: 1037SS: 930/930 and 117DC: 540/930. Fibroblasts were grown in high glucose DMEM supplemented with 10% FBS, 4 mm L-glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin. Fibroblasts were transiently transfected with Lipofectamine 2000 (Invitrogen). Briefly, 4 × 105 or 5 × 103 cells were, respectively, plated on 60 mm dishes or on 96-well plates and transfected with 20 µg or 0.75 µg of pcDNA3 or pcDNA3-huFtMt vectors (11). Experiments were performed 24 h after transfection and transfection efficiency was calculated by FtMt immunodetection on fixed cells.
Preparation of cell extracts
Cells were harvested, collected by centrifugation at 500g for 5 min and washed with Dulbecco's phosphate-buffered saline (PBS). Soluble cellular extracts, for ELISA, immunoblotting and immunoprecipitation assays, were obtained by cell lysis in 20 mm Tris–HCl pH 7.4, 0.5% Triton X-100 followed by centrifugation at 16 000g for 10 min. Immunoblotting analysis were performed in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) using specific antibodies for SDH (α-SDHA, Santa Cruz) and mtACO (α-ACO2, Sigma). For MF and PMF preparation, cells were suspended in 20 mm Tris–HCl pH 7.4, 250 mm sucrose, 0.007% digitonin. After lysis, samples were centrifuged at 1000g for 5 min to remove nuclei and unbroken cells. Then soluble fractions were centrifuged at 4000g for 10 min to obtain mitochondria-enriched pellets (MF). The resulting soluble fractions were further centrifuged at 16 000g for 10 min to obtain cytosolic soluble cellular extracts (PMF) that were used for cytochrome c and cytACO activity analysis. Cytochrome c analysis and SDH activity quantifications were performed on MF suspended in 20 mm Tris–HCl pH 7.4, 250 mm sucrose. mtACO and MDH activities were measured in MF suspended in 20 mm Tris–HCl pH 7.4, 0.5% Triton X-100 and centrifuged at 16 000g for 10 min to obtain mitochondrial soluble cellular extracts. During sample preparations, the mitochondria containing fractions were determined following SDH activity. The total protein contents were measured using BCATM protein assay (Pierce) calibrated on bovine serum albumin (BSA).
Analysis of mitochondrial ferritin content
In HeLa-tTA-FtMt clone, FtMt was determined by ELISA in soluble cellular extracts using the mouse α-huFtMt antiserum (33) calibrated on the recombinant human FtMt. Mitochondrial ferritin degradation was evaluated on 21 days induced cells, grown in glucose or galactose media. They were labeled for 18 h with 300 mCi/ml [35S]methionine, [35S]cysteine (PROMIX, Amersham), then washed twice with PBS and grown in the respective media for chase experiments. At the indicated times soluble cellular extracts were prepared and equal protein amounts immunoprecipitated using 1 µl of mouse α-huFtMt antiserum. Finally, precipitates were collected and suspended in SDS sample buffer, boiled for 10 min and 20 µl loaded on 12% SDS–PAGE, following common protocols. The gels were soaked with autoradiography enhancer (Amplify, Amersham), dried and exposed. For FtMt immunodetection, transfected fibroblasts were fixed with 4% paraformaldehyde and permeated with 0.5% Triton X-100. Cells were then incubated with mouse α-huFtMt antiserum (diluted 1:400) followed by rhodamine-conjugated rabbit anti-mouse secondary antibody.
Reactive oxygen species determination
ROS were determined by incubating cells with the redox-sensitive probe DHR 123, which could be converted, by oxidation, to the fluorescent Rhodamine 123 (37). Briefly, 6 × 104 HeLa-tTA-FtMt cells or 5 × 103 fibroblasts were plated in 48- or in 96-well plates. Cells were incubated with Hanks balanced saline solution (HBSS), 10 mm glucose and 30 µm DHR 123 for 15 min at 37°C. After two cycle washes, cells were maintained in HBSS supplemented with 10 mm glucose. Fluorescence was revealed using the Victor3 Multilabel Counter (Wallac, Perkin Elmer) at 485 nm and 535 nm for excitation and emission, respectively. Finally, results were normalized on protein content. To measure ROS generation induced by H2O2 treatments, after basal fluorescence determination, cells were incubated in HBSS, 10 mm glucose supplemented with 200 µm H2O2 at 37°C for 30 min, followed by a second fluorescence determination.
Cell viability and ATP assays
Cell viability was determined after H2O2 or Antimycin A insults following common protocols. For HeLa-tTA-FtMt cells, we used thiazolyl blue tetrazolium bromide (Sigma) assay or CellTiter-Glo® luminescent ATP assay (Promega). Fibroblasts viability, after transfection, was determined by trypan blue assay.
Labile iron pool determination
Labile Iron Pools were measured using the iron-sensitive probes Calcein-AM (42) (Molecular Probes) and RPA (Squarix), for cytosolic and mitochondrial compartments, respectively. Rhodamine B 4-[(Phenanthren-9-yl)Aminocarbonyl]benzyl ester (RPAC) (Squarix), a fluorescent probe unaffected by iron, was used to normalize mitochondrial rhodamine incorporation (41). Briefly, 6 × 104 HeLa-tTA-FtMt cells were plated in 48-well plates and incubated with αMEM, 1 mg/ml BSA supplemented with 3 µm RPA or 3 µm RPAC or 0.25 µm Calcein-AM at 37°C for 15 min. After two cycle washes with HBSS, cells were maintained in HBSS supplemented with 10 mm glucose. Basal fluorescence was revealed using the Victor3 Multilabel Counter (Wallac, Perkin Elmer) at 530 nm (excitation) and 590 nm (emission) for RPA and RPAC and at 485 nm (excitation) and 535 nm (emission) for calcein. Then cells were incubated with HBSS, 10 mm glucose supplemented with 2 mm (1 h) or 0.2 mm (5 min) pyridoxal isonicotinoyl hydrazone (PIH) for RPA and calcein-loaded cells, respectively. Fluorescence was re-measured and the increases were considered as LIP values. Finally, all the results were normalized on protein content.
Enzymatic activities
ACO, SDH and MDH activities were measured spectrophotometrically following standard protocols (52–54). ACO was assayed by measuring the disappearance of cis-aconitate at 240 nm, SDH by following the reduction of para-iodonitro-tetrazolium violet (INT) to INT-formazan at 500 nm, and MDH by monitoring disappearance of NADH at 340 nm. To determine SDH activity directly on cells, transfected fibroblasts were initially treated with 20 mm 4-Morpholinepropanesulfonic acid (MOPS) pH 7.4, 250 mm sucrose and 0.005% digitonin for 2 min. Then, cells were washed with 20 mm MOPS pH 7.4, 250 mm sucrose and 20 mm ethylene diamine tetraacetic acid (EDTA) and finally incubated with 20 mm MOPS pH 7.4, 250 mm sucrose, 1 mm EDTA, 0.2 mm phenazine methosulfate, 2 mm nitroblue-tetrazolium and 20 mm succinate. Activity was measured following violet crystals formation. Finally, cells were fixed and presence of FtMt was revealed by immunodetection as described above.
FUNDING
This study was partially supported by grants Telethon-Italia (GP0075Y01) to S.L.; PRIN-MIUR 2006 to S.L.; Ministero della Salute Progetto RF ex art56/2005/1 to F.T.; A.C. was supported by University Vita-Salute San Raffaele and PhD program of Italian Institute of Technology.
ACKNOWLEDGEMENTS
We thank Paolo Arosio for reading and editing the manuscript, Laura Silvestri for critical reading of manuscript, and Simona Allievi and Cinzia Gellera for their help in establishing FRDA cell lines. We thank Professor Des Richardson for the generous gift of PIH.
Conflict of Interest statement. We declare no conflicts of interest.
REFERENCES
- 1.Halliwell B. Antioxidant defence mechanisms: from the beginning to the end (of the beginning) Free Radic. Res. 1999;31:261–272. doi: 10.1080/10715769900300841. [DOI] [PubMed] [Google Scholar]
- 2.Harrison P.M., Arosio P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- 3.Arosio P., Levi S. Ferritin, iron homeostasis, and oxidative damage. Free Radic. Biol. Med. 2002;33:457–463. doi: 10.1016/s0891-5849(02)00842-0. [DOI] [PubMed] [Google Scholar]
- 4.Levi S., Corsi B., Bosisio M., Invernizzi R., Volz A., Sanford D., Arosio P., Drysdale J. A human mitochondrial ferritin encoded by an intronless gene. J. Biol. Chem. 2001;276:24437–24440. doi: 10.1074/jbc.C100141200. [DOI] [PubMed] [Google Scholar]
- 5.Drysdale J., Arosio P., Invernizzi R., Cazzola M., Volz A., Corsi B., Biasiotto G., Levi S. Mitochondrial ferritin: a new player in iron metabolism. Blood Cells Mol. Dis. 2002;29:376–383. doi: 10.1006/bcmd.2002.0577. [DOI] [PubMed] [Google Scholar]
- 6.Missirlis F., Holmberg S., Georgieva T., Dunkov B.C., Rouault T.A., Law J.H. Characterization of mitochondrial ferritin in Drosophila. Proc. Natl. Acad. Sci. USA. 2006;103:5893–5898. doi: 10.1073/pnas.0601471103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zancani M., Peresson C., Biroccio A., Federici G., Urbani A., Murgia I., Soave C., Micali F., Vianello A., Macri F. Evidence for the presence of ferritin in plant mitochondria. Eur. J. Biochem. 2004;271:3657–3664. doi: 10.1111/j.1432-1033.2004.04300.x. [DOI] [PubMed] [Google Scholar]
- 8.Langlois d'Estaintot B., Santambrogio P., Granier T., Gallois B., Chevalier J.M., Precigoux G., Levi S., Arosio P. Crystal structure and biochemical properties of the human mitochondrial ferritin and its mutant Ser144Ala. J. Mol. Biol. 2004;340:277–293. doi: 10.1016/j.jmb.2004.04.036. [DOI] [PubMed] [Google Scholar]
- 9.Bou-Abdallah F., Santambrogio P., Levi S., Arosio P., Chasteen N.D. Unique iron binding and oxidation properties of human mitochondrial ferritin: a comparative analysis with Human H-chain ferritin. J. Mol. Biol. 2005;347:543–554. doi: 10.1016/j.jmb.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Levi S., Arosio P. Mitochondrial ferritin. Int. J. Biochem. Cell Biol. 2004;36:1887–1889. doi: 10.1016/j.biocel.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 11.Corsi B., Cozzi A., Arosio P., Drysdale J., Santambrogio P., Campanella A., Biasiotto G., Albertini A., Levi S. Human mitochondrial ferritin expressed in HeLa cells incorporates iron and affects cellular iron metabolism. J. Biol. Chem. 2002;277:22430–22437. doi: 10.1074/jbc.M105372200. [DOI] [PubMed] [Google Scholar]
- 12.Nie G., Sheftel A.D., Kim S.F., Ponka P. Overexpression of mitochondrial ferritin causes cytosolic iron depletion and changes cellular iron homeostasis. Blood. 2005;105:2161–2167. doi: 10.1182/blood-2004-07-2722. [DOI] [PubMed] [Google Scholar]
- 13.Nie G., Chen G., Sheftel A.D., Pantopoulos K., Ponka P. In vivo tumor growth is inhibited by cytosolic iron deprivation caused by the expression of mitochondrial ferritin. Blood. 2006;108:2428–2434. doi: 10.1182/blood-2006-04-018341. [DOI] [PubMed] [Google Scholar]
- 14.Cazzola M., Invernizzi R., Bergamaschi G., Levi S., Corsi B., Travaglino E., Rolandi V., Biasiotto G., Drysdale J., Arosio P. Mitochondrial ferritin expression in erythroid cells from patients with sideroblastic anemia. Blood. 2003;101:1996–2000. doi: 10.1182/blood-2002-07-2006. [DOI] [PubMed] [Google Scholar]
- 15.Della Porta M.G., Malcovati L., Invernizzi R., Travaglino E., Pascutto C., Maffioli M., Galli A., Boggi S., Pietra D., Vanelli L., et al. Flow cytometry evaluation of erythroid dysplasia in patients with myelodysplastic syndrome. Leukemia. 2006;20:549–555. doi: 10.1038/sj.leu.2404142. [DOI] [PubMed] [Google Scholar]
- 16.Alcindor T., Bridges K.R. Sideroblastic anaemias. Br. J. Haematol. 2002;116:733–743. doi: 10.1046/j.0007-1048.2002.03378.x. [DOI] [PubMed] [Google Scholar]
- 17.Santambrogio P., Biasiotto G., Sanvito F., Olivieri S., Arosio P., Levi S. Mitochondrial ferritin expression in adult mouse tissues. J. Histochem. Cytochem. 2007;55:1129–1137. doi: 10.1369/jhc.7A7273.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Napier I., Ponka P., Richardson D.R. Iron trafficking in the mitochondrion: novel pathways revealed by disease. Blood. 2005;105:1867–1874. doi: 10.1182/blood-2004-10-3856. [DOI] [PubMed] [Google Scholar]
- 19.Pandolfo M. Iron and Friedreich ataxia. J. Neural. Transm. Suppl. 2006:143–146. doi: 10.1007/978-3-211-45295-0_22. [DOI] [PubMed] [Google Scholar]
- 20.Schoenfeld R.A., Napoli E., Wong A., Zhan S., Reutenauer L., Morin D., Buckpitt A.R., Taroni F., Lonnerdal B., Ristow M., et al. Frataxin deficiency alters heme pathway transcripts and decreases mitochondrial heme metabolites in mammalian cells. Hum. Mol. Genet. 2005;14:3787–3799. doi: 10.1093/hmg/ddi393. [DOI] [PubMed] [Google Scholar]
- 21.Bulteau A.L., O'Neill H.A., Kennedy M.C., Ikeda-Saito M., Isaya G., Szweda L.I. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science. 2004;305:242–245. doi: 10.1126/science.1098991. [DOI] [PubMed] [Google Scholar]
- 22.Michael S., Petrocine S.V., Qian J., Lamarche J.B., Knutson M.D., Garrick M.D., Koeppen A.H. Iron and iron-responsive proteins in the cardiomyopathy of Friedreich's ataxia. Cerebellum. 2006;5:257–267. doi: 10.1080/14734220600913246. [DOI] [PubMed] [Google Scholar]
- 23.Koeppen A.H., Michael S.C., Knutson M.D., Haile D.J., Qian J., Levi S., Santambrogio P., Garrick M.D., Lamarche J.B. The dentate nucleus in Friedreich's ataxia: the role of iron-responsive proteins. Acta Neuropathol. 2007;114:163–173. doi: 10.1007/s00401-007-0220-y. [DOI] [PubMed] [Google Scholar]
- 24.Rotig A., de Lonlay P., Chretien D., Foury F., Koenig M., Sidi D., Munnich A., Rustin P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 25.Wong A., Yang J., Cavadini P., Gellera C., Lonnerdal B., Taroni F., Cortopassi G. The Friedreich's ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 1999;8:425–430. doi: 10.1093/hmg/8.3.425. [DOI] [PubMed] [Google Scholar]
- 26.Ristow M., Mulder H., Pomplun D., Schulz T.J., Muller-Schmehl K., Krause A., Fex M., Puccio H., Muller J., Isken F., et al. Frataxin deficiency in pancreatic islets causes diabetes due to loss of beta cell mass. J. Clin. Invest. 2003;112:527–534. doi: 10.1172/JCI18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thierbach R., Schulz T.J., Isken F., Voigt A., Mietzner B., Drewes G., von Kleist-Retzow J.C., Wiesner R.J., Magnuson M.A., Puccio H., et al. Targeted disruption of hepatic frataxin expression causes impaired mitochondrial function, decreased life span and tumor growth in mice. Hum. Mol. Genet. 2005;14:3857–3864. doi: 10.1093/hmg/ddi410. [DOI] [PubMed] [Google Scholar]
- 28.Al-Mahdawi S., Pinto R.M., Varshney D., Lawrence L., Lowrie M.B., Hughes S., Webster Z., Blake J., Cooper J.M., King R., et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics. 2006;88:580–590. doi: 10.1016/j.ygeno.2006.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puccio H. Conditional mouse models for Friedreich ataxia, a neurodegenerative disorder associating cardiomyopathy. Handb. Exp. Pharmacol. 2007:365–375. doi: 10.1007/978-3-540-35109-2_15. [DOI] [PubMed] [Google Scholar]
- 30.Babcock M., de Silva D., Oaks R., Davis-Kaplan S., Jiralerspong S., Montermini L., Pandolfo M., Kaplan J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276:1709–1712. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- 31.Muhlenhoff U., Richhardt N., Ristow M., Kispal G., Lill R. The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins. Hum. Mol. Genet. 2002;11:2025–2036. doi: 10.1093/hmg/11.17.2025. [DOI] [PubMed] [Google Scholar]
- 32.Cavadini P., O'Neill H.A., Benada O., Isaya G. Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia. Hum. Mol. Genet. 2002;11:217–227. doi: 10.1093/hmg/11.3.217. [DOI] [PubMed] [Google Scholar]
- 33.Campanella A., Isaya G., O'Neill H.A., Santambrogio P., Cozzi A., Arosio P., Levi S. The expression of human mitochondrial ferritin rescues respiratory function in frataxin-deficient yeast. Hum. Mol. Genet. 2004;13:2279–2288. doi: 10.1093/hmg/ddh232. [DOI] [PubMed] [Google Scholar]
- 34.Popescu B.F., Pickering I.J., George G.N., Nichol H. The chemical form of mitochondrial iron in Friedreich's ataxia. J. Inorg. Biochem. 2007;101:957–966. doi: 10.1016/j.jinorgbio.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 35.Zanella I., Derosas M., Corrado M., Cocco E., Cavadini P., Biasiotto G., Poli M., Verardi R., Arosio P. The effects of frataxin silencing in HeLa cells are rescued by the expression of human mitochondrial ferritin. Biochim. Biophys. Acta. 2008;1782:90–98. doi: 10.1016/j.bbadis.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 36.Rossignol R., Gilkerson R., Aggeler R., Yamagata K., Remington S.J., Capaldi R.A. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004;64:985–993. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- 37.Royall J.A., Ischiropoulos H. Evaluation of 2′,7′-dichlorofluorescin and dihydrorhodamine 123 as fluorescent probes for intracellular H2O2 in cultured endothelial cells. Arch. Biochem. Biophys. 1993;302:348–355. doi: 10.1006/abbi.1993.1222. [DOI] [PubMed] [Google Scholar]
- 38.Malgat M.L.T., Durrieu G., Mazat J.P. Enzymatic and polarographic measurements of the respiratory chain complexes. In: Lestienne P., editor. Mitochondrial Diseases: Models and Methods. Paris: Springer-Verlag; 1999. pp. 357–377. [Google Scholar]
- 39.Hofhaus G., Johns D.R., Hurko O., Attardi G., Chomyn A. Respiration and growth defects in transmitochondrial cell lines carrying the 11778 mutation associated with Leber's hereditary optic neuropathy. J. Biol. Chem. 1996;271:13155–13161. doi: 10.1074/jbc.271.22.13155. [DOI] [PubMed] [Google Scholar]
- 40.Petrat F., de Groot H., Sustmann R., Rauen U. The chelatable iron pool in living cells: a methodically defined quantity. Biol. Chem. 2002;383:489–502. doi: 10.1515/BC.2002.051. [DOI] [PubMed] [Google Scholar]
- 41.Rauen U., Springer A., Weisheit D., Petrat F., Korth H.G., de Groot H., Sustmann R. Assessment of chelatable mitochondrial iron by using mitochondrion-selective fluorescent iron indicators with different iron-binding affinities. Chembiochem. 2007;8:341–352. doi: 10.1002/cbic.200600311. [DOI] [PubMed] [Google Scholar]
- 42.Glickstein H., El R.B., Shvartsman M., Cabantchik Z.I. Intracellular labile iron pools as direct targets of iron chelators: a fluorescence study of chelator action in living cells. Blood. 2005;106:3242–3250. doi: 10.1182/blood-2005-02-0460. [DOI] [PubMed] [Google Scholar]
- 43.Napoli E., Taroni F., Cortopassi G.A. Frataxin, iron-sulfur clusters, heme, ROS, and aging. Antioxid. Redox Signal. 2006;8:506–516. doi: 10.1089/ars.2006.8.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao G., Bou-Abdallah F., Arosio P., Levi S., Janus-Chandler C., Chasteen N.D. Multiple pathways for mineral core formation in mammalian apoferritin. The role of hydrogen peroxide. Biochemistry. 2003;42:3142–3150. doi: 10.1021/bi027357v. [DOI] [PubMed] [Google Scholar]
- 45.Zhao G., Arosio P., Chasteen N.D. Iron(II) and hydrogen peroxide detoxification by human H-chain ferritin. An EPR spin-trapping study. Biochemistry. 2006;45:3429–3436. doi: 10.1021/bi052443r. [DOI] [PubMed] [Google Scholar]
- 46.Cozzi A., Corsi B., Levi S., Santambrogio P., Albertini A., Arosio P. Overexpression of wild type and mutated human ferritin H-chain in HeLa cells: in vivo role of ferritin ferroxidase activity. J. Biol. Chem. 2000;275:25122–25129. doi: 10.1074/jbc.M003797200. [DOI] [PubMed] [Google Scholar]
- 47.Cozzi A., Corsi B., Levi S., Santambrogio P., Biasiotto G., Arosio P. Analysis of the biologic functions of H- and L-ferritins in HeLa cells by transfection with siRNAs and cDNAs: evidence for a proliferative role of L-ferritin. Blood. 2004;103:2377–2383. doi: 10.1182/blood-2003-06-1842. [DOI] [PubMed] [Google Scholar]
- 48.Culotta V.C., Yang M., O'Halloran T.V. Activation of superoxide dismutases: putting the metal to the pedal. Biochim. Biophys. Acta. 2006;1763:747–758. doi: 10.1016/j.bbamcr.2006.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang M., Cobine P.A., Molik S., Naranuntarat A., Lill R., Winge D.R., Culotta V.C. The effects of mitochondrial iron homeostasis on cofactor specificity of superoxide dismutase 2. EMBO J. 2006;25:1775–1783. doi: 10.1038/sj.emboj.7601064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bulteau A.L., Dancis A., Gareil M., Montagne J.J., Camadro J.M., Lesuisse E. Oxidative stress and protease dysfunction in the yeast model of Friedreich ataxia. Free Radic. Biol. Med. 2007;42:1561–1570. doi: 10.1016/j.freeradbiomed.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 51.Bitoun J.P., Wu G., Ding H. Escherichia coli FtnA acts as an iron buffer for re-assembly of iron-sulfur clusters in response to hydrogen peroxide stress. Biometals. 2008 doi: 10.1007/s10534-008-9154-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Drapier J.C., Hibbs J.B., Jr. Aconitases: a class of metalloproteins highly sensitive to nitric oxide synthesis. Methods Enzymol. 1996;269:26–36. doi: 10.1016/s0076-6879(96)69006-5. [DOI] [PubMed] [Google Scholar]
- 53.Munujos P., Coll-Canti J., Gonzalez-Sastre F., Gella F.J. Assay of succinate dehydrogenase activity by a colorimetric-continuous method using iodonitrotetrazolium chloride as electron acceptor. Anal. Biochem. 1993;212:506–509. doi: 10.1006/abio.1993.1360. [DOI] [PubMed] [Google Scholar]
- 54.Zeikus J.G., Fuchs G., Kenealy W., Thauer R.K. Oxidoreductases involved in cell carbon synthesis of Methanobacterium thermoautotrophicum. J. Bacteriol. 1977;132:604–613. doi: 10.1128/jb.132.2.604-613.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]