

Abstract
The aim of the study was to describe the molecular and biochemical interactions associated with amino acid biosynthesis and storage protein accumulation in the developing grains of field-grown barley. Our strategy was to analyse the transcription of genes associated with the biosynthesis of storage products during the development of field-grown barley grains using a grain-specific microarray assembled in our laboratory. To identify co-regulated genes, a distance matrix was constructed which enabled the identification of three clusters corresponding to early, middle, and late grain development. The gene expression pattern associated with the clusters was investigated using pathway-specific analysis with specific reference to the temporal expression levels of a range of genes involved mainly in the photosynthesis process, amino acid and storage protein metabolism. It is concluded that the grain-specific microarray is a reliable and cost-effective tool for monitoring temporal changes in the transcriptome of the major metabolic pathways in the barley grain. Moreover, it was sensitive enough to monitor differences in the gene expression profiles of different homologues from the storage protein families. The study described here should provide a strong complement to existing knowledge assisting further understanding of grain development and thereby provide a foundation for plant breeding towards storage proteins with improved nutritional quality.
Keywords: Amino acid metabolism, cDNA microarray, field trial, hordein, Hordeum vulgare, storage proteins
Introduction
The content and quality of proteins are major determinants of the nutritional value of cereal grains and grain-derived products. Storage protein composition is the result of a complex interaction between the plant's genetic background and its environment; the latter encompasses nutrient availability (Shewry et al., 2001; Oury et al., 2003). Nitrogen is an essential nutrient and plays a dominant role in determining the amount of protein stored in cereal grains. Intensive agriculture is driven by high inputs, in particular N fertilizer. However, increasing environmental concerns have prompted the need to improve nitrogen use efficiency (NUE) of cereals which currently only utilize approximately 30–40% of the available nitrogen (Raun and Johnson, 1998).
The study of storage protein accumulation in the cereal grain has a long history and a range of biochemical and molecular techniques have been used successfully to dissect the complex regulation of individual genes and proteins associated with storage product accumulation during grain filling (see recent reviews by Jolliffe et al., 2005; Vicente-Carbajosa and Carbonero, 2005, and references therein). While significant understanding was achieved by analysing single or a small subset of genes in isolation it also underpinned the development of molecular plant breeding techniques such as marker assisted breeding and Quantitative Trait Loci (QTL) mapping (Thomas, 2003). In recent years, a rapidly increasing number of microarray analyses have been implemented to enable ‘global’ gene expression analysis. Microarray data can readily be integrated with traditional biochemical and physiological analysis, which has apportioned function to a gene and its gene product. Therefore, microarray has the potential to enable the identification of candidate genes related to a wide range of important quality traits such as protein content and composition.
To date, microarray technology has been used to study global gene expression during grain filling and seed formation in a number species, for example, in rice (Zhu et al., 2003; Duan et al., 2005), Arabidopsis (Girke et al., 2000; Ruuska et al., 2002), wheat (Gregersen et al., 2005; Baudo et al., 2006; Kan et al., 2006), barley (Sreenivasulu et al., 2004, 2006; Radchuk et al., 2005, Druka et al., 2006), and Medicago truncatula (Firnhaber et al., 2005). Furthermore, the work of Druka et al. (2006) has provided a global framework of whole plant gene expression analysis for barley, which underpins the database named BarleyBase (http://www.plexdb.org). However, it is important to note that a common feature of the studies cited above is that the experimental plant material was grown under controlled conditions either in greenhouses or growth cabinets. Given the fact that a ‘systems approach’ must integrate the impact of the environment and since environment has a significant impact on plant performance, extrapolation of the results from glasshouse-grown material to field-grown material is not straightforward. Recent studies have demonstrated the general utility of microarray analysis of field-grown plants (Duan and Sun, 2005; Lu et al., 2005). Moreover, when comparing plant material grown in controlled conditions with field-grown material, significant differences have been illustrated (Dhanaraj et al., 2007). Therefore ‘real life’ systems analysis, where field-grown material is analysed, has a greater relevance when addressing plant performance in particular complex traits such as yield and quality.
As stated, applied nitrogen is highly important with respect to barley grain quality, however, to our knowledge, despite the very significant body of work in this area, microarray analysis has not yet been applied to barley grain development with the sole object of describing the interaction of genes associated with amino acid and protein synthesis. Our rationale was that using microarray analysis to obtain a synthesis of gene expression was a necessary first step towards studying the impact of nitrogen treatment on the expression of genes that influence grain quality traits. To achieve this; a pathway-specific analysis of microarray data derived from a custom-made cDNA microarray with 1035 genes has been performed. The array contains a comprehensive set of genes involved in nitrogen mobilization, transport, and amino acid metabolism. The results of the study will provide the opportunity to unify global gene expression with existing biochemical and molecular data with the aim of ultimately altering or regulating the nutritional quality of barley grains.
Materials and methods
Plant material
Spring barley (Hordeum vulgare L. cv. Barke) was grown in three field plots of 19.8 m2 (12 m ×1.65 m) during the summer of 2005, at the Research Centre Flakkebjerg, Denmark. After sowing, the plots were fertilized with NS24-7 (DLA Agro) which contains 12% ammonium, 12% nitrate, and 7% sulphur, at a rate of 120 kg nitrogen ha−1. After 1 week the plots were fertilized again with PK 0-4-21 (DLA Agro) at a rate of 25 kg phosphorus ha−1 and 60 kg potassium ha−1. The plots were sprayed one month after sowing with a broad-spectrum herbicide mixture containing Express ST (Tribenuron-methyl 50%; E.L. du Pont de Nemours & Co), Oxitril CM (loxynil 17.32%; Bayer Crop Science) and Starane 180s (Fluroxypyr 180 g l−1; Dow Agrosciences) herbicides. The plant material was both morphologically and chronologically staged in accordance with internationally recognized criteria (Fig. 1) (Zadoks code, Zadoks et al., 1974). Individual spikes were tagged at flowering and harvested in the morning (08.00–09.00 h) at 10, 15, 18, and 25 d after pollination (DAP). Developing grains were immediately frozen in liquid nitrogen and stored at –80 °C until analysis.
Fig. 1.

Development of barley (Hordeum vulgare cv. Barke) grains used for the expression profiling. DAP: days after pollination.
Near-infrared spectrometry
The grains harvested were analysed for water (%), starch (%), and protein content (%) using a near-infrared spectroscopy analyser (Foss Tecator, Infratec 1241, Grain Analyser v.3.40). The near-infrared spectroscopy analyser was calibrated and linked to the Danish NIT network (Buchmann et al., 2001).
Construction of barley cDNA microarrays
A set of genes (1035) was assembled from two EST libraries obtained from Clemson University Genomics Institute (termed HVSMEi and HVSMEk; (http://www.genome.clemson.edu/projects/barley/) and the microarray slides were prepared as described by Hansen et al. (2007). The list of the 1035 genes is available as Supplementary material at JXB online in Hansen et al. (2007).
RNA isolation and labelling of target material
Three biological replicates were sampled; each sample consisted of two grains collected from the midrib of independent barley spikes. After grinding the grains in liquid nitrogen the total RNA was extracted according to the manufacturer's protocol (FastRNA Pro Green Kit, Bio101 Systems, France). Messenger RNA was extracted from the total RNA using-Dynabeads (610–05, Dynal, N) according to the manufacturer's protocol. The synthesis of first and second strand cDNA and labelling with Cyanine3/Cyanine5 were performed according to Eisen and Brown (1999).
Microarray design, data pre-procession, and identification of differential expression
The hybridization protocol was performed according to Eisen and Brown (1999) with modifications according to Hansen et al. (2007).
The hybridization of the grain-specific microarray was carried out with three biological replicates. The array contained 1035 genes. Each gene was spotted in triplicate in three subgrids across the slide to control for potential sources of variation in hybridization across the area of the slide (technical replicates). The microarray experiments were performed using samples collected from field-grown barley subject to three different nitrogen regimes (50, 120, and 150 kg ha−1) at four time points (15, 18, 20, and 25 DAP). An interwoven loop experimental design was chosen (Altman and Hua, 2006) in combination with three biological replicates per treatment resulting in 18 hybridizations (see Supplementary Fig. S1 at JXB online). Data acquisition and analysis was performed using an arrayWoRx microarray scanner (BioChipReader, Applied Precision, USA) using the arrayWoRx 2.0 software suite. The spots of each individual slide were quantified using a ‘well-defined’ grid.
The experimental design dictated that two different factors (time and treatment) were combined for each hybridization, thus strengthening the statistical analysis. Although the different nitrogen regimes are not of interest in the present study, including them in a two factor set-up, such as a two-way ANOVA, allows for the inference of differential expression with a higher degree of confidence (Shrout and Fleiss, 1979). The other P-values returned by the test (for the nitrogen regimes and the conjugated P-value) were not used in the present article. A two-way ANOVA was performed treating the technical replicates as independent data points rather than means, thereby avoiding loss of variation and in turn increasing the confidence in the resulting P-values (Altman and Hua, 2006). The slides were pre-processed to ensure uniformity of hybridization before being subject to ANOVA.
The raw data files can be obtained from the ArrayExpress microarray repository at the European Bioinformatics Institute (EBI) (http://www.ebi.ac.uk/arrayexpress/, accession number: E-MEXP-1013) supplemented with the available sequences for the genes used in the experiment. Annotations of the genes can be found at http://www.genome.clemson.edu/projects/barley/. The microarray data were normalized using the non-linear Qspline algorithm (Workman et al., 2002). The data reported in this article were extracted from the interwoven loop experiment design in order to identify significantly regulated genes during grain development in the barley grown at 120 kg N ha−1.
Clustering using Partitioning Around Medoids (PAM)
Co-regulated genes were identified by generating a distance matrix using a Pearson correlation between the expression values with the highest confidence limits. The statistical package used was R (Becker et al., 1988) (http://www.r-project.org/). The distance matrix was subsequently clustered by the Partitioning Around Medoids method (PAM) (Kaufman and Rousseeuw, 1990) using the cluster package in R. The PAM algorithm is a robust version of k-means and it searches for a specified number of medoids (representatives), k, around which clusters are constructed. Minimizing the sum of the dissimilarities of all observations and assigning them to their closest medoid generated the clusters. The value of k=3 was identified by manual inspection as the optimal number of clusters and it divided the data into three categories, one having the highest expression at day 10, another at 15–18 d, while the last cluster showed the highest expression at 25 d (Fig. 2).
Fig. 2.

Cluster analysis. The gene expression profile of the first 300 most significantly regulated probes representing early-, mid-, and late phases of the field-grown barley grain. The relative expression is depicted by the Z-score (obtained for each measurement by subtracting from it the mean intensity for the given probe and dividing the result with the corresponding standard deviation) separated by the sampling time points. The red line indicated the average gene expression during development.
Heat map and supervised hierarchical clustering
The heat map diagram (Fig. 3) shows the result of the one-way hierarchical clustering of genes of the samples (Eisen et al., 1998). Every horizontal row represents an individual gene and the gene clustering tree is shown on the left. Developmental stages (days after flowering), assigned in 10, 15, 18, and 25 d intervals at the top, are represented in vertical columns. The clustering is performed on the log2 transformed expression values. A Z-score is calculated and used for the clustering by subtracting the mean value from the absolute expression value for each gene followed by the division with standard deviation across samples. The colour scale shown at the bottom illustrates the relative expression level of a gene across all samples: the red colour represents an expression level above the mean and the blue colour represents expression levels lower than the mean (Eisen et al., 1998).
Fig. 3.

Hierarchical clustering of genes. Heat map of hierarchical clustering for 55 selected differentially expressed genes: horizontal rows represent individual genes and vertical rows represent individual time points. Red and blue indicate transcript level above and below the median for that gene across all samples, respectively. Distinct clusters of differentially expressed genes can be seen for early, middle, and late developmental stages.
Real-time RT-PCR expression analysis
Total RNA was isolated from a pool of three individual grains taken from the middle of three independent barley spikes using FastRNA Pro Green Kit (Bio101, Systems, France) and resuspended in 50 μl DEPC-treated water according to the manufacturer's manual. The diluted RNA was quantified using a GeneQuant II DNA/RNA calculator (Pharmacia Biotech, Piscataway NJ, USA). First strand cDNA synthesis and real time RT-PCR were carried out as described by Hansen et al. (2007). Primer Express software (Applied Biosystems, Foster City, CA, USA) was used to design the primer to the same region as the microarray probe (see Supplementary Table S1 at JXB online). Primers for the hordein family were designed towards homologous regions identified using sequence alignments generated from accessions recovered from the GenBank database. To investigate the specificity of each primer set (see Supplementary Table S1 at JXB online) a dissociation curve analysis was implemented. Expression level units of each gene of interest were calculated relative to the housekeeping gene, actin, in the samples (Livak and Schmittgen, 2001). For the individual candidate genes, the expression at 18 DAP and 25 DAP was calibrated to the expression at 10 DAP (calibrator). The Ct value was obtained for each specific gene in the samples, followed by a quality check of linear regression (R 2) and relative expression calculation for each gene using parameters of the software REST© according to Pfaffl et al. (2002).
Results
To assess parity between our experimental material and commercially grown barley the starch and protein contents of the experimental field-grown material was determined and found to be 61.6% and 10.4%, respectively, which was in agreement with Danish field trials (http://orgprints.org/8019/01/8019.pdf). The yield of our field trial, based on a 19.8 m2 plot, was calculated to 55.5 kg ha−1, which again compared favourably to the 60.4 kg ha−1 reported for the 2005 national trials.
Gene expression profile, cluster analysis
The focus of this study was on the developmental phase between 10 DAP and 25 DAP, because, during this period, the grains undergo highly orchestrated programmed events that synchronize the synthesis and deposition of storage products (Coruzzi and Bush, 2001; Coruzzi and Zhou, 2001; Palenchar et al., 2004).
A pathway-specific analysis was conducted on a subset of data generated from the grain-specific microarray. This established the temporal expression profile of genes associated with nitrogen mobilization, transport, and amino acid metabolism and offered an insight to the basic metabolic and biosynthetic pathways in the developing grain of field-grown barley. Upon manual inspection, three gene expression clusters were created which correlated with the development stages described above (Fig. 2). In an attempt to establish the robustness of the clusters, the level of significance was iteratively increased to P <6.58E-06 (300 probes corresponding to the genes clustered have this limit). The set of data with 1200 probes corresponds to 501 genes (P <0.01328) covering 51 out of the 55 significantly expressed genes discussed in the article and is available at JXB online in Supplementary Fig. S2 and Table S2. The extra four genes labelled with an asterisk (*) had lower P values (P <0.1) and were chosen to extend the discussion. We proceeded with pathway-specific analysis of the whole data set, focusing primarily on the genes associated with carbon provision and primary metabolism of aspartate-, arginine-, ornithine-, and proline-derived amino acids. These data were correlated with transcriptional profiles of the highly significant expression of storage protein genes (Tables 1–3).
Table 1.
Genes with early expression profile of cluster 1
| Library name | Swissprot name | Annotation | P-values |
| HVSMEk0013C18 | RUBB_SECCE | RuBisCO subunit – binding-protein β-subunit | 5.68E-07 |
| HVSMEk0012K01 | PGKY_WHEAT | Phosphoglycerate kinase – cytosolic (EC 2.7.2.3) | 4.56E-04 |
| HVSMEi0008N09 | G3PX_HORVU | GAPDH; glyceraldehyde-3-phosphate dehydrogenase (GAPDH. EC 1.2.1.12) | 0.0453 |
| HVSMEk0009J22 | GLNA2_MAIZE | Glutamine synthetase – root isozyme 2 (EC 6.3.1.2) | 7.33E-05 |
| HVSMEi0007N22 | GLNA4_MAIZE | Glutamine synthetase – root isozyme 4 (EC 6.3.1.2) | 0.00510 |
| HVSMEi0002J06 | DHE2_ARATH | Glutamate dehydrogenase 2 (EC 1.4.1.3) | 7.88–04 |
| HVSMEk0019F01 | AATC_ORYSA | Aspartate aminotransferase – cytoplasmic (EC 2.6.1.1) | 0.00264 |
| HVSMEk0023I12 | AAT5_ARATH | Aspartate aminotransferase – chloroplast (EC 2.6.1.1) | 0.00939 |
| HVSMEk0002D13 | AAT1_ARATH | Aspartate aminotransferase – mitochondria (EC 2.6.1.1) | 3.42E-05 |
| HVSMEi0005N08 | LYS9_YEAST | Saccharopine dehydrogenase (EC 1.5.1.9) | 0.00747 |
| HVSMEk0002K02* | METB_ARATH | Cystathionine γ-synthase (EC 2.5.1.48) | 0.0564 |
| HVSMEk0004D10 | METK_HORVU | SAM synthetase 1; S-adenosylmethionine synthetase 1 (EC 2.5.1.6) | 1.75E-07 |
| HVSMEk0014G04 | ADK HUMAN | Adenosine kinase 2 (EC 2.7.1.20) | 8.08E-05 |
| HVSMEk0007K03 | DCAM_HORCH | SAM decarboxylase; S-adenosylmethionine decarboxylase (EC 4.1.1.50) | 2.66E-05 |
| HVSMEk0015E07 | ILV5_ARATH | Ketol-acid reductoisomerase (EC 1.1.1.86) | 0.00417 |
| HVSMEk0024A17* | P5CS_ORYSA | P5CS; delta 1-pyrroline-5-carboxylate synthetase (EC 2.7.2.11) | 0.058 |
| HVSMEk0003E21 | ASSY_ARATH | Argininosuccinate synthase (EC 6.3.4.5) | 2.71E-04 |
| HVSMEk0012K23* | ARLY_DESPS | Argininosuccinate lyase (EC 4.3.2.1) | 0.0851 |
| HVSMEk0006I09 | HOR3_HORVU | B3-hordein | 9.51E-05 |
| HVSMEk0005A14 | HOR1_HORVU | B1-hordein | 0.00165 |
| HVSMEk0006P03 | GLTB_WHEAT | LMW – B hordein | 0.00730 |
| HVSMEi0011I18 | HOG1_HORVU | Gamma-1-Hordein | 4.08E-07 |
| HVSMEk0012D09 | HOG1_HORVU | Gamma-1-Hordein | 4.22E-05 |
The listed genes corresponded to a cluster 1 profile and were significantly expressed (P <0.05). Asterisks indicate P <0.1.
Table 2.
Genes with mid expression profile of cluster 2
| Library name | Swissprot name | Annotation | P-values |
| HVSMEk0001E12* | PSBR_SPIOL | Photosystem II 10 kDa polypeptide, chloroplast precursor | 0.0796 |
| HVSMEi0011M10 | PORA_HORVU | Protochlorophyllide reductase A (EC 1.3.1.33) | 0.0165 |
| HVSMEi0008P04 | RBL_HORVU | RuBisCO – large subunit (EC 4.1.1.39) | 1.28E-07 |
| HVSMEk0004K02 | PPDK_MAIZE | Pyruvate phosphate dikinase. chloroplast precursor (EC 2.7.9.1) | 7.38E-05 |
| HVSMEk0005B15 | GLNA_PYRFU | Glutamine synthetase (EC 6.3.1.2) | 3.44E-04 |
| HVSMEk0012A15 | ASPG_LUPAR | L-asparaginase (EC 3.5.1.1) | 0.00343 |
| HVSMEk0005P10 | DHAS_CORFL | Aspartate-semialdehyde dehydrogenase (EC 1.2.1.11) | 1.59E-05 |
| HVSMEk0019N08 | METN_CAEEL | Probable S-adenosylmethionine synthetase 1 (EC 2.5.1.6) | 0.00260 |
| HVSMEk0003O05 | BCAL1_ARATH | Branched-chain-amino-acid aminotransferase-like protein 1 (EC 2.6.1.42) | 0.0361 |
| HVSMEi000P5CR | P5CR1_ARATH | P5CR; delta-(1)-pyrroline-5-carboxylate reductase (EC 1.5.1.2) | 7.55E-04 |
| HVSMEk0012H15 | GLTB_WHEAT | LMW – B hordein | 0.00159 |
| HVSMEi0011M13 | HOG3_HORVU | Gamma-3-Hordein | 0.00809 |
| HVSMEi0011I01 | HOG1_HORVU | Gamma-1-Hordein | 1.71E-07 |
| HVSMEi0004I12 | GLT3_WHEAT | Hordein D (glutenin HMW) | 1.92E-04 |
The listed corresponded to a cluster 2 profile and were significantly expressed (P <0.05). Asterisks indicate P <0.1.
Table 3.
Genes with late expression profile of cluster 3
| Library name | Swissprot name | Annotation | P-values |
| HVSMEk0022K16 | KPYA_RICCO | Pyruvate kinase A – chloroplastic (EC 2.7.1.40) | 6.96E-04 |
| HVSMEk0015E21 | ASNS_TRIVS | Asparagine synthetase (EC 6.3.5.4) | 8.20E-04 |
| HVSMEk0022N22 | DCDA_PSEAE | DAP; diaminopimelate decarboxylase(EC 4.1.1.20) | 0.00889 |
| HVSMEi0006C02 | SAHH_WHEAT | Adenosylhomocysteinase (EC 3.3.1.1) | 6.43E-09 |
| HVSMEi0015K09 | P5CS_ORYSA | P5CS; delta-(1)-pyrroline-5-carboxylate synthetase (EC 2.7.2.11) | 3.99E-05 |
| HVSMEi0006A15 | HOR3_HORVU | B3-hordein | 1.54E-04 |
| HVSMEi0016H16 | HOR8_HORVU | C-Hordein | 2.56E-04 |
| HVSMEi0003C02 | HOG1_HORVU | Gamma-1-hordein | 2.10E-05 |
| HVSMEi0002P07 | GLT1_WHEAT | Hordein D (glutenin. HMW) | 1.31E-04 |
| HVSMEk0022O15 | GLB1_MAIZE | Globulin-1 S allele precursor | 7.88E-11 |
| HVSMEk0019C20 | GLB1_MAIZE | Globulin-1 S allele precursor | 1.85E-11 |
| HVSMEi0006O15 | GLB1_MAIZE | Globulin-1 S allele precursor | 1.13E-10 |
| HVSMEk0024C24 | SSG1_AVESA | 12S seed storage globulin precursor | 1.39E-04 |
| HVSMEi0005H12 | SSG1_AVESA | 12S seed storage globulin precursor | 2.60E-07 |
| HVSMEk0019E19 | VCLA_GOSHI | Vicilin (alpha globulin) | 7.02E-08 |
| HVSMEk0003D11 | GLUB1_ORYSA | Glutelin type-B 1 precursor rice homologue) | 1.62E-07 |
| HVSMEk0015F23 | GLUB1_ORYSA | Glutelin type-B 1 precursor(rice homologue) | 1.52E-06 |
| HVSMEk0017D10 | GLUB1_ORYSA | Glutelin type-B 1 precursor(rice homologue) | 8.17E-08 |
The listed corresponded to a cluster 3 profile and were significantly expressed (P <0.05).
The use of the Partitioning Around Medoids (PAM) method for clustering allowed the gene expression profiles to be collated with early, middle, and late developmental stages of grain development. To represent individual gene expression patterns, the gene expression values of the 55 genes selected for discussion were displayed in a heat map format (Fig. 3). The heat map is a graphical representation of data where the values taken by a variable in a two-dimensional map are represented as colours. Each coloured cell in the heat map represented the gene expression value for a probe in a sample. The largest gene expression values are displayed in red, the smallest values in blue and intermediate values in shades of red (pink) or blue (Eisen et al., 1998).
The provision of carbon for storage product accumulation
In our field experiment the barley grains remained green until approximately 20 DAP whereafter they began to lose chlorophyll and exhibited signs of senescence (Fig. 1). By contrast the flag leaves showed signs of senescence around 15–18 DAP (data not shown). Dissecting the gene expression profiles that make up clusters 1 and 2 (Fig. 2) revealed differential expression of genes associated with photosynthesis (Fig. 3).
The need for reductant and ATP was supported by the expression of genes encoding a photosystem II protein (HVSMEk0001E12) and the protochlorophyllide reductase A (HVSMEi0011M10), an enzyme involved in the production of the chlorophyll antenna components (Fig. 3).
The results indicated that the gene encoding the Rubisco binding assembly protein (HVSMEk0013C18) was expressed at a high level in cluster 1; this preceded the gene encoding Rubisco large subunit (HVSMEi0008P04), which peaked later around 15–18 DAP and fell into cluster 2 (Fig. 3). The grains were green and contained substantial amounts of mRNA coding for the Rubisco large subunit, despite the absence of sufficient light for the operation of the Calvin cycle. Moreover, the genes encoding for Calvin cycle enzymes, for example, phosphoglycerate kinase (EC 2.7.2.3) or glyceraldehyde-3-phoshate dehydrogenase (GAPDH, EC 1.2.1.12) were exclusively expressed during the first phase of development while Rubisco peaked in the mid-stage (Fig. 3).
Similar to Rubisco, pyruvate orthophosphate dikinase (PPDK, EC 2.7.9.1) was highly up-regulated during the second phase of development and corresponds to cluster 2, pyruvate kinase (PK, EC 2.7.1.40) belonged to cluster 3 (Fig. 3).
Amino acid biosynthesis related genes of primary metabolism
Glutamate, aspartate, and serine are the most abundant amino acids that are translocated in the phloem of barley and thus provide the primary source of nitrogen from the leaves to the sink tissue (grain) during barley grain filling (Winter et al., 1992).
Glutamate is assimilated principally by the cytosolic glutamine synthetase (GS, EC 6.3.1.2) which catalyses the ATP-dependent conversion of glutamate and ammonia into glutamine (Miflin and Habash, 2002; Kichey et al., 2006). The microarray used in this study included three GS probes and sequence analysis suggests these homologues were cytosolic. The steady-state level of two of the cytosolic GS (HVSMEk0009J22; HVSMEi0007N22) genes was high at the beginning of grain filling (10 DAP), whereas the third GS (HVSMEk0005B15) appears to obtain the highest steady-state level of expression at 18 DAP (Fig. 3). The product of GS, glutamine, reacts subsequently with 2-oxoglutarate from the Krebs’ cycle leading to the creation of two molecules of glutamate, a step catalysed by glutamine 2-oxoglutarate amino transferase which is also named glutamate synthase (NADH-GOGAT, EC 1.4.1.14; Fd-GOGAT, EC 1.4.7.1). In our field experiment it was found that GOGAT expression was not temporally regulated (data not shown).
As an alternative, glutamate can be formed by glutamate dehydrogenase (GDH, EC 1.4.1.2–1.4.1.4) via reductive amination of 2-oxoglutarate, although the reaction is known to be reversible (Purnell et al., 2005). In our field experiment, the temporal expression profiles of the two homologues of GS which were expressed early (HVSMEk0009J22; HVSMEi0007N22) were accompanied by glutamate dehydrogenase 2 (GDH2, EC 1.4.1.3) (Fig. 3).
Aspartate aminotransferases (AspAT, EC 2.6.1.1), which convert glutamate and oxaloacetate to aspartate, exhibited a similar temporal expression profile to the early expressing homologues of GS and GDH (Fig. 3). The steady-state mRNA level was highest at 10 DAP and declined thereafter.
Aspartate produced via AspAT is the substrate for asparagine synthetase (AS, EC 6.3.5.4), which transfers the amide group of glutamine to aspartate, generating asparagine and glutamate in a reaction driven by ATP. There was an AS2 probe (HVSMEk0015E21) in the array and the steady-state level of the AS2 transcript was high at the beginning of the experiment (10 DAP) and declined until 18 DAP where upon it increased again from 18 DAP to 25 DAP (Fig. 3).
L-Asparaginase (ASase, EC 3.5.1.1) hydrolyses the amide group of asparagine to produce aspartate and ammonia and thus provides a route where asparagine is utilized for the synthesis of amino acids and proteins. The mRNA level of the gene encoding ASase (potassium-independent isoform) increased from 10 DAP to 18 DAP in this experiment (Fig. 3).
Aspartate-semialdehyde dehydrogenase (AspSD; EC 1.2.1.11) produces L-aspartate-semialdehyde by the reductive dephosphorylation of L-β-aspartyl phosphate utilizing NADPH. This enzyme lies at the first branch point in the biosynthetic pathway from L-aspartic acid in plants, leading to the formation of the amino acids lysine, isoleucine, methionine, and threonine (Cohen, 1983). The expression profile of the AspSD gene belonged to cluster 2 (Fig. 3).
Aspartate-derived amino acids
The ‘aspartate family’ of amino acids takes its name from aspartate, the main precursor in the biosynthetic pathway towards lysine, methionine, threonine, and isoleucine.
Lysine metabolism is highly controlled at both the anabolic and catabolic level. During development, the catabolism of lysine is thought to be controlled by saccharopine dehydrogenase (SDH; EC 1.5.1.9) while lysine itself regulates biosynthesis through product feedback inhibition (Stepansky et al., 2006). A high steady-state mRNA level was measured for SDH (HVSMEi0005N08) at 10 DAP, the mRNA level decreased dramatically until 18 DAP when it again increased (Fig. 3). Unfortunately, the feedback sensitive aspartate kinase was not present in the cDNA library. The gene involved in the terminal step of the lysine pathway encoding diaminopimelate (DAP) decarboxylase (Lys A, EC 4.1.1.20), increased up to 18 DAP and thereafter remained constant (Fig. 3), while diaminopimelate epimerase 2 (EC 5.1.1.7) was not differentially regulated over time (data not shown).
The biosynthesis of methionine is initiated from the ‘aspartate family’ intermediate o-phosphohomoserine (OPH) and is further metabolized through to methionine and S-adenosylmethionine (SAM) via a suite of enzymes. Cluster 1 included a gene encoding cystathionine γ-synthase (EC 2.5.1.48), which is linked to methionine biosynthesis and S-adenosylmethionine synthetase (SAM-S, EC 2.5.1.6) (Fig. 3). The second SAM-S gene present in the microarray has an expression profile characteristic for cluster 2, emphasizing the importance of SAM in the later stage of development as well (Fig. 3). Similarly, the gene encoding S-adenosylmethionine decarboxylase (EC 4.1.1.50), which facilitates many important methylation events (Poulton, 1981) showed an expression profile characteristic of cluster 1 (Fig. 3). Adenosine kinase (ADK 2, EC 2.7.1.20) and S-adenosylhomocysteine hydrolase (SAHH, EC 3.3.1.1), which are both required for the maintenance and recycling of S-adenosylmethionine-dependent methylation in plants, showed contrasting expression patterns; ADK gene belonged to cluster 1, while SAHH gene belonged to cluster 3 (Fig. 3; Table 3).
The precursor aspartate is further allocated through the threonine biosynthesis pathway towards the production of leucine, isoleucine, and valine. Significant temporal regulation of the gene coding for acetolactate synthase (ALS; EC 2.2.1.6) was not observed, the first common enzyme in the biosynthetic pathway of branched-chain amino acids. However, the genes coding ketol-acid reductoisomerase (EC 1.1.1.86) and the branched-chain-amino-acid aminotransferase-like protein 1 (EC 2.6.1.42) exhibited significant temporal expression (Fig. 3).
Arginine, ornithine, and proline amino acids
In higher plants, there are two possible pathways for proline biosynthesis: one utilizes glutamine as a precursor, the other one uses ornithine (Delauney and Verma, 1993). The precise contribution of the glutamine pathway and the ornithine pathway to proline biosynthesis, especially during fruit or seed development, remains uncertain (Hare et al., 1998, and references therein; Stines et al., 1999).
In the field experiment, the expression profile of two allelic isoforms encoding delta 1-pyrroline-5-carboxylate synthetase (P5CS; EC 2.7.2.11), the rate-limiting enzymes of the proline biosynthesis pathway differed. One P5CS homologue (HVSMEk0024A17) was apportioned to cluster 1 due to the apparent high initial steady-state level of mRNA, which thereafter decreased during development (Fig. 3). The expression profile of the second P5CS homologue (HVSMEi0015K09) fell into cluster 3, where the expression continued to increase from 15 DAP through to 25 DAP, the period of the experiment (Fig. 3). The genes coding for the enzymes of the ornithine pathway, acetylornithine aminotransferases (EC 2.6.1.11) and ornithine carbamoyltransferase (EC 2.1.3.3) did not change significantly during the period studied (data not shown). Delta 1-pyrroline-5-carboxylate reductase (P5CR; EC 1.5.1.2), the last enzyme of the proline biosynthetic pathway, is situated at the confluence of both proline biosynthesis pathways (Delauney and Verma, 1993). The expression pattern of the gene encoding P5CR belonged to cluster 2, with the mRNA level peaking around 15 DAP (Fig. 3).
It is noteworthy that the transcriptional data for one of the P5CS homologues (HVSMEk0024A17) belonging to cluster 1 in our experiments does not compare with the similar homologue deposited at http://www.plexdb.org. Further analysis of the Affymetrix Barley1 22K GeneChip® (Close et al., 2004) probe sets illustrated errors (the last two out of the 11 probes are outside of the 3’ untranslated region and may be related to activities of a gene other than P5CR).
The gene corresponding to the arginine pathway, encoding argininosuccinate synthase (EC 6.3.4.5) exhibited a high level of expression at 10 DAP and belonged to cluster 1, while argininosuccinate lyase (EC 4.3.2.1) had a more constant level of expression and belonged to cluster 1 with lower P value (0.08) only.
The transcriptional profile of storage proteins
Barley storage protein is made up of glutelin, albumins, globulin, and hordeins that are encoded by multi-gene families (for review see Shewry and Halford, 2002). To assess the expression of these families, homologues of barley storage proteins were included in the microarray and considerable variation was observed in the temporal gene expression profile of members of the family (Fig. 3). For example, three out of the five B hordein genes represented on the array showed an expression profile within cluster 1, a single probe exhibited expression characteristics of cluster 2, while one gene appeared to be expressed late in development and corresponded to cluster 3 (Fig. 3). Similar differences were observed for the expression patterns of the five γ-hordein genes, two were present in cluster 1, two others belonged to cluster 2, and one to cluster 3. The two D hordein genes were represented in both cluster 2 and cluster 3 (Fig. 3). All six significantly expressed globulin genes belonged to cluster 3 (Fig. 3). The expression patterns of genes coding for a hordein C homologue and the lysine-rich glutelin genes were all associated with cluster 3, where the respective mRNA levels increased late in development (Fig. 3).
When some of our expression patterns were compared with those reported in the BarleyBase expression library (http://www.plexdb.org), further mistakes were found in the Affymetrix probe sets. The probe sets are wrongly assigned in the case of one of the D hordeins and the C-hordein in the Affymetrix Barley1 22K GeneChip®.
Validation by real-time RT-PCR
The gene expression profiles obtained from the microarray experiments were validated by real-time RT-PCR for a selection of genes (Fig. 4). Primers homologous to all members of the appropriate genes families present on the microarray were used (see Supplementary Table S1 at JXB online), so the real-time RT-PCR results represented an average expression level among the family members. This was confirmed when an average profile for the different homologues was created from the microarray absolute expression values (Fig. 5). The profile of the C-hordein and the three glutelin homologues indicated that transcription of the respective genes continued to increase up to 25 DAP, which correlated with the results of the cDNA microarray and confirmed that the genes belong to cluster 3 (Fig. 5). Similarly the real-time PCR results matched the average profile pattern for the B-, D- and gamma-hordeins (Fig. 5). In addition, the profiles of a Rubisco large subunit unigene were analysed by real-time RT-PCR (Fig. 4). The profiles indicated a comparative trend to the microarray results, although suggested sharper increase from 10 to 18 DAP, while the real-time RT-PCR showed a stronger decrease at 25 DAP (Fig. 5). No changes were detected in the expression profiles of SAM-S genes by real-time PCR. This result was very different from the microarray results where one SAM-S gene present in the microarray had an expression profile characteristic for cluster 1 and a second for cluster 2. The real-time PCR did not match either of the SAM-S gene expression profiles detected in the microarray experiments, but it was in good agreement with the average expression profile for the two SAM-S genes (Fig. 5).
Fig. 4.

Genes of interest validated by real-time RT-PCR. Values of fold changes calculated relatively to 10 DAP are presented in logarithmic scale. Gene expression profiles: (open diamonds) C-hordein; (filled circles) glutelin; (filled squares) B-hordein; (filled triangles) D-hordein; (crosses) γ-hordein; (open triangles) SAM-S; (open circles) Rubisco large subunit.
Fig. 5.
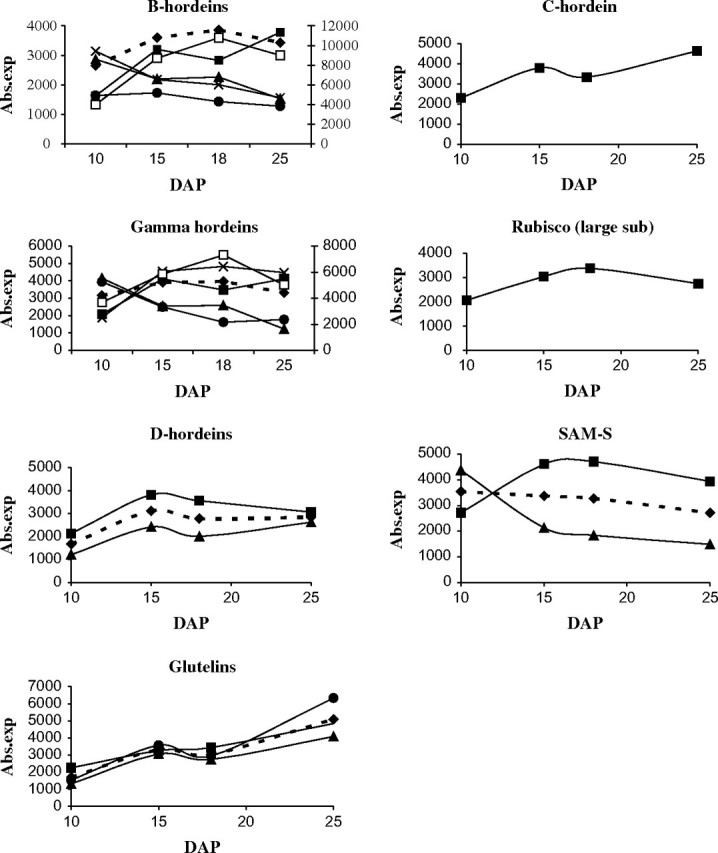
The absolute expression profiles of the genes mentioned in the real-time RT-PCR experiments: absolute expression values and the created average profile for the different homologous of the genes present on the microarray chips. The (filled diamonds, dashed line) represents average gene expression. The B-hordein diagrams shows profiles for: (filled squares) HVSMEi0006A15, (filled triangles) HVSMEk0006I09, (crosses) HVSMEk0005A14, (filled circles) HVSMEk0006P03, (open squares) HVSMEk0012H15 (right y-axis). The gamma hordein profiles are: (filled triangles) HVSMEi0011I18, (crosses) HVSMEi0011I01, (filled squares) HVSMEi0003C02, (filled circles) HVSMEk0012D09, (open squares) HVSMEi0011M13 (right y-axis). The gene expression profiles for D-hordein: (filled squares) HVSMEi0004I12, (filled triangles) HVSMEk0002P07. The glutelins expressions: (filled squares) HVSMEk0015F23, (filled triangles) HVSMEk0003D11, (filled circles) HVSMEk0017D10. The profiles of SAM-S: (filled squares) HVSMEk0019N08, (filled triangles) HVSMEk0004D10.
Discussion
The objective of the study was to correlate global gene expression analysis with the molecular and biochemical interactions associated with amino acid biosynthesis and storage protein accumulation in the developing grains of field-grown barley. Some of our results are in disagreement with the reported expression patterns in the barley database (http://www.plexdb.org). The analysis of the Affymetrix Barley1 22K GeneChip® probe sets (Close et al., 2004) revealed errors in the design, resulting in specific probes lying outside the 3’ untranslated region of target genes. Mistakes were found in the Affymetrix Barley1 22K GeneChip® in connection with P5CS and some of the hordein coding genes. In the recent microarray literature, similar problems with the probe set were described for rat and human Affymetrix arrays (Cambon et al., 2007; Lu et al., 2007).
Gene clusters correlated with real life gene expression prediction
The development of barley grains can be divided into three broad stages: (Phase 1) an initial phase of approximately 2 weeks post-anthesis where the endosperm cellularizes and organelles proliferate; (Phase 2) a phase characterized by the rapid synthesis of storage products; (Phase 3) a phase, which occurs around 30 d after anthesis, where the dry matter accumulation rate decreases and grains begin to desiccate (Ma and Smith, 1992; Goldberg et al., 1994).
The choice of an appropriate clustering algorithm is a complex one, since no given method is universally superior (Fraley and Raftery, 1998; Jain et al., 1999). The best choice will depend on the size of data set. As hierarchical methods generally scale poorly with increasing-sized data sets and the resulting dendrograms become harder to interpret, the partitioned algorithm was chosen for the larger data set. The three clusters created by manual inspections correlated with the developmental stages described above (see Supplementary Fig. S2 and Table S2 at JXB online). Based on these three major clusters, 55 genes of interest, showing the three distinct gene expression profiles, were represented by hierarchical methods (Fig. 3).
The provision of carbon for storage product accumulation: differential expression of genes associated with photosynthesis
Developing barley grains source photosynthate from the flag leaf, awns of the spike, and the pericarp (Frey-Wyssling and Buttrose 1959; Thorne, 1963; Duffus and Rosie, 1973; Kjack and Witters, 1974; Watson and Duffus, 1988; Ma and Smith 1992). Therefore, the level of photosynthesis is assumed to be high in the green grains during the early and middle stages of development. Our findings showed elevated expression of a number of photosynthesis-related genes in the early stage of development (Fig. 3). A similar pattern was reported in the developing seeds of Arabidopsis, where the major group of photosynthesis-related genes represented by light-harvesting complex II, photosystem II, and a Rubisco small subunit peaked around 11 DAP (Ruuska et al., 2002). In Arabidopsis seeds, the decline in gene expression did not correlate with photosynthetic activity. It was reported that the system was still functional until the seeds began to desiccate around 17 DAP (Fait et al., 2006).
Interestingly, the expression pattern of genes encoding the Rubisco large subunit and pyruvate orthophosphate dikinase (PPDK) belonged to cluster 2 (Fig. 2). The recent work of Schwender et al. (2004) using B. napus has demonstrated a metabolic route, not previously described, that accounts for Rubisco activity in the absence of Calvin cycle-related enzymes thus increasing the efficiency of carbon partitioning into oil. The net result was improved carbon efficiency of the developing green seeds. The authors concluded that this might explain why seeds of many species are green and contain substantial Rubisco activity during development, despite the absence of sufficient light for the operation of the Calvin cycle. Our results perhaps support this as the genes encoding for Calvin cycle enzymes were exclusively expressed during the first phase of development, while Rubisco peaked in the mid-stage.
While PPDK is known to play an important role as a photosynthetic enzyme in C4 plants, it has been suggested to have a non-photosynthetic function in C3 plants (Chastain et al., 2006). In C4 maize the PPDK enzyme peak occurs at the end of starch accumulation (21 DAP) and suggests a critical role in the starch–protein balance (Mechin et al., 2007). In C3 plants, expression of PPDK is detected in the early stages of grain development similar to our results (wheat, Aoyagi and Chua, 1988; rice, Chastain et al., 2006; Arabidopsis, Parsley and Hibberd, 2006). It is concluded that cytoplasmic PPDK serves as the major means of producing cellular ATP during early grain development by competing with ADP-dependent cytoplasmic pyruvate kinase (PK) and thus bypassing the default route of phosphoenolpyruvate (PEP) to pyruvate (Pyr) via PK (Chastain et al., 2006).
Amino acid biosynthesis related genes of the primary metabolism are interconnected
It was shown for leaves, roots, and seeds of Arabidopsis that the glutamate and aspartate pathways are interconnected and GDH, GS, GOGAT, AS, AspAT, and ASase interact in the synthesis of glutamine/glutamate and asparagine/aspartate (Lam et al., 1995; Zhu et al., 2003). A similar picture can be drawn from our study of developing barley grains. The recycled nitrogen from transported amino acids, together with the photorespiratory NH4 (reactions catalysed by GDH and GS), would appear to be available to enter the aspartate- and glutamate-pathways. The importance of these routes is supported by the early expression of genes coding for GDH, the GS1 (two isoforms), AS2, and AspAT (all three isoforms). These genes are highly expressed at the early stage of seed development, as their encoded proteins are involved in producing intermediates for the synthesis of other amino acids and proteins. ASase expression peaked during the mid-stage, suggesting that ASase catalysed asparagine catabolism starts to be important at this point in grain development. The increase in the steady-state level of ASase observed was concomitant with the decreased level of AS mRNA, further suggesting that the high level of ASase is involved in the mobilization of asparagine during the mid-stage of development The expression of the third GS isoform was similar to ASase and therefore might indicate the involvement of the gene product in NH4 detoxification. Similarly, the increased steady-state level of AS2 mRNA at the late stage of seed development might accommodate increased demand on aspartate and glutamine assimilation.
The fate of aspartate-derived amino acids
In the metabolite study of Arabidopsis seeds, the pool of free lysine significantly declined from 10 DAP to 17 DAP before dramatically increasing during the desiccation period (Fait et al., 2006). Our results were in good agreement with the reported results, as at 10 DAP a high steady-state mRNA level of saccharopine dehydrogenase was measured, a gene thought to be involved in the catabolism of lysine, while the gene encoding diaminopimelate (DAP) decarboxylase, involved in the terminal step of the lysine pathway, increased up to 15 DAP and thereafter remained constant.
SAM-S carries out the terminal step producing SAM, a major methyl donor in plants (Azevedo et al., 1997). The significant differential expression of genes associated with methionine and SAM biosynthesis and metabolism can be explained by their essential and perhaps ubiquitous nature supporting homeostasis of the grain cell during development. As an example in plants, the metabolite SAM is used as the methyl donor for the synthesis of ethanolamine, pectins, chlorophyll, lipids, and nucleic acids (Ravanel et al., 1998).
Pereira et al. (2007) reported a co-ordinated and probably transcriptional regulation of ADK and SAHH genes in most organs of Arabidopsis, while SAHH abundance was distinctly higher in seeds and roots, which suggests that it may have a non-methyl-related role in these organs. In our case, ADK and SAHH transcript amounts were shown to fluctuate independently (Fig. 3). ADK was expressed at an early stage of grain development, while SAHH was expressed in the late stage, indicating that SAHH may have a non-methyl-related role during the late stage of barley seed development.
Arginine, ornithine, and proline amino acids: glutamate versus ornithine pathways
In addition to its incorporation into polypeptides, free proline may have a role in osmoprotection, both maintaining homeostasis and acting as an osmoprotectant in response to water and NaCl stress (Hare and Cress, 1997; Zhu et al., 1998; Sawahel and Hassan, 2002).
The expression profile of the second P5CS homologue (HVSMEi0015K09) fell into cluster 3, while the gene encoding P5CR belonged to cluster 2, therefore suggesting a decoupling of expression of the gene from the major phase of desiccation. The synthesis of glutamate pathway intermediates, glutamic-γ-semialdehyde (GSA) and pyrroline-5-carboxylate (P5C), would seem to indicate additional diverse metabolic roles (Hua et al., 2001). Interestingly, it has been reported that P5C/GSA triggers a salicylic acid–mediated signalling cascade and if not metabolized rapidly, leads to cell death. Furthermore, an increase in the capacity to metabolize P5C/GSA leads to protection from cell death (Deuschle et al., 2004). Therefore P5C is not only involved in proline biosynthesis and degradation, but also in the metabolism of ornithine and arginine and citrulline.
Argininosuccinate synthase and argininosuccinate lyase, two genes corresponding to the arginine pathway, belonged to cluster 1 (Fig. 3). This may support the importance of the ornithine pathway, as their substrate is ornithine. Similarly, the recent metabolomic study carried out by Fait et al. (2006) found a high proportion of arginine accumulation during the period of desiccation of Arabidopsis seeds, which again supports a role for the ornithine pathway.
The developmental transcriptional profile of storage proteins: cross-talk between the primary metabolism and storage product pathways
It is widely suggested that storage product accumulation occurs in the later phase of grain or seed development in preparation for a period of dormancy before germination, which would be fuelled by the utilization of the storage products (Shewry and Halford, 2002). However, in our studies, storage product gene expression was observed not only in the later period of development but also in the early stages. This is in line with a recent study which reported the early expression of hordeins in microspore-derived embryos (Pulido et al., 2006). Pulido and colleagues (2006) suggested that these proteins might be synthesized and consumed according to the requirements of the embryogenic microspores and early embryos. Therefore, the presence of storage products, albeit transiently in some cases, early in development may not reflect a genetically programmed response, but a metabolic response to the significant flux of metabolites into the developing grain from the host plant i.e. the flag leaf, the awns and the pericarp. This hypothesis is perhaps supported by our observation that cluster 1 was dominated by genes associated with primary metabolism, for example, genes coding TCA cycle enzymes as well as genes involved in glycolysis (TCA cycle enzymes encoding genes, e.g. pyruvate carboxylase, ATP-citrate synthase, isocitrate dehydrogenase, succinyl-CoA ligase, succinate dehydrogenase, malate dehydrogenase; genes involved in glycolysis, e.g. aldolases, glyceraldehyde phosphate dehydrogenase, enolase, glucose-6-phosphate isomerase, triosephosphate isomerase; see Supplementary Table S2 available at JXB online). Combined with genes associated with photosynthesis described above, a picture emerges which suggests a significant flux of carbon skeletons, which are sequestered albeit temporally in a storage ‘vehicle’. This intense activity is likely to produce free radicals and cause REDOX stress. Free proline accumulates in plant tissues during abiotic stresses (Skopelitis et al., 2006) and contributes to the scavenging of surplus free radicals (Kaul et al., 2006). This could explain the high steady-state level of the P5CR gene expression at 10 DAP, as P5CR is the terminal gene of the proline biosynthetic pathway.
The temporal expression profiles of the homologues observed within the storage protein families seem to coincide with protein production reported by Rahman et al. (1984). The interplay among the differential temporal expression, suggests that the genes of each family of proteins are subject to different transcriptional regulation, implying that the regulatory units of the genes respond to different developmental and environmental stimuli. This opens the intriguing possibility of breeding selectively for specific alleles/homologues to confer enhanced amino acid profile of the barley storage proteins.
Validation by real-time RT-PCR: pros and cons of the primer designs
Real-time PCR is commonly used for validation of microarray results (Mackay et al., 2002). A review of the literature illustrates that microarray data generally are in good agreement, but not always confirmed by real-time RT PCR (Jason et al., 2008; Linton et al., 2008). In their fruit development study of watermelon, Wechter et al. (2008) reported that 72 of the 750 (9.6%) tissue-type quantitative-reactions were in conflict with the microarray results, thus 90.4% were in agreement. The failure to validate microarray data with real-time PCR is frequently explained by the possibility of the use of the tissue sources at different developmental stages (Gregersen et al., 2005; Lee et al., 2008).
The real-time RT PCR validation performed as part of this study was conducted with primers designed to homologous regions within selected gene families. Our rationale was that it was desirable to capture the ‘average’ expression level within a gene family; hence the real-time PCR results were compared with an average of the microarray data which combined the expression of the alleles. Amplification of multiple family members which may have slightly different melting curves (depending on the level of sequence conservation) could affect the calculation of the relative expression levels. In spite of this possible error, adopting such an approach resulted in a good correlation between the microarray and the real-time PCR results, although the allelic variation observed and reported as part of this study was lost (Fig. 5). Extending this argument, we would like to urge caution when designing primers for real-time PCR validation, as it is clear that to design primers to a gene requires full knowledge of the allelic complement in any given genome. Without full sequence information and carefully considering the region used for primer design, gene expression observed using real-time PCR can be an over or underestimation of relevant gene expression, thus compromising the attempt to get valid microarray-derived results.
Concluding remarks
The data presented here provides a comprehensive, publicly accessible transcriptomic analysis of cereal grain development of field-grown material. It is based on a set of genes chosen from cDNA libraries of developing barley seeds. Although the available microarray data set deposited in the BarleyBase (http://www.plexdb.org) is very comprehensive, it is limited to greenhouse material with 20 DAP being the oldest developmental stage reported. Coupled to this, a number of errors were identified in the primers used. It is beyond the scope of this article to describe the inconsistency between the data sets in greater detail except to say that data derived from Affymetrix probe sets should be treated with care given the reported problems with the probe sets in rat and human databases (Cambon et al., 2007; Lu et al., 2007). The temporal expression profiles of a range of genes involved in photosynthesis, amino acid metabolism, and storage protein accumulation are described and discussed. It is concluded that the grain-specific microarray coupled pathway-specific analysis is a fast, reliable, and cost-effective tool for monitoring temporal changes in the transcriptome of the major metabolic pathways in the barley grain. Therefore, microarray analysis could provide the knowledge required for the rational design of an optimal AA profile with the intriguing possibility of breeding selectively for specific alleles/homologues to confer an enhanced amino acid profile of the barley storage proteins and so increase its utility as animal feed.
Supplementary data
The supplementary data, which can be found at JXB online, consists of two tables and two figures.
Table S1. Specific primers used for real-time RT-PCR.
Table S2. Significantly regulated genes from the developing grain-specific microarray. The 501 genes (P <0.05) are described with a specific library name from Clemson University (http://www.genome.clemson.edu/projects/barley/).
Fig. S1. The array design of hybridizations.
Fig. S2. Cluster analysis. The gene expression profile of the first 501 most significantly regulated genes (1200 probes) representing the early-, mid- and late phases of the field-grown barley grain.
Acknowledgments
We would like to thank KB Nellerup and OB Hansen for their excellent technical support and HB Rasmussen for the photographic work. This work was supported by a grant (93S-943-F07-00047) from The Danish Directorate for Food, Fisheries and Agri Business.
References
- Altman N, Hua J. Extending the loop design for two-channel microarray experiments. Genet Research. 2006;88:153–163. doi: 10.1017/S0016672307008476. [DOI] [PubMed] [Google Scholar]
- Aoyagi K, Chua N-H. Cell-specific expression of pyruvate, Pi dikinase. In situ mRNA hybridization and immunolocalization labeling of protein in wheat seed. Plant Physiology. 1988;86:364–368. doi: 10.1104/pp.86.2.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo RA, Arruda P, Turner WL, Lea PJ. The biosynthesis and metabolism of the aspartate derived amino acids in higher plants. Phytochemistry. 1997;46:395–419. doi: 10.1016/s0031-9422(97)00319-1. [DOI] [PubMed] [Google Scholar]
- Baudo MM, Lyons R, Powers S, Pastori GM, Edwards KJ, Holdsworth MJ, Shewry PR. Transgenesis has less impact on the transcriptome of wheat grain than conventional breeding. Plant Biotechnology Journal. 2006;4:369–380. doi: 10.1111/j.1467-7652.2006.00193.x. [DOI] [PubMed] [Google Scholar]
- Becker RA, Chambers JM, Wilks AR. The new S language. Wadsworth & Brooks/Cole; 1988. [Google Scholar]
- Buchmann NB, Josefsson H, Cowe IA. Performance of European artificial neural network (ANN) calibrations for moisture and protein in cereals using the Danish near-infrared transmission (NIT) network. Cereal Chemistry. 2001;78:572–577. [Google Scholar]
- Cambon AC, Khalyfa A, Nigel GF, Cooper NGF, Thompson CM. Analysis of probe level patterns in Affymetrix microarray data. BMC Bioinformatics. 2007;8:146. doi: 10.1186/1471-2105-8-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chastain CJ, Heck JW, Colquhoun TA, Voge DG, Gu XY. Posttranslational regulation of pyruvate, orthophosphate dikinase in developing rice (Oryza sativa) seeds. Planta. 2006;224:924–934. doi: 10.1007/s00425-006-0259-3. [DOI] [PubMed] [Google Scholar]
- Close TJ, Wanamaker SI, Caldo RA, Turner SM, Ashlock DA, Dickerson JA, Wing RA, Muehlbauer GJ, Kleinhofs A, Wise R. A new resource for cereal genomics: 22K Barley GeneChip comes of age. Plant Physiology. 2004;134:960–968. doi: 10.1104/pp.103.034462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen GN. The common pathway to lysine, methionine, and threonine. In: Herrmann KM, Somerville RL, editors. Amino acids: biosynthesis and genetic regulation. Reading, MA: Addison-Wesley Publishing Co.; 1983. pp. 147–171. [Google Scholar]
- Coruzzi G, Bush DR. Nitrogen and carbon nutrient and metabolite signalling in plants. Plant Physiology. 2001;125:61–64. doi: 10.1104/pp.125.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coruzzi GM, Zhou L. Carbon and nitrogen sensing and signalling in plants: emerging ‘matrix effects’. Current Opinion in Plant Biology. 2001;4:247–253. doi: 10.1016/s1369-5266(00)00168-0. [DOI] [PubMed] [Google Scholar]
- Delauney AJ, Verma DPS. Proline biosynthesis and osmoregulation in plants. The Plant Journal. 1993;4:215–223. [Google Scholar]
- Deuschle K, Funck D, Forlani G, Stransky H, Biehl A, Leister D, van der Graaff E, Kunzee R, Frommer WB. The role of delta-(1)-pyrroline-5-carboxylate dehydrogenase in proline degradation. The Plant Cell. 2004;16:3413–3425. doi: 10.1105/tpc.104.023622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanaraj AL, Alkharouf NW, Beard HS, Chouikha IB, Matthews BF, Wei H, Arora R, Rowland LJ. Major differences observed in transcript profiles of blueberry during cold acclimation under field and cold room conditions. Planta. 2007;225:735–751. doi: 10.1007/s00425-006-0382-1. [DOI] [PubMed] [Google Scholar]
- Druka A, Muehlbauer G, Druka I, et al. An atlas of gene expression from seed to seed through barley development. Functional Integrated Genomics. 2006;6:202–211. doi: 10.1007/s10142-006-0025-4. [DOI] [PubMed] [Google Scholar]
- Duan K, Luo Y-H, Luo D, Xu Z-H, Xue H-W. New insights into the complex and co-ordinated transcriptional regulation networks underlying rice seed development through cDNA chip-based analysis. Plant Molecular Biology. 2005;57:785–804. doi: 10.1007/s11103-005-1803-4. [DOI] [PubMed] [Google Scholar]
- Duan MJ, Sun SSM. Profiling the expression of genes controlling rice grain quality. Plant Molecular Physiology. 2005;59:165–178. doi: 10.1007/s11103-004-7507-3. [DOI] [PubMed] [Google Scholar]
- Duffus CM, Rosie R. Some enzyme activities associated with the chlorophyll containing layers of the immature barley pericarp. Planta. 1973;114:219–226. doi: 10.1007/BF00389037. [DOI] [PubMed] [Google Scholar]
- Eisen MM, Brown PO. DNA arrays for analysis of gene expression. Methods in Enzymology. 1999;303:79–205. doi: 10.1016/s0076-6879(99)03014-1. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proceedings of the National Academy of Sciences. USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fait A, Angelovici R, Less H, Ohad I, Urbanczyk-Wochniak E, Fernie AR, Galili G. Arabidopsis seed development and germination is associated with temporally distinct metabolic switches. Plant Physiology. 2006;142:839–854. doi: 10.1104/pp.106.086694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firnhaber C, Puhler A, Kuster H. EST sequencing and time-course microarray hybridizations identify more than 700 Medicago truncatula genes with developmental expression regulation in flowers and pods. Planta. 2005;222:269–283. doi: 10.1007/s00425-005-1543-3. [DOI] [PubMed] [Google Scholar]
- Fraley C, Raftery AE. How many clusters? Which clustering methods? Answers via model-based cluster analysis. Computer Journal. 1998;41:578–588. [Google Scholar]
- Frey-Wyssling A, Buttrose MA. Photosynthesis in the ear of barley. Nature. 1959;184:2031–2032. [Google Scholar]
- Girke T, Todd J, Ruuska S, White J, Benning C, Ohlrogge J. Microarray analysis of developing Arabidopsis grains. Plant Physiology. 2000;124:1570–1581. doi: 10.1104/pp.124.4.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg RB, Depaiva G, Yadegari R. Plant embryogenesis: zygote to seed. Science. 1994;266:605–614. doi: 10.1126/science.266.5185.605. [DOI] [PubMed] [Google Scholar]
- Gregersen PL, Brinch-Pedersen H, Holm PB. A microarray-based comparative analysis of gene expression profiles during grain development in transgenic and wild type wheat. Transgenic Research. 2005;14:887–905. doi: 10.1007/s11248-005-1526-y. [DOI] [PubMed] [Google Scholar]
- Hansen M, Lange M, Friis C, Dionisio G, Holm' PB, Vincze E. Antisense mediated suppression of C-hordein biosynthesis in the barley grain results in correlated changes in the transcriptome, protein profile and amino acid composition. Journal of Experimental Botany. 2007;58:3987–3995. doi: 10.1093/jxb/erm254. [DOI] [PubMed] [Google Scholar]
- Hare PD, Cress WA. Metabolic implications of stress-induced proline accumulation in plants. Plant Growth Regulation. 1997;21:79–102. [Google Scholar]
- Hare PD, Cress WA, van Standen J. Dissecting the roles of osmolyte accumulation during stress. Plant, Cell and Environment. 1998;21:535–551. [Google Scholar]
- Hua XJ, Van de Cotte B, Van Montagu M, Verbruggen N. The 5’ untranslated region of the At-P5R gene is involved in both transcriptional and post-transcriptional regulation. The Plant Journal. 2001;26:157–169. doi: 10.1046/j.1365-313x.2001.01020.x. [DOI] [PubMed] [Google Scholar]
- Jain AK, Murty MN, Flynn PJ. Data clustering: a review. ACM Computing Surveys (CSUR) 1999;31:264–323. [Google Scholar]
- Jason A, Holliday SG, White RR, Bohlmann J, Aitken SN. Global monitoring of autumn gene expression within and among phenotypically divergent populations of Sitka spruce (Picea sitchensis) New Phytologist. 2008;178:103–122. doi: 10.1111/j.1469-8137.2007.02346.x. [DOI] [PubMed] [Google Scholar]
- Jollife NA, Craddock CP, Frigerio L. Pathways for protein transport to seed storage vacuoles. Biochemical Society Transactions. 2005;33:1016–1018. doi: 10.1042/BST20051016. [DOI] [PubMed] [Google Scholar]
- Kan YC, Wan YF, Beaudoin F, Leader DJ, Edwards K, Poole R, Wang DW, Mitchell RAC, Shewry PR. Transcriptome analysis reveals differentially expressed storage protein transcripts in seeds of Aegilops and wheat. Journal of Cereal Sciences. 2006;44:75–85. [Google Scholar]
- Kaul S, Sharma SS, Mehta IK. Free radical scavenging potential of L-proline: evidence from in vitro assays. Amino Acids. 2006;34:315–320. doi: 10.1007/s00726-006-0407-x. [DOI] [PubMed] [Google Scholar]
- Kaufman L, Rousseeuw PJ. Finding groups in data: an introduction to cluster analysis. New York: John Wiley & Sons; 1990. [Google Scholar]
- Kichey T, Heumez E, Pocholle D, Pageau K, Vanacker H, Dubois F, Le Gouis J, Hirel B. Combined agronomic and physiological aspects of nitrogen management in wheat highlight a central role for glutamine synthetase. New Physiologist. 2006;169:265–278. doi: 10.1111/j.1469-8137.2005.01606.x. [DOI] [PubMed] [Google Scholar]
- Kjack JL, Witters RE. Physiological activity of awns in isolines of Atlas barley. Crop Science. 1974;14:243–248. [Google Scholar]
- Lam H-M, Coschigano K, Schultz C, Melo-Oliveira R, Tjaden G, Oliveira I, Ngai N, Hsieh M-H, Coruzzi G. Use of Arabidopsis mutants and genes to study amideamino acid biosynthesis. The Plant Cell. 1995;7:887–898. doi: 10.1105/tpc.7.7.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Kim B, Song SK, et al. Large-scale analysis of the GRAS gene family in Arabidopsis thaliana. Plant Molecular Biology. 2008;67:659–670. doi: 10.1007/s11103-008-9345-1. [DOI] [PubMed] [Google Scholar]
- Linton KM, Hey Y, Saunders E, et al. Acquisition of biologically relevant gene expression data by Affymetrix microarray analysis of archival formalin-fixed paraffin-embedded tumours. British Journal of Cancer. 2008;98:1403–1414. doi: 10.1038/sj.bjc.6604316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lu C, Hawkesford MJ, Barraclough BJ, Poulton PR, Wilson ID, Barker GL, Edwards KJ. Markedly different gene expression in wheat grown with organic or inorganic fertilizer. Proceedings of the Royal Societyof London, Biological Sciences. 2005;272:1901–1908. doi: 10.1098/rspb.2005.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Lee JC, Salit ML, Cam MC. Transcript-based redefinition of grouped oligonucleotide probe sets using AceView: high-resolution annotation for microarrays. BMC Bioinformatics. 2007;8:108. doi: 10.1186/1471-2105-8-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay IM, Arden KE, Nitsche A. Real-time PCR in virology. Nucleic Acids Research. 2002;30:1292–1305. doi: 10.1093/nar/30.6.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma BL, Smith DL. Apical development of spring barley under field conditions in north-eastern North America. Crop Science. 1992;32:144–149. [Google Scholar]
- Mechin V, Thevenot C, Le Guilloux M, Prioul J-L, Damerval C. Developmental analysis of maize endosperm proteome suggests a pivotal role for pyruvate orthophosphate dikinase. Plant Physiology. 2007;143:1203–1219. doi: 10.1104/pp.106.092148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miflin BJ, Habash DZ. The role of glutamine synthetase and glutamate dehydrogenase in nitrogen assimilation and possibilities for improvement in the nitrogen utilization of crops. Journal of Experimental Botany. 2002;53:979–987. doi: 10.1093/jexbot/53.370.979. [DOI] [PubMed] [Google Scholar]
- Oury FX, Berard P, Brancourt-Hulmel M, et al. Yield and grain protein concentration in bread wheat: a review and study of multi-annual data from a French breeding program. Journal of Genetic Breeding. 2003;57:59–68. [Google Scholar]
- Palenchar PM, Kouranov A, Lejay LV, Coruzzi GM. Genome-wide patterns of carbon and nitrogen regulation of gene expression validate the combined carbon and nitrogen (CN)-signaling hypothesis in plants. Genome Biology. 2004;5:R91. doi: 10.1186/gb-2004-5-11-r91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsley K, Hibberd JM. The Arabidopsis PPDK gene is transcribed from two promoters to produce differentially expressed transcripts responsible for cytosolic and plastidic proteins. Plant Molecular Biology. 2006;62:339–349. doi: 10.1007/s11103-006-9023-0. [DOI] [PubMed] [Google Scholar]
- Pereira LAR, Todorova M, Cai X, Makaroff CA, Emery RJN, Moffatt BA. Methyl recycling activities are co-ordinately regulated during plant development. Journal of Experimental Botany. 2007;58:1083–1098. doi: 10.1093/jxb/erl275. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST©) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Research. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulton JE. Transmethylation and demethylation reactions in the metabolism of secondary plant products. In: Coon EE, editor. The biochemistry of plants: a comprehensive treatise. New York: Academic Press; 1981. pp. 667–672. [Google Scholar]
- Pulido A, Hernando A, Bakos F, Méndez E, Devic M, Barnabás B, Olmedilla A. Hordeins are expressed in microspore-derived embryos and also during male gametophytic and very early stages of seed development. Journal of Experimental Botany. 2006;57:2837–2846. doi: 10.1093/jxb/erl042. [DOI] [PubMed] [Google Scholar]
- Purnell MP, Roubelakis-Angelakis KA, Botella JR. Modulation of higher-plant NAD(H)-dependent glutamate dehydrogenase activity in transgenic tobacco via alteration of beta subunit levels. Planta. 2005;222:167–180. doi: 10.1007/s00425-005-1510-z. [DOI] [PubMed] [Google Scholar]
- Radchuk VV, Sreenivasulu N, Radchuk RU, Wobus U, Weschke W. The methylation cycle and its possible functions in barley endosperm development. Plant Molecular Physiology. 2005;59:289–307. doi: 10.1007/s11103-005-8881-1. [DOI] [PubMed] [Google Scholar]
- Rahman S, Kreis M, Forde BG, Shewry PR, Miflin BJ. Hordein-gene expression during development of the barley (Hordeum vulgare) endosperm. Biochemical Journal. 1984;223:315–322. doi: 10.1042/bj2230315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raun WR, Johnson GV. Improving nitrogen use efficiency for cereal production. Agronomy Journal. 1998;91:357–363. [Google Scholar]
- Ravanel S, Gakiere B, Job D, Douce R. The specific features of methionine biosynthesis and metabolism in plants. Proceedings of the National Academy of Sciences, USA. 1998;95:7805–7812. doi: 10.1073/pnas.95.13.7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruuska SA, Girke T, Benning C, Ohlrogge JB. Contrapuntal networks of gene expression during Arabidopsis seed filling. The Plant Cell. 2002;14:1191–1206. doi: 10.1105/tpc.000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawahel WA, Hassan AH. Generation of transgenic wheat plants producing high levels of the osmoprotectant proline. Biotechnology Letters. 2002;24:721–725. [Google Scholar]
- Schwender J, Goffman F, Ohlrogge JB, Shachar-Hill Y. Rubisco without the Calvin cycle improves the carbon efficiency of developing green seeds. Nature. 2004;432:779–782. doi: 10.1038/nature03145. [DOI] [PubMed] [Google Scholar]
- Shewry PR, Tatham AS, Halford NG. Nutritional control of storage protein synthesis in developing grain of wheat and barley. Plant Growth Regulation. 2001;34:105–111. [Google Scholar]
- Shewry PR, Halford NG. Cereal seed storage proteins: structures, properties and role in grain utilization. Journal of Experimental Botany. 2002;53:947–58. doi: 10.1093/jexbot/53.370.947. [DOI] [PubMed] [Google Scholar]
- Shrout PE, Fleiss JL. Intraclass correlations: uses in assessing rater reliability. Psychological Bulletin. 1979;86:420–428. doi: 10.1037//0033-2909.86.2.420. [DOI] [PubMed] [Google Scholar]
- Skopelitis DS, Paranychianakis NV, Paschalidis KA, Pliakonis ED, Delis ID, Yakoumakis DI, Kouvarakis A, Papadakis AK. Abiotic stress generates ROS that signal expression of anionic glutamate dehydrogenases to form glutamate for proline synthesis in tobacco and grapevine. The Plant Cell. 2006;18:2767–2781. doi: 10.1105/tpc.105.038323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreenivasulu N, Altschmeid L, Radchuk V, Gubatz S, Wobus U, Weschke W. Transcript profiles and deduced changes of metabolic pathways in maternal and filial tissues of developing barley grains. The Plant Journal. 2004;37:539–553. doi: 10.1046/j.1365-313x.2003.01981.x. [DOI] [PubMed] [Google Scholar]
- Sreenivasulu N, Radchuk V, Strickert M, Miersch O, Weschke W, Wobus U. Gene expression patterns reveal tissue-specific signalling networks controlling programmed cell death and ABA-regulated maturation in developing barley seeds. The Plant Journal. 2006;47:310–327. doi: 10.1111/j.1365-313X.2006.02789.x. [DOI] [PubMed] [Google Scholar]
- Stepansky A, Less H, Angelovici R, Aharon R, Zhu X, Galili G. Lysine catabolism, an effective versatile regulator of lysine level in plants. Amino Acids. 2006;30:121–125. doi: 10.1007/s00726-005-0246-1. [DOI] [PubMed] [Google Scholar]
- Stines AP, Naylor DJ, Hoj PB, van Heeswijck R. Proline accumulation in developing grapevine fruit occurs independently of changes in the levels of Delta(1)-pyrroline-5-carboxylate synthetase mRNA or protein. Plant Physiology. 1999;120:923–931. doi: 10.1104/pp.120.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas WTB. Prospects for molecular breeding in barley. Annals of Applied Biology. 2003;142:1–12. [Google Scholar]
- Thorne GN. Varietal differences in photosynthesis of ears and leaves of barley. Annals of Botany. 1963;27:155–174. [Google Scholar]
- Vicente-Carbajosa J, Carbonero P. Seed maturation: developing an intrusive phase to accomplish a quiescent state. International Journal of Developmental Biology. 2005;49:645–651. doi: 10.1387/ijdb.052046jc. [DOI] [PubMed] [Google Scholar]
- Watson PA, Duffus CM. Carbon dioxide fixation by detached cereal caryopses. Plant Physiology. 1988;87:504–509. doi: 10.1104/pp.87.2.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechter WP, Levi A, Harris KR, et al. Gene expression in developing watermelon fruit. BMC Genomics. 2008;9:275. doi: 10.1186/1471-2164-9-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter H, Lohaus G, Heldt HW. Phloem transport of amino acids in relation to their cytosolic levels in barley leaves. Plant Physiology. 1992;99:996–1004. doi: 10.1104/pp.99.3.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman C, Jensen LJ, Jarmer H, Berka R, Gautier L, Nielsen HB, Saxild HH, Nielsen C, Brunak S, Knudsen S. A new non-linear normalization method for reducing variability in DNA microarray experiments. Genome Biology. 2002;30:1–16. doi: 10.1186/gb-2002-3-9-research0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadoks JC, Chang TT, Konzak CF. A decimal code for the growth stages of cereals. Weed Research. 1974;14:415–421. [Google Scholar]
- Zhu BC, Su J, Chan MC, Verma DPS, Fan YL, Wu R. Overexpression of a delta-(1)-pyrroline-5-carboxylate synthetase gene and analysis of tolerance to water- and salt-stress in transgenic rice. Plant Science. 1998;139:41–48. [Google Scholar]
- Zhu T, Budworth P, Chen W, Provart N, Guimil S, Su W, Estes B, Zou G, Wang X. Transcriptional control of nutrient portioning during rice grain filling. Plant Biotechnology Journal. 2003;1:59–70. doi: 10.1046/j.1467-7652.2003.00006.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.