

Abstract
Background and Objectives:
Glutaric aciduria Type-I (GA-I) has characteristic clinical and neuroimaging features, which clinches the diagnosis in a majority of patients. However, there have been few case reports on GA-I from India. This study was undertaken to study the clinical presentations, metabolic profile, neuroimaging findings and outcome of patients with GA-I.
Study Design:
The present study was a retrospective study.
Materials and Methods:
Retrospective review of charts of patients with a diagnosis of GA-I was carried out from March 2008 to April 2010. The clinical, laboratory and neuroimaging findings were extracted in a predesigned proforma and the data was analyzed.
Results:
Eleven cases were found to have GA-1. Clinical presentation was quite varied. Follow-up of patients revealed that one patient with macrocephaly as the only clinical finding was developmentally normal. One patient with encephalitis-like illness steadily improved and started walking at 2 years. Two patients were bed ridden and had severe dystonia. One patient died during follow-up. The remaining six patients had dystonia and other abnormal movements, but had attained sitting without support and were not ambulatory.
Conclusion:
GA-I is not an uncommon disorder and diagnosis can be made easily based on clinical, laboratory investigations and neuroimaging findings. It is one of the treatable metabolic disorders and, if managed appropriately, favorable prognosis can be given.
Keywords: Encephalitis, glutaric aciduria type 1, macrocephaly, neuroimaging
Introduction
Glutaric aciduria type I (GA-I), caused by glutaryl-CoA dehydrogenase (GCDH) deficiency, is an autosomal-recessive neurometabolic disorder.[1] Although rare in incidence, one can easily diagnose this condition based on characteristic clinical and neuroimaging findings.[2] In developing countries like India, very few cases of this disorder have been reported mainly in the form of isolated case reports[2] without much details on the treatment, prognosis and outcome. During the last three decades, attempts have been made to establish and optimize therapy for GA-I. Many recommendations on diagnosis and management have been published and revised in the recent past,[3,4] and these recommend use of special lysine-free, tryptophan-reduced amino acid supplements. Because of the non-availability of special diets, many of the patients in developing countries are advised a non-specific protein-restricted diet, carnitine supplementation and emergency-care management measures. Hence, the outcome of patients from GA-I from developing countries may be different from the developed nations. We herein present our experience with 11 cases of GA-I over a period of 2 years.
Materials and Methods
Retrospective review of charts of patients with a diagnosis of organic acidemia was performed at the Pediatric Neurology Clinic attached to a Tertiary Care Hospital over a period of 3 years from March 2008 to April 2011. Diagnosis of GA-I was suspected based on clinical and neuroimaging findings and confirmed by blood tandem mass spectrometry (TMS) and/or urine gas chromatography and mass spectrometry (GC/MS). Genetic analysis was also obtained for one patient. The clinical, laboratory and neuroimaging findings were extracted in a predesigned proforma and the data was analyzed. The outcome was assessed from the follow-up notes in the chart and telephonic interview of the patients who did not come for follow-up visits.
Results
Thirty-three cases of organic acidemia were diagnosed during the study period, of which 11 cases were found to have GA-I (33%). Nine of them were males. History of consanguinity was observed in eight patients (73%). Six patients had symptoms before 6 months of age; four between 6 months and 1 year of age and one patient presented after 1 year of age. One patient presented only with macrocephaly and four patients had encephalitis-like crisis at the onset (36.36%). Abnormal movements (choreiform, athetoid, dystonia) were noted in 10 out of 11 patients (90.9%) and developmental delay in nine patients. On laboratory evaluation, all patients had normal serum electrolytes, ammonia and random blood sugar values. Urine ketone bodies were not present in any of the cases. All of them had elevated arterial lactate, with a mean of 2.8 mmol/L (range: 2.5–3.8). In neuroimaging, characteristic widening of Sylvian fissure (Bat-wing appearance) was observed in all patients, and six patients had bilateral subdural frontotemporal atrophy with subdural collection, diffuse white matter signal abnormality and high signal in basal ganglia [Figure 1]. Nine patients’ blood sample was sent for TMS, and all showed elevated Glutaryl Carnitine levels (>1.5-times the normal laboratory cut-off for C5DC levels). Five patients (including three with abnormal TMS) showed increased urinary excretion of both glutaric acid (GA) and 3-OH glutaric acid on GC/MS. Complete gene sequencing (amplification by polymerase chain reaction and sequencing using BigDye Terminator v3.1 and capillary electrophoresis) was performed for one of the patients, and this revealed a novel mutation in the gene for GA-1, i.e. P217S (proline to serine mutation) on chromosome 19p13.2.
Figure 1.
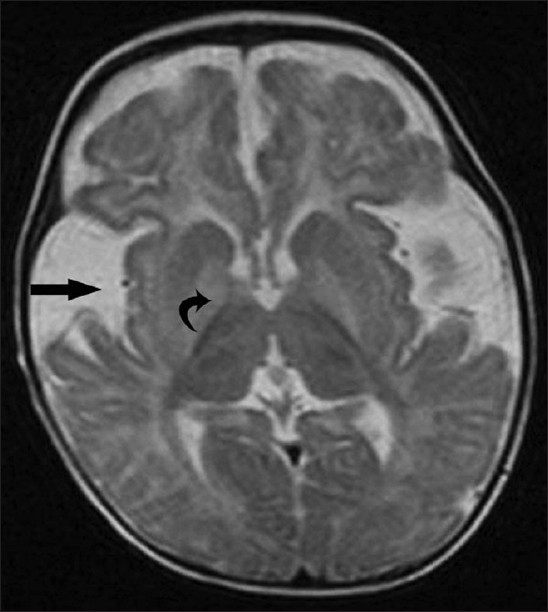
Axial T2W magnetic resonance image reveals fronto-temporal atrophy, dilated sylvian fissures with open opercula (straight arrow), diffuse white matter signal abnormality and bilateral high signal in the basal ganglia (curved arrow). Widening of the sylvian fissure gives the characteristic “bat-wing” appearance
All patients were started on riboflavin and carnitine supplements along with a protein-restricted diet especially lacking lysine and tryphophan. Care of patients during infections was advised as per the recent guidelines. Follow-up of patients revealed that one patient with macrocephaly only and subdural effusions on neuroimaging was developmentally normal. One patient with a single encephalitis-like illness steadily improved and started walking at 2 years. At the last follow-up, the patient had minimal choreiform movement and clumsy walking. Two patients were bed ridden and had severe dystonia. Both these patients had recurrent episodes of worsening of symptoms with febrile illness at follow-up. They were from remote rural places and had poor access to medical treatment and could not follow the emergency care advice. One patient died during follow-up at the age of 7 years. The remaining six patients had dystonia and other abnormal movements, had attained sitting without support and were not ambulatory.
Discussion
GA-I was first described in 1975 by Goodman et al.[5] and has an estimated overall prevalence of 1 in 100,000 newborns.[6] The causative gene is localized on chromosome 19p13.2, and more than 150 disease-causing mutations are known. Biochemically, GA-I is characterized by an accumulation of GA, 3-hydroxyglutaric acid (3-OH-GA), glutaconic acid (less frequently) and glutarylcarnitine (C5DC). These can be detected in body fluids (urine, plasma, cerebrospinal fluid) and tissues by gas chromatography-mass spectrometry[7] of organic acids or by electrospray-ionization tandem mass spectrometry of acylcarnitines.
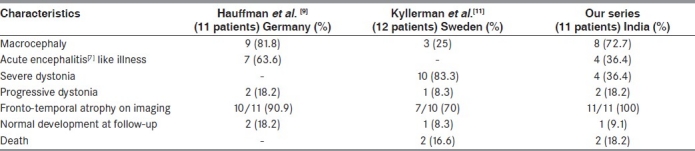
Clinically, the disease course is usually determined by acute encephalopathic crises precipitated by infectious diseases, immunizations and surgery during infancy or childhood. The characteristic neurological sequel is acute striatal injury and, subsequently, dystonia. Patients develop normally until the initial neurologic presentation, which may be at 2–37 months (mean, 14 months).[8] Recovery from acute episode is slow and incomplete with developmental delay, dystonia or dyskinesia as sequelae. The findings of our study are comparable to other studies. Macrocephaly was seen in three-fourths of our patients, which is comparable to the study by Hauffmann et al.[9] and this may be an early sign before other neurologic alterations [Table 1]. Rarely, large head can be the only manifestation and children can be otherwise neurologically normal.[10] In our study too, we found one such patient. They do have neuroradiographic evidence of frontotemporal atrophy. Abnormal movements was the most common finding in our study (90.9%), which was similar to the study done by Hoffmann et al. and Kyllerman et al.[9,11] During the study period of over 2 years, we found that patients presenting with recurrent episodes of encephalopathic crisis had higher morbidity and mortality, which is comparable to the study done by Kölker et al.[12] and Kyllerman et al.[11] On the other hand, patients who presented with only macrocephaly or a single episode of encephalopathic crisis and those who followed a strict advice regarding diet and therapy have got better outcome than those who were on irregular follow-up and inappropriate treatment.
Table 1.
Comparison of characteristics of patients with glutaric aciduria type-I
Because of the lack of specialized centers to deal with neurometablic disorders and due to the lack of knowledge in developing countries, many cases are being misdiagnosed or may be on inappropriate treatment. During the last three decades, attempts have been made to establish and optimize therapy for GA-I. Treatment consists of carnitine supplementation (100 mg/kg/day) along with a low protein diet, especially deficient in lysine and tryptophan, and careful management of these children during stressful episodes.[3,4] The role of riboflavin supplementation is not very clear. This treatment strategy has considerably reduced the frequency of acute encephalopathic crises and thus morbidity and mortality in early-diagnosed patients. Therefore, GA-I deficiency is now considered to be a treatable neurometabolic disorder. During crisis like in infections, trauma and surgeries, there is risk of encephalopathy and irreversible brain injury. Management includes[1] the prevention or reversal of a catabolic state by administration of a high-energy intake (plus insulin to control for hyperglycemia if required);[2] reduction of GA and 3-OH-GA production by transient reduction or omission of natural protein for 24–48 h;[3] amplification of physiological detoxification mechanisms and prevention of secondary carnitine depletion by L-carnitine supplementation (doubling the maintenance dose of 100 mg/kg day) and[4] maintenance of normal fluid, electrolytes and pH status via enteral of IV fluids. Non-adherence to previously described emergency treatment recommendations has been associated with a high probability of developing striatal injury.[4]
GA-I is an autosomal-recessive disorder, and there is a 25% chance of recurrence in future pregnancies. Hence, if a child is diagnosed with this disorder in a family, then prenatal diagnosis can be offered in the subsequent pregnancy. If there are, already, asymptomatic siblings then they should be screened for GA-I and, if positive, initiated on appropriate treatment. This will prevent the occurrence or reduce the severity of encephalopathic crises and ensure good outcome.
Glutaric aciduria is one of the inborn errors of metabolism where neuroimaging can give a clue to the underlying etiological diagnosis. The finding of very widely open opercula suggests GA-1 and, if combined with basal ganglia, lesions are almost pathognomonic, especially in a child with macrocephaly.[13] Diagnosis of GA-I should be considered whenever a child has include macrocephaly, acute encephalopathy, basal ganglia injury, white matter disease, movement disorders, subdural and retinal hemorrhage and isolated elevation of GA, 3-OH-GA and glutarylcarnitine in body fluids. Most of these in isolation have a wide differential diagnosis, whereas a combination of two or more abnormalities increases the probability for GA-I.
Conclusion
GA-I is not an uncommon disorder, and diagnosis can be made easily based on clinical, laboratory investigations and neuroimaging findings. Timely diagnosis and start of treatment, i.e. before an acute encephalopathic crisis, is likely to result in a better outcome than diagnosis and start of treatment after the onset of neurological disease. It is one of the treatable neurometabolic disorder and, if managed appropriately, favorable prognosis can be given.
Acknowledgments
The authors wish to thank Dr. Usha Dave from the Preventine Life Care Private Limited, Mumbai, for the urine gas chromatography/mass spectrometry analysis, Dr. Rita Christopher, Professor and Head, Department of Neurochemistry, NIMHANS, Bangalore, for the tandem mass spectrometry analysis and Dr. Parag Tamankar, Genetic Research Center, NIRRH, Mumbai, for the genetic analysis.
Footnotes
Source of Support: Nil,
Conflict of Interest: Nil
References
- 1.Strauss KA, Puffenberger EG, Robinson DL, Morton DH. “Type I glutaric aciduria, part 1: Natural history of 77 patients”. Am J Med Genet C Semin Med Genet. 2003;121C:38–52. doi: 10.1002/ajmg.c.20007. [DOI] [PubMed] [Google Scholar]
- 2.Kamate M, Patil VD, Chetal V, Hattiholi V. Glutaric aciduria Type I- An easily diagnosable and treatable metabolic disorder. Indian J Pediatr. 2009;76:562–3. doi: 10.1007/s12098-009-0080-7. [DOI] [PubMed] [Google Scholar]
- 3.Kölker S, Christensen E, Leonard JV, Greenberg CR, Burlina AB, Burlina AP, et al. Guideline for the diagnosis and management of glutaryl-CoA dehydrogenase deficiency (glutaric aciduria type I) J Inherit Metab Dis. 2007;30:5–22. doi: 10.1007/s10545-006-0451-4. [DOI] [PubMed] [Google Scholar]
- 4.Köolker S, Christensen E, Leonard JV, Greenberg CR, Boneh A, Burlina AB, et al. Diagnosis and management of glutaric aciduria type I – revised recommendations. J Inherit Metab Dis. 2011;34:677–94. doi: 10.1007/s10545-011-9289-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodman SI, Markey SP, Moe PG, Miles BS, Teng CC. Glutaric aciduria: A ‘new’ disorder of amino acid metabolism. Biochem Med. 1975;12:12–21. doi: 10.1016/0006-2944(75)90091-5. [DOI] [PubMed] [Google Scholar]
- 6.Lindner M, Kölker S, Schulze A, Christensen E, Greenberg CR, Hoffmann GF. Neonatal screening for glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2004;27:851–9. doi: 10.1023/B:BOLI.0000045769.96657.af. [DOI] [PubMed] [Google Scholar]
- 7.Baric I, Wagner L, Feyh P, Liesert M, Buckel W, Hoffmann GF. Sensitivity of free and total glutaric and 3-hydroxyglutaric acid measurement by stable isotope dilution assays for the diagnosis of glutaric aciduria type I. J Inherit Metab Dis. 1999;22:867–82. doi: 10.1023/a:1005683222187. [DOI] [PubMed] [Google Scholar]
- 8.Bjugstad KB, Goodman SI, Freed CR. Age at symptom onset predicts severity of motor impairment and clinical onset of glutaric aciduria type I. J Pediatr. 2000;137:681–6. doi: 10.1067/mpd.2000.108954. [DOI] [PubMed] [Google Scholar]
- 9.Hoffmann GF, Trefz FK, Barth PG, Böhles HJ, Biggemann B, Bremer HJ, et al. Glutaryl-coenzyme A dehydrogenase deficiency: A distinct encephalopathy. Pediatrics. 1991;88:1194–203. [PubMed] [Google Scholar]
- 10.Martinez Granero MA, Garcia Perez A, Martinez-Pardo M, Parra E. “Macrocephaly the first manifestation of glutaric aciduria type I: The importance of early diagnosis”. Neurologia. 2005;20:255–60. [PubMed] [Google Scholar]
- 11.Kyllerman M, Skjeldal OH, Lundberg M, Holme I, Jellum E, von Döbeln U, et al. Dystonia and dyskinesia in glutaric aciduria type I: Clinical heterogeneity and therapeutic considerations. Mov Disord. 1994;9:22–30. doi: 10.1002/mds.870090105. [DOI] [PubMed] [Google Scholar]
- 12.Kolker S, Garbade SF, Greenberg CR, Leonard JV, Saudubray JM, Ribes A, et al. “Natural history, outcome, and treatment efficacy in children and adults with glutaryl-CoA dehydrogenase deficiency”. Pediatr Res. 2006;59:840–7. doi: 10.1203/01.pdr.0000219387.79887.86. [DOI] [PubMed] [Google Scholar]
- 13.Brismar J, Ozand P. CT and MR of the brain in glutaric academia type I. A review of 59 published cases and report of 5 new patients. Am J Neuroradiol. 1995;16:675–83. [PMC free article] [PubMed] [Google Scholar]