

Abstract
Hypertrophic pachymeningitis (HP) is a rare chronic inflammatory disease of the dura mater, described in association with various infections, systemic vasculitides such as Wegener's granulomatosis and giant cell arteritis. However, HP in association with Takayasu arteritis (TA) has not been described. We report a young woman who presented with headache, seizures, and right third and fourth cranial neuropathy. Magnetic resonance imaging of the brain showed HP in bifrontal and right temporal region extending to cavernous sinus. She was also found to have systemic hypertension, stenosis of left subclavian, and left renal artery with narrowing of abdominal aorta, satisfying the diagnostic criteria for TA. A detailed evaluation for secondary causes of HP failed to reveal an alternative etiology. This report describes an unusual association of HP in a patient with TA, also emphasizing that seizures and cranial neuropathy may further expand the spectrum of neurological manifestations in patients with TA.
Keywords: Cranial pachymeningitis, cranial nerve palsy, Hypertrophic pachymeningitis, large vessel vasculitis, Takayasu arteritis
Introduction
Hypertrophic pachymeningitis (HP) is a chronic progressive inflammatory disease of the dura mater. It has been described in association with various diseases including infections, trauma, or tumors. Its association with autoimmune or vasculitic disorders including Wegener's granulomatosis, giant cell arteritis, Sjogren syndrome, and sarcoidosis is well known.[1,2] However, in literature, till now, there are no reports of HP described in association with Takayasu arteritis (TA), which is also a chronic inflammatory disease affecting large vessels. We report a young woman, who presented with right third and fourth cranial neuropathy secondary to HP, who also satisfied the diagnostic criteria for TA.
Case Report
A 24-year-old woman presented with history of recurrent episodic right hemicranial headache and seizures of 3 years duration. She also conceded that she used to get vertiginous sensations before the onset of generalized tonic clonic seizures each time. This time she presented with right hemicranial headache of different character, which was more severe and associated with retro-orbital pain 15 days before admission. Ten days after the onset of headache with different character, she noticed double vision followed by progressive drooping of right eye lid. General examination showed asymmetrical upper limb pulses, left radial, and brachial pulse being feeble. Her blood pressure was 170/110 mm Hg on recording from the right upper limb, while on left it was 90/60 mm Hg. In addition, there was evidence for bilateral renal bruit (left more than right). Neurological examination revealed right third and fourth cranial nerve palsies, and the remainder of the examination was normal.
Routine urine analysis, complete blood counts, and serum biochemistry (liver and kidney functions, glucose level, and lipid panel) were normal, except for an elevated erythrocyte sedimentation rate (ESR) of 88 mm at the end of an hour. Vasculitic work up in the form of rheumatoid factor, antinuclear antibodies, anti-double stranded DNA antibody, antiphospholipid antibody, proteinase-3-antineutrophil cytoplasmic antibody (ANCA) and myeloperoxidase-ANCA were negative. Serology for syphilis (Venereal Disease Research Laboratory [VDRL] test), HIV, and hepatitis B and C was negative. Chest X-ray, serum calcium, and angiotensin-converting enzyme levels were normal. Tuberculin skin test as well as Quantiferon-TB test were negative. Cerebrospinal fluid (CSF) analysis showed 2 lymphocytes/mm3, protein 75 mg/dl, and glucose 78 mg/dl (serum glucose-112 mg/dl). Cytological examination of the CSF did not show any malignant cell. CSF microbiology in the form of gram staining, acid fast bacilli (AFB) staining, VDRL, panfungal antigen, and cultures for AFB and fungi did not yield positive results, so was polymerase chain reaction for mycobacterium tuberculosis.
Magnetic resonance imaging of the brain revealed abnormal, thickened enhancing pachymeninges in bilateral frontal and right temporal region extending to cavernous sinus [Figures 1 a and b]. In addition, there was T2 and FLAIR (Fluid Attenuated Inversion Recovery) hyperintensity in right temporal lobe [Figure 2]. In view of feeble left radial pulse and renal bruit, patient underwent conventional digtal subtraction angiography (DSA), for better characterization of the vascular system. DSA revealed stenosis of left subclavian artery distal to the origin of vertebral artery, osteal stenosis of left renal artery, and narrowing of abdominal aorta at the level of the origin of renal arteries with normal terminal aorta and bilateral common iliac arteries [Figure 1 c and d]. Transthoracic two-dimensional echocardiography revealed mild aortic regurgitation with normal left ventricular function. Electroencephalogram showed intermittent epileptiform discharges over the right temporal region. Meningeal biopsy could not be performed as the patient declined the procedure. A diagnosis of TA was considered as patient satisfied the American College of Rheumatology (ACR) criteria.[3]
Figure 1.
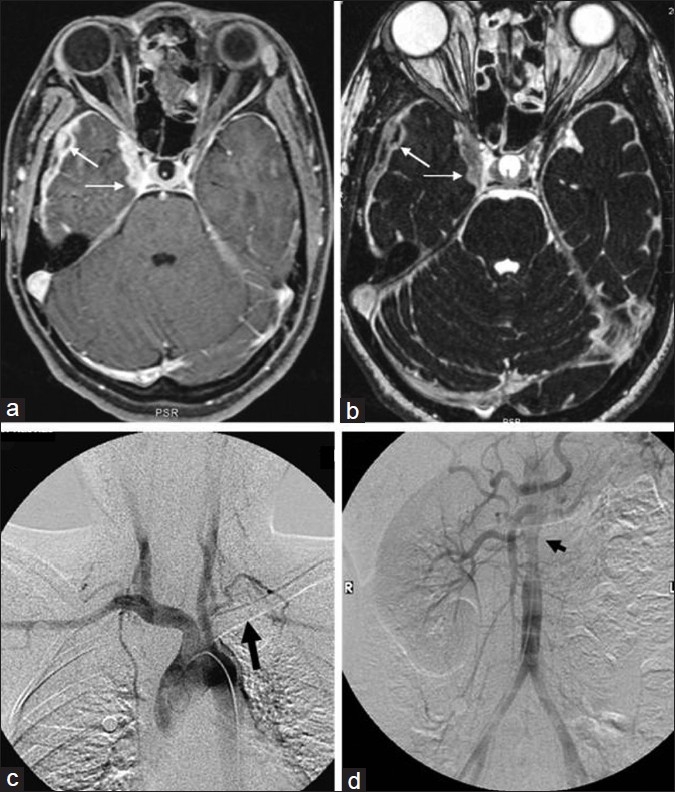
Axial contrast-enhanced T1-weighted fat saturated image shows thickened enhancing pachymeninges in the right middle cranial fossa and paracavernous region (a-thin arrows). Thickened pachymeninges can also be seen on T2-weighted constructive interference at steady state (CISS) images also (b-thin arrows). Digital subtraction angiography (DSA) of the aortic arch (c) and abdominal aorta (d) shows the left subclavian occlusion (thick arrow) and the non-visualization of the left renal artery with narrowing of the juxta renal abdominal aorta (arrow head), respectively
Figure 2.
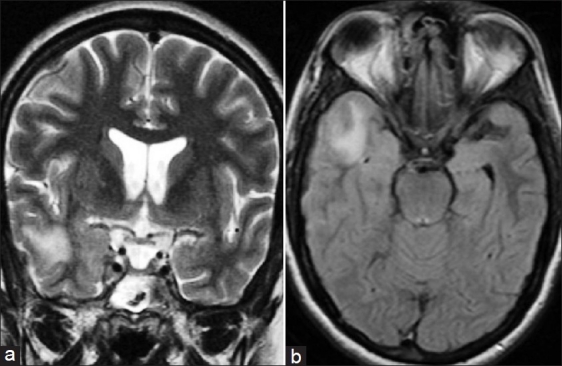
Coronal T2-weighted image (a) and axial Fluid Attenuated Inversion Recovery (FLAIR) sequence (b, shows subcortical white matter hyperintensity in right temporal lobe extending in to the overlying cortex
After starting prednisolone (1 mg/kg/day), there was complete recovery from right third and fourth cranial nerve palsy over a period of 3 weeks along with normalization of ESR. Her blood pressure was well controlled on atenolol, while seizures on 300 mg of phenytoin. However, asymptomatic blood pressure difference in upper limbs was persistent. She was continued on phenytoin, atenolol, and low-dose prednisolone (tapered over a period of 12 weeks to 10 mg alternate days). She remained asymptomatic on these medications at 1-year follow-up. A postcontrast computed tomography (CT) scan at 6 months follow-up was normal and there was no evidence for meningeal enhancement. Her laboratory evaluation including ESR and renal function tests were within normal limits at the last follow-up. As patient remained asymptomatic and did not show signs of vascular insufficiency, a repeat angiogram, being an invasive test, was not considered.
Discussion
HP is a rare chronic inflammatory condition characterized by localized or diffuse thickening of the dura mater, first described by Charcot and Joffroy with respect to spinal meninges.[4] HP has been described in association with infections such as mycobacterium tuberculosis, syphilis, lyme disease, fungal infections, cysticercosis, and rarely with HTLV-1. It has also been described with meningioma and malignancies such as dural carcinomatosis or lymphoma.[1] A well-known association of HP is with systemic autoimmune or vasculitic disorders such as Wegener's granulomatosis, rheumatoid arthritis, sarcoidosis, Behcet's disease, Sjogren syndrome, and temporal arteritis.[2,5] A diagnosis of idiopathic hypertrophic pachymeningitis is considered, when the evaluation fails to reveal any cause.
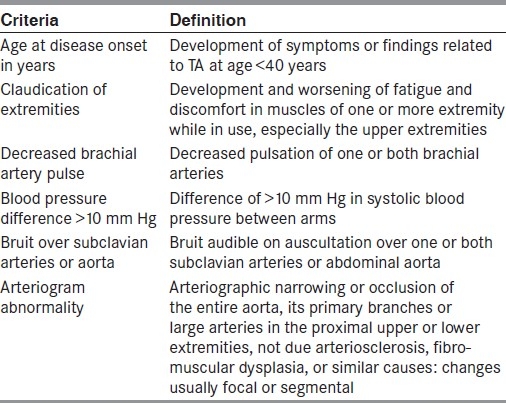
TA is also well known, yet rare form of chronic inflammatory arteritis affecting large vessels, especially the aorta and its main branches.[6] First described by Japanese ophthalmologist, Dr. Mikito Takayasu in 1905,[7] TA commonly affects individuals in their second or third decade of life.[8] In its classic form, three phases have been described in the disease evolution, first the early systemic phase, characterized by non-specific systemic manifestations such as fever, arthralgia, weight loss, and night sweats, second dominated by vessel pain due to vessel inflammation and finally the fibrotic phase, in which bruits and vascular insufficiency predominate.[9,10] However, initial systemic manifestations may be absent in majority of patients and the disease may remain undiagnosed, before vascular symptoms or signs become apparent, as in our patient.[8] As the natural history of TA is quite variable, clinical diagnosis of TA is always a challenge to the clinicians, especially in early phase. The diagnosis of the TA is based on a set of criteria suggested by ACR in 1990 as shown in Table 1.[3] The presence of any three or more criteria suggests high probability of TA yielding a sensitivity of 90.5% and a specificity of 97.8%.[3] Our patient satisfied five of these six criteria, thus establishing the diagnosis of TA.
Table 1.
1990 criteria of American College of Rheumatology for the classification of TA. A patient shall be said to have TA if at least three of these six criteria are present.[3]
Neurological manifestations of TA results either from decreased blood flow due to a steno-occlusive lesion and/or shunting of blood flow, thromboembolism, or hypertension. The common neurologic manifestations include headache, visual disturbances, syncope, transient ischemic attack, cerebral infarction, cerebral hemorrhage, hypertensive encephalopathy, seizures, and paraplegia.[6,8,11] Rarely, compression cranial neuropathy can occur secondary to aneurysmal dilatation of the intracranial vessels.[12] Seizures as presenting symptoms are rare in patients with TA.[13] Cranial neuropathy in our patient was secondary to HP involving cavernous sinus, while seizures were due to cerebral parenchymal edema in the temporal lobe adjacent to the affected dura complicating HP. However, till now, to the best of our knowledge, HP has not been described in patients with TA.
TA and HP both are chronic inflammatory disease, pathologically characterized by infiltration by lymphocytes, plasma cells, epithelioid cells, and occasionally formation of granulomas, which are replaced by fibrosis in late stages.-[1,14] Although the etiology in patients with TA still remains elusive, autoimmune disturbances have been suspected in its pathogenesis for a long time. Literature suggests that TA, rather than being a restricted form of vasculitis, is a generalized systemic disorder affecting various organs. Moreover, its association with various diseases such as arthritis, episcleritis, pericarditis, and pleuritis further strengthens the common underlying immunological mechanisms.[6] The inflammatory lesions in TA originate in the vasa vasorum which is followed by cellular infiltration invading the media and adventitia of the large blood vessels.[15] We believe that similar inflammatory process involving the small vessels of the dura mater might have resulted in HP in our patient. However, exactly how the dura was affected in this case remains uncertain. Infections especially tuberculosis have since long been implicated in the etiopathogenesis of TA in view of the high prevalence of these infections, past or present, in affected patients.[14] It is also one of the leading causes of HP in endemic areas. However, the possibility of chronic infections including tuberculosis was fairly excluded in our patient. Although a meningeal biopsy to exclude any infectious process or sarcoidosis could not be performed, the spontaneous resolution of our patient's symptoms and major signs on treatment, and an interval brain CT showing no abnormality or meningeal enhancement on steroid taper have been remarkable, thereby substantially excluding any infection or other inflammatory process, which is known to occur in association with HP. The aforementioned clinical recovery suggests that HP in our patient is most likely related to or secondary to TA. In conclusion, our case emphasizes that neurological manifestations in patients with TA does not limit to vascular compromise, and one should be vigilant for such a rare presentation in these patients.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil
References
- 1.Kupersmith MJ, Martin V, Heller G, Shah A, Mitnick HJ. Idiopathic hypertrophic pachymeningitis. Neurology. 2004;62:686–94. doi: 10.1212/01.wnl.0000113748.53023.b7. [DOI] [PubMed] [Google Scholar]
- 2.Joelson E, Ruthrauff B, Ali F, Lindeman N, Sharp F. Multifocal dural enhancement associated with temporal arteritis. Arch Neurol. 2000;57:119–22. doi: 10.1001/archneur.57.1.119. [DOI] [PubMed] [Google Scholar]
- 3.Arend WP, Michel BA, Bloch DA, Hunder GG, Calabrese LH, Edworthy SM, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum. 1990;33:1129–34. doi: 10.1002/art.1780330811. [DOI] [PubMed] [Google Scholar]
- 4.Charcot JM, Joffroy A. Deux cas d‘atrophie musculaire progressive avec lesions de la substance grise et des faisceaux anterolateraux de la moelle epiniere. Arch Physiol Norm Pathol. 1869;2:354–67. [Google Scholar]
- 5.Ashraf VV, Bhasi R, Kumar RP, Girija AS. Primary Sjögren's syndrome manifesting as multiple cranial neuropathies: MRI findings. Ann Indian Acad Neurol. 2009;12:124–6. doi: 10.4103/0972-2327.53083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma BK, Jain S, Sagar S. Systemic manifestations of Takayasu arteritis: The expanding spectrum. Int J Cardiol. 1996;(Suppl 54):S121–6. doi: 10.1016/s0167-5273(96)88784-5. [DOI] [PubMed] [Google Scholar]
- 7.Takayasu M. A case with peculiar changes of the retinal central vessels [in Japanese] Acta Opthalmic Soc Jpn. 1908;12:554–5. [Google Scholar]
- 8.Sharma BK, Sagar S, Aminder PS, Suri S. Takayasu arteritis in India. Heart Vessels. 1992;7(Suppl 1):37–43. doi: 10.1007/BF01744542. [DOI] [PubMed] [Google Scholar]
- 9.Hall S, Barr W, Lie JT, Stanson AW, Kazmier FJ, Hunder GG. Takayasu arteritis.A study of 32 North American patients. Medicine (Baltimore) 1985;64:89–99. [PubMed] [Google Scholar]
- 10.Veda H, Morooka S, Ito I, Yamaguchi H, Takeda T, Saito Y. Clinical observation of 52 cases of aortitis syndrome. Jpn Heart J. 1969;10:277–88. doi: 10.1536/ihj.10.277. [DOI] [PubMed] [Google Scholar]
- 11.Kim HJ, Suh DC, Kim JK, Kim SJ, Lee JH, Choi CG, et al. Correlation of neurological manifestations of Takayasu's arteritis with cerebral angiographic findings. Clin Imaging. 2005;29:79–85. doi: 10.1016/j.clinimag.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 12.Stepieñ A, Durka-Kesy M, Warczyñska A. Compression neuropathy of cranial nerves in the course of Takayasu arteritis. Neurol Neurochir Pol. 2007;41:557–61. [PubMed] [Google Scholar]
- 13.Ioannides MA, Eftychiou C, Georgiou GM, Nicolaides E. Takayasu arteritis presenting as epileptic seizures: A case report and brief review of the literature. Rheumatol Int. 2009;29:703–5. doi: 10.1007/s00296-008-0747-9. [DOI] [PubMed] [Google Scholar]
- 14.Johnston SL, Lock R J, Gompels MM. Takayasu arteritis: A review. J Clin Pathol. 2002;55:481–6. doi: 10.1136/jcp.55.7.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noris M. Pathogenesis of Takayasu's arteritis. J Nephrol. 2001;14:506–13. [PubMed] [Google Scholar]