

Abstract
Huntingtin (htt) is a 350 kDa protein of unknown function, with no homologies with other known proteins. Expansion of a polyglutamine stretch at the N-terminus of htt causes Huntington's disease (HD), a dominant neurodegenerative disorder. Although it is generally accepted that HD is caused primarily by a gain-of-function mechanism, recent studies suggest that loss-of-function may also be part of HD pathogenesis. Huntingtin is an essential protein in the mouse since inactivation of the mouse HD homolog (Hdh) gene results in early embryonic lethality. Huntingtin is widely expressed in embryogenesis, and associated with a number of interacting proteins suggesting that htt may be involved in several processes including morphogenesis, neurogenesis and neuronal survival. To further investigate the role of htt in these processes, we have inactivated the Hdh gene in Wnt1 cell lineages using the Cre-loxP system of recombination. Here we show that conditional inactivation of the Hdh gene in Wnt1 cell lineages results in congenital hydrocephalus, implicating huntingtin for the first time in the regulation of cerebral spinal fluid (CSF) homeostasis. Our results show that hydrocephalus in mice lacking htt in Wnt1 cell lineages is associated with increase in CSF production by the choroid plexus, and abnormal subcommissural organ.
INTRODUCTION
Huntingtin (htt) is a 350 kDa protein of unknown function, and with no homologies with other known proteins. Expansion of a polymorphic polyglutamine stretch at the N-terminus of htt causes Huntington's disease (HD), an adult-onset dominant neurodegenerative disorder characterized by selective neuronal cell death in cortex and striatum (1). Since a gain-of-function mechanism is widely accepted as a cause of HD, little attention has been focused on elucidating the normal function of wild-type htt. Recent studies however suggest that loss-of-function may also be part of HD pathogenesis (2–4). Therefore, understanding the normal function of htt is likely to provide important information regarding HD pathogenesis.
Huntingtin is widely expressed in the early developing mouse embryo and becomes restricted to the central nervous system (CNS) later in gestation (5,6). Huntingtin has an essential role during embryogenesis since inactivation of the mouse HD homolog gene (Hdh) results in developmental retardation and death of nullizygous embryos between E7.5 and E10.5 (7–10). Although null embryos gastrulate, their development is stalled by E7.5 accompanied with extensive apoptosis. Further analyses of huntingtin-deficient embryos revealed that huntingtin is essential in the early patterning of the embryo for the formation of the anterior region of the primitive streak (11). In vitro analyses showed htt has anti-apoptotic functions in neuronal cell cultures (2). Htt is also associated with a large number of interacting partners, implicating htt in processes that include transcriptional regulation, cell signaling and morphogenesis (12).
The early lethality of the Hdh nullizygous embryos, however, precludes the analyses of the function of huntingtin in vivo in later developmental events. To further define the roles of huntingtin in embryonic development, brain patterning and in neuronal cells, our research group is pursuing a systematic analyses of the effects of loss of the Hdh gene at different stages and different cell lineages in the mouse, using the Cre-loxP system of recombination. With this approach, we have previously reported that conditional inactivation of Hdh in the forebrain results in progressive neurodegeneration (3), indicating that htt is required postnatally for neuronal survival in cortex and striatum.
To investigate the role of htt in brain development, we conditionally inactivated the Hdh gene in Wnt1 cell lineages, which contribute to the midbrain, hindbrain, granular cells of the cerebellum, as well as to dorsal midline-derived ependymal secretory structures (13–16). Our results show that the loss of htt in Wnt1 cell lineages results in congenital hydrocephalus associated with abnormalities in the choroid plexus and subcommissural organ (SCO).
RESULTS
Inactivation of the mouse Hdh gene in Wnt1 cell lineages causes hydrocephalus
To generate the experimental progeny of interest, Wnt1-Cre/+ mice, expressing the Cre recombinase under control of the Wnt1 regulatory sequences (14,15,17) were crossed with Hdh+/− mice (10). The resulting Wnt1-Cre/+; Hdh+/− mice were then crossed with Hdh flox/flox mice (3). Wnt1-Cre/+; Hdhflox/− mice were born at the expected Mendelian ratio, and were normal in size and appearance at birth. However, by postnatal day 6 (P6), they were easily distinguishable from their littermates—they developed an enlarged domed cranium, reminiscent of mice with hydrocephalus (Fig. 1A and B). Skeletal analyses also demonstrated domed skull and, in addition, calvarium bone suture fusion defects which are likely to be secondary to the enlargement of the brain prior to physiological closure of the sutures (Fig. 1C–H). Consistent with observations of other mouse mutants with congenital hydrocephalus (18,19), Wnt1-Cre/+; Hdhflox/− mutant mice displayed reduced growth (Fig. 1B) and progressive wasting accompanied by ruffled coats, ataxic gait and dehydration, leading to death between P6 and P18.
Figure 1.
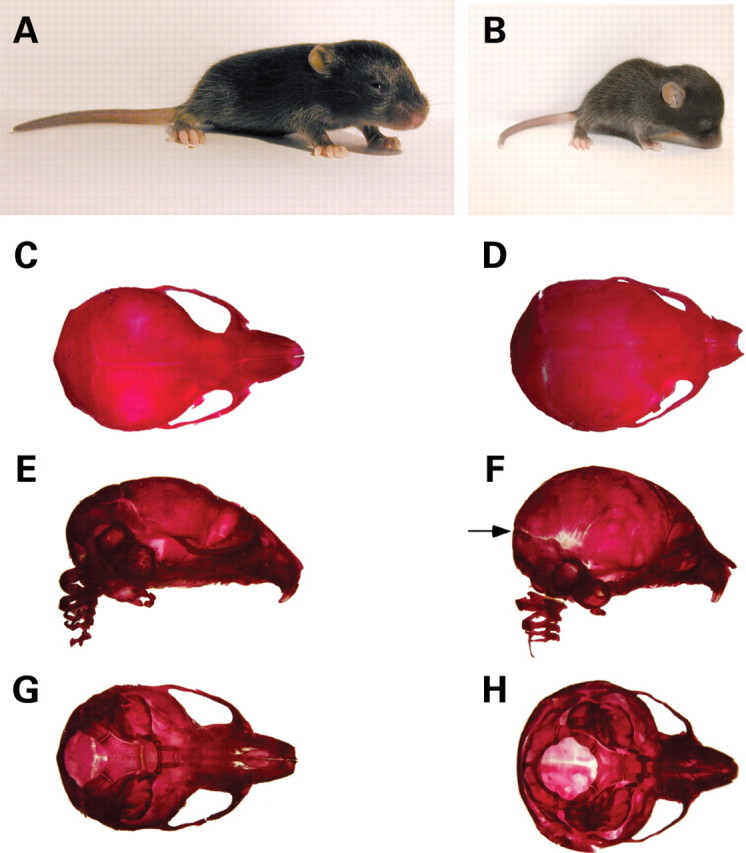
Hydrocephalus in Wnt1-Cre/+; Hdhflox/− mutants. (A and B) Appearance of WT (A) and mutant (B) mice at P13. Note that the mutant mouse (B) is small, runted and displays a domed cranium. (C–H) Skeletal head preparations of WT (C, E, G) and mutant (D, F, H) mice at P10. (C, D) Dorsal view; (E, F) lateral view and (G, H) ventral view. For both WT and mutant, the lower jaw has been removed prior to photographic documentation. Note the enlarged domed cranium in the mutant. Although all skeletal bones appear well formed, suture fusion defects are apparent (arrow in F).
Ventricular dilation and tissue loss in hydrocephalic Wnt1-Cre/+; Hdhflox/− mutant mice
Anatomical analyses of mutant brains at P4, P10 and P15 revealed the presence of excessive cerebral spinal fluid (CSF). Histological analyses demonstrated enlargement of the lateral and third ventricles in mutant brains (Fig. 2A–E), and tissue loss in the cortex (Fig. 2C), likely to be secondary to CSF accumulation. In more severe cases, compression of the cerebellum and other brain regions was also observed but no gross brain developmental abnormalities were detected. Notably, midbrain, hindbrain and cerebellum appeared grossly normal at all stages analyzed (Fig. 2E, and data not shown). To investigate whether ‘leaky’ Cre-mediated recombination could be responsible for tissue loss in forebrain regions in Wnt1-Cre/+; Hdhflox/− mutant mice, we verified the pattern of recombination of Wnt1Cre transgenic line by crossing Wnt1-Cre/+ mice with the Rosa26 reporter line (R26R) (20), which expresses β-galactosidase upon Cre-mediated recombination. X-Gal staining of Wnt1-Cre/+; R26R/+ brains confirmed that recombination of the R26R allele was always confined to midbrain, hindbrain and cerebellum (Fig. 3A). Also, western analyses on total protein extracts from forebrain and cerebellum of P10 mutant and control mice revealed that htt levels in the mutant forebrain were comparable to that of controls, while a 70% reduction was observed in cerebellum protein extracts (Fig. 3B). In addition, TUNEL analysis in the cortices of mutant and control brains, preceding the onset of hydrocephalus and tissue loss, did not reveal any differences in apoptotic cell death (data not shown). These results indicate that tissue loss in the forebrain in Wnt1-Cre/+; Hdhflox/− mutant mice is secondary to CSF accumulation.
Figure 2.

Ventricular dilation and tissue loss in Wnt1-Cre/+; Hdhflox/− mutant mice. (A–C) H&E histological examinations of coronal sections of P4 WT (A) and mutant (B, C) brains. Extensive dilation of the lateral ventricles (lv) is evident in the mutants at P4 (B), and tissue loss in the cortex (ctx) is observed in more caudal sections (arrowheads in C). (D and E) H&E histological examinations of sagital sections of P14 WT (D) and mutant (E) brains. Note that the progressive hydrocephalus leads to secondary compression of the cerebellum (cb) and more pronounced tissue loss in the mutant brain (E).
Figure 3.
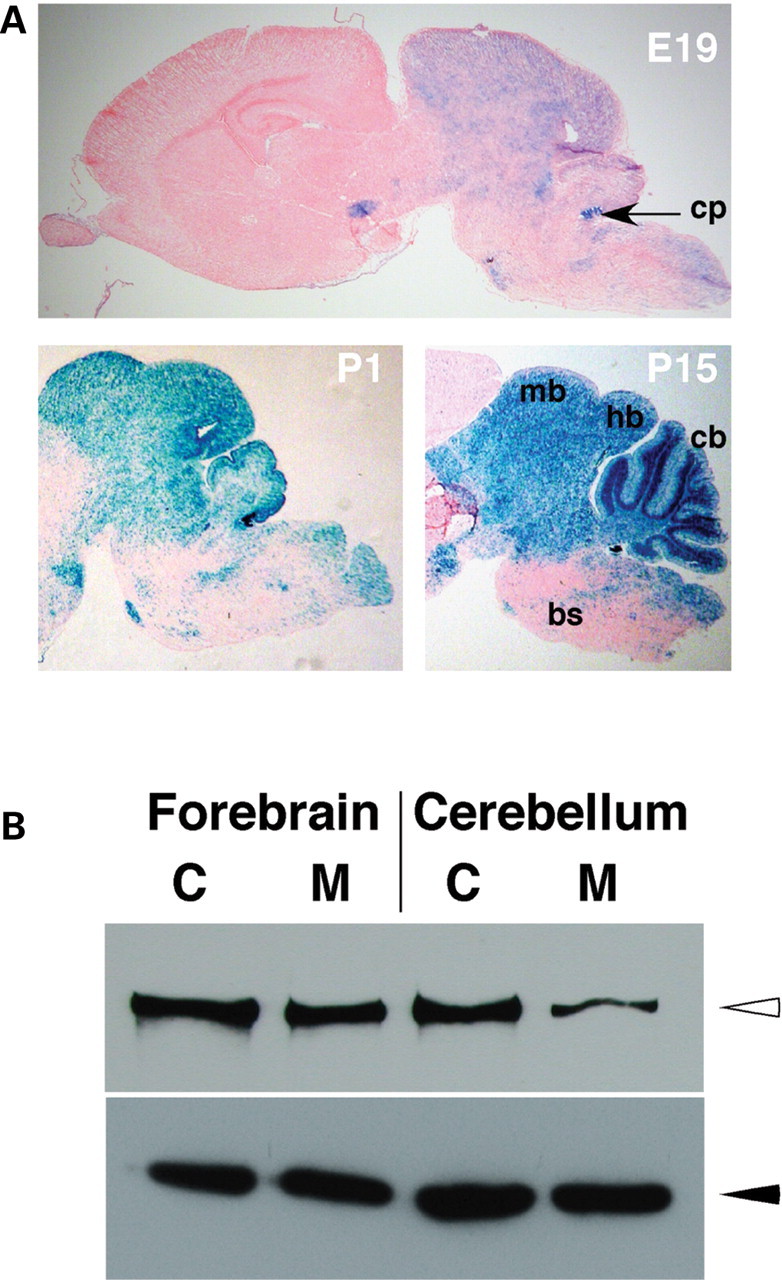
Pattern of Wnt1-Cre-mediated recombination. (A) X-Gal staining of sagital sections of E19, P1 and P15 Wnt1-Cre/+; R26R/+ brains. Note that in all cases, X-Gal staining (blue color) is restricted to the midbrain (mb), hindbrain (hb) and cerebellum (cb). Sparse and scattered staining is observed in the brain stem (bs) and almost no staining is present in the forebrain. cp=choroid plexus. (B) Western analyses of total protein lysates from forebrain (lanes 1 and 2) and cerebellum (lanes 3 and 4) of control (C) Hdhflox/− (lanes 1 and 3) and mutant (M) Wnt1-Cre/+; Hdhflox/− (lanes 2 and 4) mice. Upper panel shows detection of htt (open arrowhead) with the monoclonal anti-htt antibody 2166, and lower panel shows anti-beta-tubulin staining (filled arrowhead), for loading control.
Hydrocephalus in Wnt1-Cre/+; Hdhflox/− mutant mice is congenital
To determine the timing of onset of hydrocephalus in the Wnt1-Cre/+; Hdhflox/− mutant mice, we performed anatomical and histological analyses on brains from late gestation embryos (E17.5) and postnatal mice (P0 to P15). Although the cerebral cortex and lateral ventricles appeared normal at E17.5, enlargement of the lateral ventricles was already observed in a significant fraction of mutants at P0 and was present in 100% of the mutants after P4 (Table 1). The presence of ventricular enlargement at birth indicates that hydrocephalus in Wnt1-Cre/+; Hdhflox/− mice is congenital.
Table 1.
Onset and frequency of hydrocephalus in Wnt1-Cre/+; Hdhflox/− mice
| Stage | Total number of mutant mice analyzed | % hydrocephalic |
|---|---|---|
| E17.5 | 3 | 0 |
| P0 | 5 | 40 |
| P4 | 4 | 75 |
| P10–P12 | 7 | 100 |
| P14–P16 | 5 | 100 |
Hydrocephalus in Wnt1-Cre/+; Hdhflox/− mutant mice is associated with choroid plexus and SCO abnormalities
Congenital hydrocephalus in mutant mice and rats is often found in association with abnormalities of the choroid plexus and of the SCO (18,21–24). Since the SCO and the choroid plexus of the third and fourth ventricles are derived from Wnt1-expressing cell populations of the dorsal midline (13,25,26), we examined whether these structures were affected in Wnt1-Cre/+; Hdhflox/− mice.
Gross anatomical examination of choroid plexus from the third and fourth ventricles did not reveal significant differences in size between Wnt1-Cre/+; Hdhflox/− and control littermates. Corroborating these findings, immunohistochemistry for ki-67, a marker for cell proliferation, did not show increase in cell proliferation in mutant choroid plexus compared to control littermates (data not shown). However, histological analyses of Wnt1-Cre/+; Hdhflox/− choroid plexus of third and fourth ventricles at P2, P9 and P15 revealed that there was a 2-fold increase in cytoplasmic volume accompanied by decreased electron-density in a large fraction of choroid plexus epithelial cells compared to control littermates (Fig. 4A–D). Consistent with a potential role for huntingtin in choroid plexus and CSF production, immunohistochemistry for htt in P1 and P12 brain sections revealed that htt is abundantly expressed in choroid plexus of WT mice, and that its expression is completely abolished in Wnt1-Cre/+; Hdhflox/− mutants (Fig. 4 E–F, and data not shown). The epithelial cells of the choroid plexus secrete CSF by a process which involves unidirectional transport of ions, achieved due to both the polarity of the epithelium [i.e. the ion transport proteins in the blood-facing (basolateral) membrane are different to those in the ventricular (apical) membrane] and to the barrier formed by junctional complexes between the epithelial cells (27,28). Immunohistochemistry for the tight junction associated protein zonula occludens1 (ZO-1) and the tight junction protein Claudin-1, however, did not show significant differences between mutants and controls, indicating that tight junctions are normal in mutant CP (data not shown). Immunohistochemistry for Aquaporin-1 (AQP1), a water channel expressed strongly at the apical surface of choroid plexus epithelium (29–31) also did not show any differences in the distribution or levels of expression indicating that polarity is also maintained in the choroid plexus epithelium of Wnt1-Cre/+; Hdhflox/− mutants (data not shown). These results suggest that the apparent overproduction of CSF by the CP in Wnt1-Cre/+; Hdhflox/− mutants is probably not due to a gross structural abnormality of the CP, but may be a consequence of abnormal physiological control of CSF production and secretion.
Figure 4.
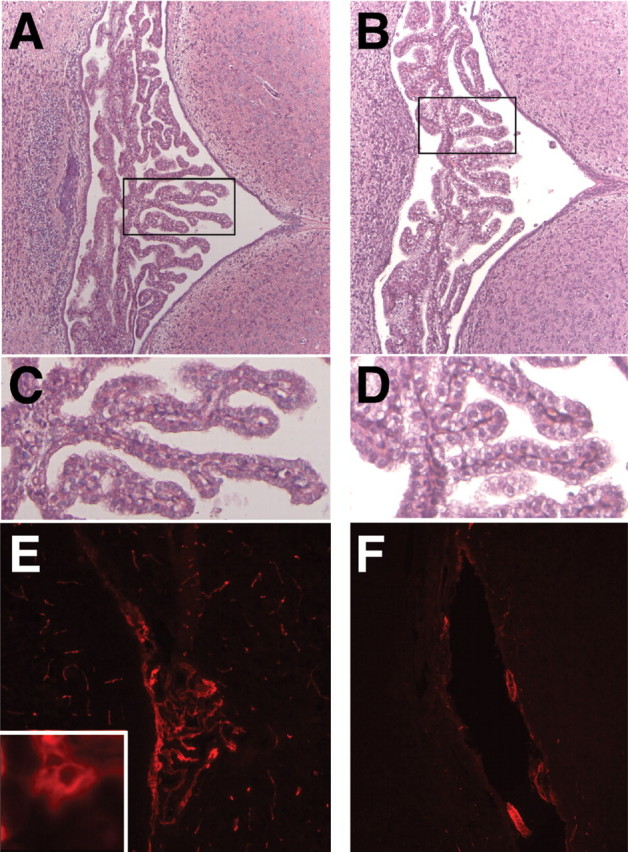
Abnormal choroid plexus in Wnt1-Cre/+; Hdhflox/− mice. (A and B) H&E staining of coronal sections through the fourth ventricle of WT (A) and Wnt1-Cre/+; Hdhflox/− (B) brains at P4. Note that the overall morphology of the choroid plexus appears normal in mutants. (C and D) High magnification of the areas marked by the boxes in (A) and (B), respectively. Note that although the structure of mutant cp (D) is similar to controls (C), a large fraction of the epithelial cells in the mutants display enlarged cytoplasmic volume. (E and F) Immunohistochemistry for htt in the fourth ventricle choroid plexus of P9 WT (E) and Wnt1-Cre/+; Hdhflox/− (F) brains. Note that htt is expressed in choroid plexus epithelial cells of WT mice (see high magnification, inset in E), and that expression is absent in the mutants.
We next examined whether the SCO was affected in Wnt1-Cre/+; Hdhflox/− mutants. In WT mice, the SCO is located in the roof of the third ventricle at the entrance of the aqueduct of Sylvius, and spans the caudal portion of the third ventricle and rostral part of the aqueduct (22,32). Analyses of serial coronal sections from Wnt1-Cre/+; Hdhflox/− and control littermate brains at P0 and P4 revealed that although the rostral portion of the SCO was present in mutant mice, no structure resembling the SCO was visible in more caudal sections (Fig. 5A–F), indicating that the SCO is significantly reduced in size in mutant brains. Comparison of the SCO length of mutant and control brains at P0, P4 and P9 revealed that the SCO in the mutants is ∼40% the size of the SCO of its control littermates. In addition, stenosis of the aqueduct was also observed in caudal sections (Fig. 5F, and data not shown). To determine if the SCO ependymal cells were properly differentiated in Wnt1-Cre/+; Hdhflox/− mutants, we performed immunostaining for SCO-spondin, the major glycoprotein expressed by the secretory ependymal cells of the SCO that aggregates to form Reissner Fiber (RF) (32). Immunostaining using the anti-RF antibody AFRU (33) showed normal expression of SCO-spondin in the rostral portion of the SCO in the mutants (Fig. 6B and C). However, although in WT mice, RF-immunoreactivity is restricted to the SCO and absent from other ependymal cells (18,34) (Fig. 6D), high levels of RF-immunoreactivity was detected in the ependyma lining the aqueduct in mutant mice (Fig. 6F) in a pattern corresponding to that of dorsal-midline-derived Wnt1 cell lineages (Fig. 6A).
Figure 5.
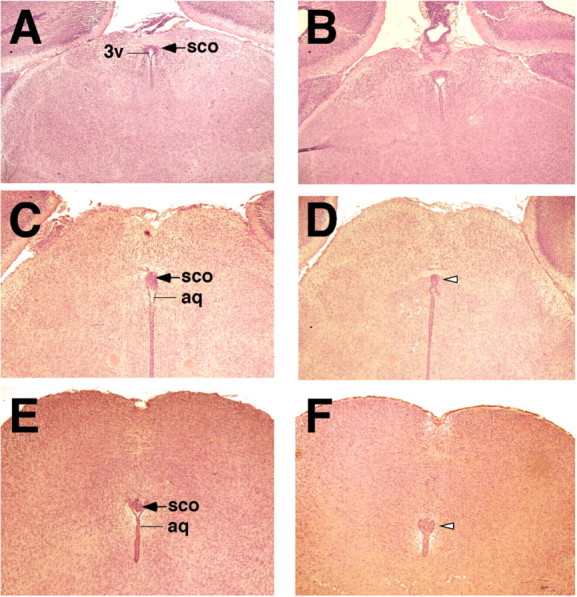
Abnormalities of the SCO in Wnt1-Cre/+; Hdhflox/− mutant mice. (A–F) H&E-stained rostral to caudal coronal sections from newborn littermates WT (A, C, E) and Wnt1-Cre/+; Hdhflox/− (B, D, F) brains, prior to onset of overt hydrocephalus. Note that the rostral portion of the SCO is well developed in the mutant (B), but no structure resembling the SCO is observed in more caudal sections (open arrowheads in D and F). In addition, discontinuities are also observed at distinct levels in the aqueduct (aq), and stenosis of the aqueduct is seen at more caudal level (F). 3v=third ventricle.
Figure 6.

SCO-spondin expression in Wnt1-Cre/+; Hdhflox/− mutant mice. (A) X-Gal staining of coronal brain sections from Wnt1-Cre/+; R26R/+ mice confirms extensive cre-mediated recombination in cells forming the SCO (blue staining). In addition, X-Gal staining is also observed in the dorsal population of ependymal cells of the aqueduct. (B–F) Immunohistochemistry for SCO-spondin using anti-RF antibody at P18. (B and D) WT and (C, E, F) Wnt1-Cre/+; Hdhflox/− brain sections. Note that progressive hydrocephalus leads to significant dilation of the third ventricle (3v) and aqueduct (aq) in mutant mice (C and E). (B and C) Expression of SCO-spondin is normal in the rostral portion of the SCO in mutants (C) compared to control mice (B). Note that at more caudal levels, RF immunoreactivity is discontinuous in the mutant SCO (E), that SCO is absent in further caudal sections (F), while ectopic RF immunoreactivity is observed in the dorsal ependymal layer of the aqueduct (open arrowhead in F), in a pattern corresponding to the X-Gal staining seen in (A).
These results indicate that abnormalities in the SCO and in the regulation of the CSF production by the choroid plexus underlie the congenital hydrocephalus of Wnt1-Cre/+; Hdhflox/− mice.
DISCUSSION
Congenital hydrocephalus is a human disorder with high mortality rate, occurring in 1/1000 live births (35–37). Alterations affecting the flow, synthesis or absorption of CSF can all lead to net accumulation of CSF and consequently to hydrocephalus (35,38). Despite the high prevalence of this disorder, the molecular basis of hydrocephalus and the mechanisms regulating CSF homeostasis are still largely unknown. A growing body of evidence, however, suggests that genetic factors play a major role in the pathogenesis of hydrocephalus. Although it is estimated that ∼40% of congenital hydrocephalus cases in humans have a possible genetic cause (39), so far only one gene, the X-linked L1-CAM (L1 cell adhesion molecule), has been identified, and accounts for ∼5% of the congenital cases with genetic cause (24,39). Over the past decade, the characterization of genetically modified animal models that recapitulate this disorder has led to the identification of more than 10 other genes resulting in hydrocephalus in rodents (19,22,24,40).
Here we show that conditional inactivation of the Hdh gene in Wnt1 cell lineages leads in congenital hydrocephalus in mice, implicating huntingtin for the first time in the regulation of CSF homeostasis. Our results show that hydrocephalus in mice lacking htt in Wnt1 cell lineages is associated with abnormalities of the SCO, abnormal expression of SCO-spondin on ependymal cells of the cerebral aqueduct and apparent increase in CSF production by the choroid plexus.
Lack of SCO or abnormal SCO development is a common feature in rats and mice that display congenital hydrocephalus (22,41). This developmental abnormality is observed in mice carrying a null mutation for the transcription factors RFX3 or RFX4 (18,42); in mice overexpressing engrailed-1 (En-1) in Wnt1 cell lineages (26,43,44); as well as in mice carrying null or hypomorphic mutations in genes whose expression is restricted to the dorsal midline at early stages of development, such as the transcription factors Pax6 and Msx1 (25,45,46), and the secreted signaling molecule Wnt1 (26,47).
In Wnt1-Cre/+; Hdhflox/− mutants, the SCO is smaller than in WT mice, and although it appears to be normally developed rostrally, its middle and caudal portions are either grossly malformed, or missing (Fig. 5A–F). Interestingly, while a null mutation in the gene encoding the transcription factor Msx1 leads to complete agenesis of the SCO, heterozygous mice display features similar to those seen in Wnt1-Cre/+; Hdhflox/− mutants (25), suggesting that loss of htt in the dorsal midline may lead to downregulation of intracellular pathways required for SCO development.
Htt, an essential protein in mouse development with unknown function(s), is believed to act as a scaffold molecule that orchestrates the assembly of large protein complexes by modulating binding activities of accessory factors (12,48–50). This has been best demonstrated in neuronal cells, where htt, acting as a scaffold protein, regulates the action of proteins involved in neuronal function and in expression of genes essential for neuronal survival (51–53). During mouse embryonic development, absence of htt results in early embryonic lethality and impaired anterior streak development (8–11), while reduction of htt expression leads to aberrant neurogenesis (54). Although a scaffold function for htt in these processes has not been demonstrated, the transcriptional deregulation observed in Hdh knockout embryos suggests that htt may either play a direct role in transcriptional regulation or may act in pathways that regulate the expression of essential genes (11). The fact that htt interacts with several members of transcription complexes, including transcription co-repressors and co-activators, as well as basal transcription factors, supports the notion that htt may play a role in transcriptional regulation by acting as a scaffold protein that modulates the activity of the transcription machinery. A similar role for htt may apply in the development of the SCO. Among the basal transcription factors, htt interacts with the transcriptional activator Sp1, and the co-activator TAFII130, which interacts with Sp1 to stimulate transcription (55,56). In particular, Sp1 binding sites have been shown to be essential for the transcription of Pax6 and Msx1 (57,58), two genes that are required for proper development of the SCO. Although the role of htt in this interaction is still not clear, it has been proposed that htt may strengthen Sp1 and TAFII130 binding therefore ensuring increased gene transcription (56).
The mechanism by which lack of SCO results in hydrocephalus is not fully understood. It has been suggested that the secretion of SCO-spondin by the SCO and its aggregation to form the RF is required to maintain the aqueduct of Sylvius open. Absence of SCO or absence of RF along the aqueduct correlates with stenosis of the aqueduct and hydrocephalus in a large number of mouse and rat mutants (41,42). Also injections of SCO-spondin antibodies in rats during embryonic development result in hydrocephalus (59). However, growing amount of evidence suggest that in many of these models the first signs of hydrocephalus occur prior to stenosis of the aqueduct, and therefore that stenosis of the aqueduct may be a secondary event (22). Although aqueduct stenosis has been observed in our Wnt1-Cre/+; Hdhflox/− mice, the abnormally enlarged cytoplasm of the epithelial cells of the CP (Fig. 4D) suggest that increased CSF production may also contribute to hydrocephalus of our mutant mice. About 70–80% of CSF is secreted by the CP (60,61), and its secretion is inhibited by sympathetic innervation, and stimulated by cholinergic innervation (62,63). Htt is expressed in CP epithelial cells and although we did not detect any gross changes in polarity or tight junction complexes, other structural or physiological abnormalities may underlie CSF overproduction by mutant CP. Alternatively, increased CSF production may be an indirect consequence of the dysgenesis of the SCO. It has been suggested that soluble SCO secretion present in the CSF may regulate CSF homeostasis by either removing monoamines present in the CSF or by directly binding to RF-receptors present in the apical surface of CP epithelial cells (64,65).
Differentiation of other ependymal cells also appears to be affected in Wnt1-Cre/+; Hdhflox/− mutants. Immunohistochemistry on coronal sections spanning the aqueduct of Sylvius revealed high levels of ectopic RF immunoreactivity in ependymal cells lining the aqueduct of mutant brains (Fig. 6F). Significantly the pattern of distribution of these ependymal cells (in the dorsal portion of the wall) also corresponds to the distribution of Wnt1 cell lineages in the ependyma (Fig. 6A). In mice, the aqueduct is an irregularly shaped cavity with three distinct regions (rostral, middle and caudal) lined by various types of ependyma (66). Ependymal cells develop late in gestation and their complete differentiation occurs in the first two postnatal weeks, with differential expression of genes in dorsal and ventral walls of the aqueduct (67). Our results indicate that Wnt1 cell lineages contribute to dorsal ependymal populations of the aqueduct wall, and suggest that huntingtin is required for proper differentiation of these cells. Interestingly, ectopic SCO-spondin-immunoreactivity in ependymal cells of the aqueduct has also been observed in other hydrocephalic models (18).
The implication of htt in hydrocephalus in humans is currently unknown. Although ventricular dilation is observed at late stages in HD, as well as in other neurodegenerative disorders, hydrocephalus in these cases appears to be secondary, due to tissue loss and brain atrophy; a condition known as ex vacuo hydrocephalus. So far, only one case of HD associated with obstructive hydrocephalus of familiar origin has been reported (68). As for mutations that abolish htt function, not much attention has been focused on, there is only one case report of a truncation in heterozygosity of the HD gene that has been reported in humans (69). Our findings suggest that the HD gene could be a candidate gene for hydrocephalus in humans, and that the search for more subtle mutations may confirm this hypothesis.
MATERIALS AND METHODS
Genetic crosses
Wnt1-Cre/+ mice (15) (Jax Lab stock number 003829) were backcrossed onto the C57BL/6 background, and the resulting Wnt1-Cre/+ progeny was then crossed with Hdh+/− mice (10). To obtain the progeny of interest, Wnt1-Cre/+; Hdh+/− mice were crossed with Hdh flox/flox (3). Mutant and control mice were in C57BL/6 genetic background.
For verification of Wnt1Cre pattern of recombination, Wnt1-Cre/+ mice were crossed with the Rosa26 Reporter (20) (R26R; JaxLab stock number 003474) strain and Wnt1-Cre/+; R26R/+ progeny were used for the analyses.
Genotyping of mice
For routine genotyping of progeny, we used PCR analysis (30 cycles consisting of 45 s denaturation at 94°C, 45 s annealing at 61°C and 1 min extension at 72°C). The Hdh null allele was detected using the primers Neo 3 (Neo reverse): 5′- AGAGCAGCCGATTGTCTGTTGT-3′ and Neo 6 (MC1 forward): 5′- AACACCGAGCGACCCTGCAG-3′, which generate a 130 bp product.
The cre transgene was detected using the primers Cre 1 (Cre forward): 5′-CTGCCACGACCAAGTGACAGC-3′ and Cre 2 (Cre reverse): 5′-CTTCTCTACACCTGCGGTGCT-3′ to generate a 325 bp product corresponding to a portion of the cre coding region. The Hdh floxed allele was detected using the primers pgk (pgk promoter sequence): 5′-GCCCGGCATTCTGCACGCTT-3′ and Hdhpr13 (Hdh promoter sequence): 5′-CTGTCTGGAGGTGATCCATGC-3′, which generate a 150 bp PCR product. For detection of the R26R allele, the primers Lac 1 (LacZ forward): 5′-CGGCGGTGAAATTATCGATGAG-3′ and Lac 2 (LacZ reverse): 5′-ATCAGCAGGATATCCTGCACCA-3′ were used to generate a 315 bp PCR product.
Histological analyses
For histology, brains were fixed in 4% (wt/vol) paraformaldehyde in phosphate-buffered saline (PBS); incubated for 24 h at 4°C in PBS containing 0.25 M sucrose, 0.2 M glycine; dehydrated; cleared with toluene; and embedded in paraffin. Paraffin blocks were sectioned at 7 µm and stained with hematoxylin and eosin.
X-gal staining
Staining of sections to visualize lacZ expression was performed as described (70). Brains were first fixed overnight at 4°C in 0.1 M PIPES pH 6.9, 2 mm MgCl2, 5 mm EGTA containing 0.2% paraformaldehyde and then cryopreserved in PBS containing 30% sucrose and 2 mm MgCl2. Brains were then embedded in OCT compound (TissueTek), and frozen at −80°C. Brains were sectioned on a sagital or coronal plane (9 µm thickness) in a cryostat and mounted on superfrost slides (Fisher). Sections were stained with X-Gal at 37°C overnight, washed with PBS and counterstained with eosin.
Western blotting
Protein extracts from forebrain and cerebellum were obtained by homogenizing tissue in protein extraction buffer (10 mm HEPES, pH 8.3; 1.5 mm MgCl2; 1 mm DTT) containing protease inhibitor cocktail (Roche). Insoluble debris was discarded after centrifugation, and protein concentration was determined by the Bradford assay (Bio-Rad). Approximately 20 µg of protein was separated by SDS–PAGE (6% gel) and transferred into nitrocellulose membranes. Membranes were blocked in 5% non-fat milk for 1 h at room temperature and incubated overnight at 4°C with mouse monoclonal antibodies against huntingtin (mAb2166, Chemicon, 1:3,000), or β-tubulin (Chemicon, 1:5,000). Membranes were washed and incubated with secondary antibodies for 1 h at room temperature. Protein bands were visualized by chemiluminescence (PIERCE or Amersham) followed by exposure to autoradiographic film.
Skeletal preparations
For skeletal preparations skin and viscera were removed, and carcasses were dehydrated in 95% ethanol for 24 h. Skeletons were then differentially stained with alcian blue (cartilage) and alizarin red (bone). Skeletons were transferred to 1% KOH, monitored for clearing, with changes of KOH as needed. Following clearance of tissue, skeletons were washed with glycerol containing 1% KOH and stored in glycerol (71).
Immunohistochemistry
Immunohistochemistry was performed as described (15). Briefly, brains were rapidly dissected in PBS, fixed overnight at 4°C in 4% paraformaldehyde (4% PFA), equilibrated in 30% sucrose in phosphate buffer (PBS) for 24 h at 4°C, embedded in OCT freezing compound (TissueTek) and frozen at −80°C. Blocks were sectioned coronally or sagitally (9 µm thickness) in a cryostat, and sections were mounted in superfrost slides (Fisher). For fluorescent immunohistochemistry, sections were then postfixed in 4% PFA at room temperature, and blocked at room temperature for 30 min with 1% BSA and 0.1% Triton X-100 in PBS. Primary antibodies mouse monoclonal anti-htt (Mab2166, Chemicon); rabbit polyclonal anti-ZO-1 (Zymed), or anti-Claudin1 (Zymed) were applied after 1:200 dilution in 0.1% BSA, 0.1% Triton X-100 prepared in PBS, and slides were incubated overnight at 4°C in a humidified chamber. After three washes in PBS, slides were incubated for 1 h with secondary anti-mouse or anti-rabbit Cy3-fluorescent-conjugated antibody (Jackson ImmunoResearch), washed in PBS and mounted in Vectashield (Vector Laboratories).
For immunodetection using DAB substrate, slides were air-dried, post-fixed with 4% PFA and incubated with 0.3% H202 in methanol for 20 min, to quench endogenous peroxidase. Sections were then blocked for 1 h with 1% BSA, 0.1% triton in PBS and incubated with primary rabbit polyclonal antibody anti-ki-67 (DAKO); anti-aquaporin 1 (Chemicon); or anti-RF (AFRU, a gift from Dr E.M. Rodriguez) overnight at 4°C in a humidified chamber. After several washes in PBS, primary antibody detection was carried out using the Vector ABC kit according to manufacturers' instructions, followed by incubation with DAB (BD Biosciences) or Fast-DAB with metal enhancer (Sigma).
ACKNOWLEDGEMENTS
We are grateful to Dr E.M. Rodríguez (Instituto de Anatomia, Hystologia y Patologia; Faculdad de Medicina, Universidad Austral de Chile) for the gift of the anti-RF antibody. We thank Dr R.K. Rao for insights in tight junctions and sharing reagents. We also thank members of the Dragatsis’ laboratory for valuable discussions.
Conflict of interest: none declared.
REFERENCES
- 1.The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 2.Cattaneo E., Rigamonti D., Goffredo D., Zuccato C., Squitieri F., Sipione S. Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci. 2001;24:182–188. doi: 10.1016/s0166-2236(00)01721-5. [DOI] [PubMed] [Google Scholar]
- 3.Dragatsis I., Levine M.S., Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y., Li M., Drozda M., Chen M., Ren S., Mejia Sanchez R.O., Leavitt B.R., Cattaneo E., Ferrante R.J., Hayden M.R., Friedlander R.M. Depletion of wild-type huntingtin in mouse models of neurologic diseases. J. Neurochem. 2003;87:101–106. doi: 10.1046/j.1471-4159.2003.01980.x. [DOI] [PubMed] [Google Scholar]
- 5.Dragatsis I., Efstratiadis A., Zeitlin S. Mouse mutant embryos lacking huntingtin are rescued from lethality by wild-type extraembryonic tissues. Development. 1998;125:1529–1539. doi: 10.1242/dev.125.8.1529. [DOI] [PubMed] [Google Scholar]
- 6.Schmitt I., Bachner D., Megow D., Henklein P., Epplen J.T., Riess O. Expression of the Huntington disease gene in rodents: cloning the rat homologue and evidence for downregulation in non-neuronal tissues during development. Hum. Mol. Genet. 1995;4:1173–1182. doi: 10.1093/hmg/4.7.1173. [DOI] [PubMed] [Google Scholar]
- 7.Dragatsis I., Zeitlin S. A method for the generation of conditional gene repair mutations in mice. Nucleic Acids Res. 2001;29:E10. doi: 10.1093/nar/29.3.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duyao M.P., Auerbach A.B., Ryan A., Persichetti F., Barnes G.T., McNeil S.M., Ge P., Vonsattel J.P., Gusella J.F., Joyner A.L., et al. Inactivation of the mouse Huntington's disease gene homolog Hdh. Science. 1995;269:407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- 9.Nasir J., Floresco S.B., O'Kusky J.R., Diewert V.M., Richman J.M., Zeisler J., Borowski A., Marth J.D., Phillips A.G., Hayden M.R. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- 10.Zeitlin S., Liu J.P., Chapman D.L., Papaioannou V.E., Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat. Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- 11.Woda J.M., Calzonetti T., Hilditch-Maguire P., Duyao M.P., Conlon R.A., MacDonald M.E. Inactivation of the Huntington's disease gene (Hdh) impairs anterior streak formation and early patterning of the mouse embryo. BMC Dev. Biol. 2005;18:5–17. doi: 10.1186/1471-213X-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harjes P., Wanker E.E. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem. Sci. 2003;28:425–433. doi: 10.1016/S0968-0004(03)00168-3. [DOI] [PubMed] [Google Scholar]
- 13.Awatramani R., Soriano P., Rodriguez C., Mai J.J., Dymecki S.M. Cryptic boundaries in roof plate and choroid plexus identified by intersectional gene activation. Nat. Genet. 2003;35:70–75. doi: 10.1038/ng1228. [DOI] [PubMed] [Google Scholar]
- 14.Danielian P.S., Echelard Y., Vassileva G., McMahon A.P. A 5.5-kb enhancer is both necessary and sufficient for regulation of Wnt-1 transcription in vivo. Dev. Biol. 1997;192:300–309. doi: 10.1006/dbio.1997.8762. [DOI] [PubMed] [Google Scholar]
- 15.Danielian P.S., Muccino D., Rowitch D.H., Michael S.K., McMahon A.P. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- 16.Nichols D.H., Bruce L.L. Migratory routes and fates of cells transcribing the Wnt-1 gene in the murine hindbrain. Dev. Dyn. 2006;235:285–300. doi: 10.1002/dvdy.20611. [DOI] [PubMed] [Google Scholar]
- 17.Brault V., Moore R., Kutsch S., Ishibashi M., Rowitch D.H., McMahon A.P., Sommer L., Boussadia O., Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 18.Baas D., Meiniel A., Benadiba C., Bonnafe E., Meiniel O., Reith W., Durand B. A deficiency in RFX3 causes hydrocephalus associated with abnormal differentiation of ependymal cells. Eur. J. Neurosci. 2006;24:1020–1030. doi: 10.1111/j.1460-9568.2006.05002.x. [DOI] [PubMed] [Google Scholar]
- 19.Merchant M., Evangelista M., Luoh S.M., Frantz G.D., Chalasani S., Carano R.A., van Hoy M., Ramirez J., Ogasawara A.K., McFarland L.M., et al. Loss of the serine/threonine kinase fused results in postnatal growth defects and lethality due to progressive hydrocephalus. Mol. Cell. Biol. 2005;25:7054–7068. doi: 10.1128/MCB.25.16.7054-7068.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 21.Lindeman G.J., Dagnino L., Gaubatz S., Xu Y., Bronson R.T., Warren H.B., Livingston D.M. A specific, nonproliferative role for E2F-5 in choroid plexus function revealed by gene targeting. Genes Dev. 1998;12:1092–1098. doi: 10.1101/gad.12.8.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meiniel A. The secretory ependymal cells of the subcommissural organ: which role in hydrocephalus? Int. J. Biochem. Cell. Biol. 2007;39:463–468. doi: 10.1016/j.biocel.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 23.Utriainen A., Sormunen R., Kettunen M., Carvalhaes L.S., Sajanti E., Eklund L., Kauppinen R., Kitten G.T., Pihlajaniemi T. Structurally altered basement membranes and hydrocephalus in a type XVIII collagen deficient mouse line. Hum. Mol. Genet. 2004;13:2089–2099. doi: 10.1093/hmg/ddh213. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J., Williams M.A., Rigamonti D. Genetics of human hydrocephalus. J. Neurol. 2006;253:1255–1266. doi: 10.1007/s00415-006-0245-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernandez-Llebrez P., Grondona J.M., Perez J., Lopez-Aranda M.F., Estivill-Torrus G., Llebrez-Zayas P.F., Soriano E., Ramos C., Lallemand Y., Bach A., Robert B. Msx1-deficient mice fail to form prosomere 1 derivatives, subcommissural organ, and posterior commissure and develop hydrocephalus. J. Neuropathol. Exp. Neurol. 2004;63:574–586. doi: 10.1093/jnen/63.6.574. [DOI] [PubMed] [Google Scholar]
- 26.Louvi A., Wassef M. Ectopic engrailed 1 expression in the dorsal midline causes cell death, abnormal differentiation of circumventricular organs and errors in axonal pathfinding. Development. 2000;127:4061–4071. doi: 10.1242/dev.127.18.4061. [DOI] [PubMed] [Google Scholar]
- 27.Brown P.D., Davies S.L., Speake T., Millar I.D. Molecular mechanisms of cerebrospinal fluid production. Neuroscience. 2004;129:957–970. doi: 10.1016/j.neuroscience.2004.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emerich D.F., Skinner S.J., Borlongan C.V., Vasconcellos A.V., Thanos C.G. The choroid plexus in the rise, fall and repair of the brain. Bioessays. 2005;27:262–274. doi: 10.1002/bies.20193. [DOI] [PubMed] [Google Scholar]
- 29.Boassa D., Yool A.J. Physiological roles of aquaporins in the choroid plexus. Curr. Top. Dev. Biol. 2005;67:181–206. doi: 10.1016/S0070-2153(05)67005-6. [DOI] [PubMed] [Google Scholar]
- 30.Mao X., Enno T.L., Del Bigio M.R. Aquaporin 4 changes in rat brain with severe hydrocephalus. Eur. J. Neurosci. 2006;23:2929–2936. doi: 10.1111/j.1460-9568.2006.04829.x. [DOI] [PubMed] [Google Scholar]
- 31.Oshio K., Watanabe H., Song Y., Verkman A.S., Manley G.T. Reduced cerebrospinal fluid production and intracranial pressure in mice lacking choroid plexus water channel Aquaporin-1. FASEB J. 2005;19:76–78. doi: 10.1096/fj.04-1711fje. [DOI] [PubMed] [Google Scholar]
- 32.Rodríguez E.M., Rodríguez S., Hein S. The subcommissural organ. Microsc. Res. Tech. 1998;41:98–123. doi: 10.1002/(SICI)1097-0029(19980415)41:2<98::AID-JEMT2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 33.Rodríguez E.M., Oksche A., Hein S., Rodríguez S., Yulis C. Comparative immunocytochemical study of the SCO. Cell. Tissue Res. 1984;237:427–441. doi: 10.1007/BF00228427. [DOI] [PubMed] [Google Scholar]
- 34.Gonçalves-Mendes N., Simon-Chazottes D., Creveaux I., Meiniel A., Guénet J.L., Meiniel R. Mouse SCO-spondin, a gene of the thrombospondin type 1 repeat (TSR) superfamily expressed in the brain. Gene. 2003;312:263–270. doi: 10.1016/s0378-1119(03)00622-x. [DOI] [PubMed] [Google Scholar]
- 35.Bruni J.E., Del Bigio M.R., Cardoso E.R., Persaud T.V. Hereditary hydrocephalus in laboratory animals and humans. Exp. Pathol. 1988;35:239–246. doi: 10.1016/s0232-1513(88)80094-x. [DOI] [PubMed] [Google Scholar]
- 36.Garton H.J., Piatt J.H., Jr Hydrocephalus. Pediatr. Clin. North. Am. 2004;51:305–325. doi: 10.1016/j.pcl.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Rizvi R., Anjum Q. Hydrocephalus in children. J. Pak. Med. Assoc. 2005;55:502–507. [PubMed] [Google Scholar]
- 38.Koh L., Zakharov A., Johnston M. Integration of the subarachnoid space and lymphatics: is it time to embrace a new concept of cerebrospinal fluid absorption? Cerebrospinal Fluid Res. 2005;20:2–6. doi: 10.1186/1743-8454-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haverkamp F., Wölfle J., Aretz M., Krämer A., Höhmann B., Fahnenstich H., Zerres K. Congenital hydrocephalus internus and aqueduct stenosis: aetiology and implications for genetic counselling. Eur. J. Pediatr. 1999;158:474–478. doi: 10.1007/s004310051123. [DOI] [PubMed] [Google Scholar]
- 40.Zhang W., Yi M.J., Chen X., Cole F., Krauss R.S., Kang J.S. Cortical thinning and hydrocephalus in mice lacking the immunoglobulin superfamily member CDO. Mol. Cell. Biol. 2006;26:3764–3772. doi: 10.1128/MCB.26.10.3764-3772.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez-Figares J.M., Jimenez A.J., Rodríguez E.M. Subcommissural organ, cerebrospinal fluid circulation, and hydrocephalus. Microsc. Res. Tech. 2001;52:591–607. doi: 10.1002/1097-0029(20010301)52:5<591::AID-JEMT1043>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 42.Blackshear P.J., Graves J.P., Stumpo D.J., Cobos I., Rubenstein J.L., Zeldin D.C. Graded phenotypic response to partial and complete deficiency of a brain-specific transcript variant of the winged helix transcription factor RFX4. Development. 2003;130:4539–4552. doi: 10.1242/dev.00661. [DOI] [PubMed] [Google Scholar]
- 43.Danielian P.S., McMahon A.P. Engrailed-1 as a target of the Wnt-1 signalling pathway in vertebrate midbrain development. Nature. 1996;383:332–334. doi: 10.1038/383332a0. [DOI] [PubMed] [Google Scholar]
- 44.Rowitch D.H., Danielian P.S., McMahon A.P., Zec N. Cystic malformation of the posterior cerebellar vermis in transgenic mice that ectopically express Engrailed-1, a homeodomain transcription factor. Teratology. 1999;60:22–28. doi: 10.1002/(SICI)1096-9926(199907)60:1<22::AID-TERA7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 45.Estivill-Torrus G., Vitalis T., Fernandez-Llebrez P., Price D.J. The transcription factor Pax6 is required for development of the diencephalic dorsal midline secretory radial glia that form the subcommissural organ. Mech. Dev. 2001;109:215–224. doi: 10.1016/s0925-4773(01)00527-5. [DOI] [PubMed] [Google Scholar]
- 46.Stoykova A., Gruss P. Roles of Pax-genes in developing and adult brain as suggested by expression patterns. J. Neurosci. 1994;14:1395–1412. doi: 10.1523/JNEUROSCI.14-03-01395.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas K.R., Capecchi M.R. Targeted disruption of the murine int-1 proto-oncogene resulting in severe abnormalities in midbrain and cerebellar development. Nature. 1990;346:847–850. doi: 10.1038/346847a0. [DOI] [PubMed] [Google Scholar]
- 48.Andrade M.A., Bork P. HEAT repeats in the Huntington's disease protein. Nat. Genet. 1995;11:115–116. doi: 10.1038/ng1095-115. [DOI] [PubMed] [Google Scholar]
- 49.Takano H., Gusella J.F. The predominantly HEAT-like motif structure of huntingtin and its association and coincident nuclear entry with dorsal, an NF-kB/Rel/dorsal family transcription factor. BMC Neurosci. 2002;3:15. doi: 10.1186/1471-2202-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MacDonald M.E. Huntingtin: alive and well and working in middle management. Sci. STKE. 2003;207:e48. doi: 10.1126/stke.2003.207.pe48. [DOI] [PubMed] [Google Scholar]
- 51.Marcora E., Gowan K., Lee J.E. Stimulation of NeuroD activity by huntingtin and huntingtin-associated proteins HAP1 and MLK2. Proc. Natl Acad. Sci. USA. 2003;100:9578–9583. doi: 10.1073/pnas.1133382100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zuccato C., Tartari M., Crotti A., Goffredo D., Valenza M., Conti L., Cataudella T., Leavitt B.R., Hayden M.R., Timmusk T., Rigamonti D., Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 53.Zuccato C., Belyaev N., Conforti P., Ooi L., Tartari M., Papadimou E., MacDonald M., Fossale E., Zeitlin S., Buckley N., Cattaneo E. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington's disease. J. Neurosci. 2007;27:6972–6983. doi: 10.1523/JNEUROSCI.4278-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.White J.K., Auerbach W., Duyao M.P., Vonsattel J.P., Gusella J.F., Joyner A.L., MacDonald M.E. Huntingtin is required for neurogenesis and is not impaired by the Huntington's disease CAG expansion. Nat. Genet. 1997;17:404–410. doi: 10.1038/ng1297-404. [DOI] [PubMed] [Google Scholar]
- 55.Dunah A.W., Jeong H., Griffin A., Kim Y.M., Standaert D.G., Hersch S.M., Mouradian M.M., Young A.B., Tanese N., Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 56.Li S.H., Cheng A.L., Zhou H., Lam S., Rao M., Li H., Li X.J. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol. Cell. Biol. 2002;22:1277–1287. doi: 10.1128/mcb.22.5.1277-1287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takahashi T., Guron C., Shetty S., Matsui H., Raghow R. A minimal murine Msx-1 gene promoter. Organization of its cis-regulatory motifs and their role in transcriptional activation in cells in culture and in transgenic mice. J. Biol. Chem. 1997;272:22667–22678. doi: 10.1074/jbc.272.36.22667. [DOI] [PubMed] [Google Scholar]
- 58.Zheng J.B., Zhou Y.H., Maity T., Liao W.S., Saunders G.F. Activation of the human PAX6 gene through the exon 1 enhancer by transcription factors SEF and Sp1. Nucleic Acids Res. 2001;29:4070–4078. doi: 10.1093/nar/29.19.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vio K., Rodríguez S., Navarrete E.H., Perez-Figares J.M., Jimenez A.J., Rodríguez E.M. Hydrocephalus induced by immunological blockage of the subcommissural organ-Reissner's fiber (RF) complex by maternal transfer of anti-RF antibodies. Exp. Brain Res. 2000;135:41–52. doi: 10.1007/s002210000474. [DOI] [PubMed] [Google Scholar]
- 60.Davson H., Segal M.B. Physiology of the CSF and Blood-Brain-Barriers. Boca Raton, FL: CRC Press; 1996. [Google Scholar]
- 61.Welch K. Secretion of cerebrospinal fluid by choroid plexus of the rabbits. Am. J. Physiol. 1963;205:617–624. doi: 10.1152/ajplegacy.1963.205.3.617. [DOI] [PubMed] [Google Scholar]
- 62.Haywood J.R., Vogh B.P. Some measurements of the autonomic nervous system influence on production of cerebrospinal fluid in cat. J. Pharmacol. Exp. Ther. 1979;208:341–346. [PubMed] [Google Scholar]
- 63.Lindvall M., Edvinsson L., Owman C. Sympathetic nervous control of cerebrospinal fluid production from the choroid plexus. Science. 1978;201:176–178. doi: 10.1126/science.663649. [DOI] [PubMed] [Google Scholar]
- 64.Caprile T., Hein S., Rodriguez S., Montecinos H., Rodriguez E. Reissner fiber binds and transports away monoamines present in the cerebrospinal fluid. Brain Res. Mol. Brain. Res. 2003;110:177–192. doi: 10.1016/s0169-328x(02)00565-x. [DOI] [PubMed] [Google Scholar]
- 65.Rodríguez E., Yulis C.R. Subcommissural organ. Cellular, molecular, physiological, and pathological aspects: one 100 years of subcommissural organ research. Microsc. Res. Tech. 2001;52:459–460. doi: 10.1002/1097-0029(20010301)52:5<459::AID-JEMT1031>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 66.Wagner C., Batiz L.F., Rodríguez S., Jiménez A.J., Páez P., Tomé M., Pérez-Fígares J.M., Rodríguez E.M. Cellular mechanisms involved in the stenosis and obliteration of the cerebral aqueduct of hyh mutant mice developing congenital hydrocephalus. J. Neuropathol. Exp. Neurol. 2003;62:1019–1040. doi: 10.1093/jnen/62.10.1019. [DOI] [PubMed] [Google Scholar]
- 67.Silva-Alvarez C., Carrasco M., Balmaceda-Aguilera C., Pastor P., García Mde L., Reinicke K., Aguayo L., Molina B., Cifuentes M., Medina R., Nualart F. Ependymal cell differentiation and GLUT1 expression is a synchronous process in the ventricular wall. Neurochem. Res. 2005;30:1227–1236. doi: 10.1007/s11064-005-8794-z. [DOI] [PubMed] [Google Scholar]
- 68.Lorenzo N.Y., Leibrock L.G., Lorenzo A.S. A single case of Huntington's disease simultaneously occurring with obstructive hydroc ephalus. Surg. Neurol. 1991;35:136–138. doi: 10.1016/0090-3019(91)90265-b. [DOI] [PubMed] [Google Scholar]
- 69.Gusella J.F., MacDonald M.E. Huntington's disease and repeating trinucleotides. N. Engl. J. Med. 1994;330:1450–1451. doi: 10.1056/NEJM199405193302011. [DOI] [PubMed] [Google Scholar]
- 70.Hogan B., Beddington R., Costantini F., Lacy E. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- 71.Kaufman M.H. The Atlas of Mouse Development. Academic Press; 1992. [Google Scholar]