

Abstract
Incidences of oral tongue, base of the tongue, and tonsil cancers have been increasing steadily in many parts of the world in spite of declining rates of tobacco use over the last four decades. A better understanding of the etiology, interactions between risk factors, and new approaches to prevention and treatment are necessary to change this course. This paper will present evidence supporting a potential role of chronic inflammation in the etiologies of oral human papillomavirus infection and head and neck squamous cell carcinoma, and it will discuss the implications for prevention and treatment.
1. Introduction
Over 500 000 new cases of head and neck squamous cell carcinoma (HNSCC) occur annually in the world [1]. The National Cancer Institute's Surveillance, Epidemiology, and End Results (SEER) has reported a steady increase in the incidence of oropharyngeal cancers since 1973 despite the significant decline in tobacco use since 1965 [2]. Similar trends are observed in many parts of the world [3–5].
HNSCC is largely preventable, yet the majority of current research has focused on treatment and diagnosis. Most of the existing prevention strategies for HNSCC have been secondary and tertiary with limited success [6, 7]. Chemoprevention, including retinoids, selenium, vitamin E, interferon-α (IFN-α), cyclo-oxygenase-2 (COX-2), and epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors, usually have obstacles including toxicity and reversal of their effects after cessation of treatment [8, 9]. Primary prevention strategies, focused mainly on smoking cessation, have not lead to a visible reduction in HNSCC incidence [1, 2]. Oral human papillomavirus (HPV) infection has emerged as an independent etiological factor for at least a subset of HNSCC, and is another target for primary prevention [10–12]. A highly effective vaccine is available to prevent cervical HPV infection which is currently recommended for females aged 9 to 26 years and males aged 9 to 21 years [13]. However, oral HPV infection can be transmitted at or any time after birth, and the target population for the vaccine to prevent oral HPV infection has not been defined [14]. A large percentage of the general population who are already exposed to the virus or those who already developed an HPV-related disease do not benefit from the current vaccine. A better understanding of the etiology of HNSCC, especially the interactions among risk factors, is critical for effective prevention strategies.
This short review will (1) present evidence supporting a potential role of chronic inflammation on the etiologies of oral HPV infection and HNSCC and (2) discuss the implications for prevention and treatment.
2. Chronic Inflammation and Head and Neck Squamous Cell Carcinoma
Clinical, epidemiologic, and animal studies support an association between chronic inflammation and cancer in several organs [15–18]. Chronic cervicitis and cervical cancer, ulcerative colitis and colorectal cancer, pancreatitis and pancreatic cancer, skin inflammation and skin cancer, esophagitis and esophageal cancer, and hepatitis and liver cancer are a few examples (Table 1).
Table 1.
Chronic inflammatory diseases and associated cancers.
| Chronic inflammation | Associated neoplasm |
|---|---|
| Chronic bronchitis | Lung carcinoma |
| Chronic gastritis | Gastric adenocarcinoma |
| Chronic cervicitis, pelvic inflammatory disease | Cervical carcinoma, ovarian carcinoma |
| Asbestosis, silicosis | Mesothelioma, lung carcinoma |
| Chronic ulcerative colitis | Colorectal cancer |
| Chronic pancreatitis, hereditary pancreatitis | Pancreatic carcinoma |
| Chronic sunburned skin, skin inflammation | Melanoma, basal-cell carcinoma, squamous cell carcinoma |
| Reflux esophagitis, Barrett's esophagus | Esophageal carcinoma |
| Chronic hepatitis | Hepatocellular carcinoma |
| Mononucleosis | Burkitt's, Hodgkin's and non-Hodgkin's lymphoma. |
| Chronic cystitis | Bladder cancer |
| Chronic osteomyelitis | Skin carcinoma in draining sinuses |
In the oral cavity, periodontitis is a chronic inflammatory disease of the structures around teeth associated with dental biofilm [19]. The ensuing chronic inflammation leads to local pathologic anatomic changes, namely, periodontal pocket formation, clinical attachment loss (CAL), and alveolar bone loss (ABL) [20]. As the disease progresses and more bone is lost, junctional epithelium at the bottom of the periodontal pocket migrates apically along the root surface and the pocket depth increases. Untreated periodontitis eventually leads to tooth loss. The pocket epithelium is characterized by continuous proliferation, formation of rete-ridges, and ulcerations. In the connective tissue, there is increased angiogenesis, chronic inflammatory infiltrate, fibrosis, and connective tissue loss. The average prevalence of periodontitis in the general population is 30% [21]. Periodontitis results in a continuous release of inflammatory cytokines, enzymes, and toxins into saliva. The levels of these inflammatory markers in saliva is directly associated with the extent and severity of periodontitis [22, 23]. In addition, periodontal pathogens and inflammatory cytokines can travel with saliva and blood causing inflammation and tissue injury at distant sites [24–31]. Periodontal treatment has been shown to reverse some of these systemic adverse effects [32–34].
A link between poor oral hygiene and HNSCC has been suggested for over two decades [35–46] but the underlying mechanism of this association was not clear. Studies from our group have suggested that chronic periodontitis, an outcome of poor oral hygiene, is associated with oral premalignant lesions [47] and HNSCC [48, 49]. In a secondary analysis of the Third National Health and Nutrition Examination Survey (NHANES III), a large nationwide cross-sectional study, including 13,798 men and women of age 20 and older, a history of periodontitis, measured by clinical attachment loss (CAL), was significantly associated with the prevalence of oral premalignant lesions (OR = 1.55, 95% CI = 1.06–2.27) after adjusting for age, gender, race/ethnicity, education, tobacco, alcohol, and occupational hazard [47]. Periodontitis was not associated with the presence of any oral soft tissue lesion (OR = 1.09, 95% CI = 0.91–1.31) (Table 2) [47].
Table 2.
Risk of oral premalignant lesions from periodontitis.
| Premalignant lesions* | Any lesion | |
|---|---|---|
| N (%)† | 323 (2.3%) | 3,421 (24.8%) |
| Periodontitis‡ | 1.55 [1.06–2.27]§ | 1.09 [0.91–1.31] |
*Premalignant lesions were defined as erythroplakia, leukoplakia (homogeneous and nonhomogeneous), nonspecific ulcer, and smokeless tobacco-associated lesions.
†Count (percentage).
‡Periodontitis was defined as CAL >1.5 mm.
§Odds ratios (OR) and their 95% confidence intervals derived from multiple logistic regression analysis adjusting for age, gender, race/ethnicity, education, tobacco, alcohol, and occupational hazard.
In a recent hospital-based case-control study with histologically confirmed 266 primary incident HNSCC cases and 207 controls in Upstate New York, ABL was significantly higher in cases compared to controls (4.04 mm versus 2.44 mm, P < 0.001). After adjustment for age, gender, race/ethnicity, marital status, smoking status, alcohol use, and missing teeth, each millimeter of ABL was associated with >4-fold increased odds of HNSCC (OR = 4.36, 95% CI = 3.16–6.01). The strength of the association was greatest in the oral cavity (OR = 4.52, 95% CI = 3.03–6.75), followed by oropharynx (OR = 3.64, 95% CI = 2.54–5.22) and larynx (OR = 2.72, 95% CI = 1.78–4.16) (Table 3). Furthermore, patients with periodontitis were more likely than those without periodontitis to have poorly differentiated oral cavity SCC (32.8% versus 11.5%, P = 0.038) [49].
Table 3.
Association between periodontitis and head and neck cancer stratified by tumor site (N = 473).
| Alveolar bone loss (per millimeter) | N (cases/controls) | Crude OR [95% CI]* | Adjusted† OR [95% CI] |
|---|---|---|---|
| All HNSCC patients | 266/207 | 3.85 [2.96–5.01] | 4.36 [3.16–6.01] |
| Oral cavity SCC patients | 100/207 | 3.26 [2.44–4.36] | 4.52 [3.03–6.75] |
| Oropharyngeal SCC patients | 115/207 | 3.06 [2.29–4.07] | 3.64 [2.54–5.22] |
| Laryngeal SCC patients | 51/207 | 3.75 [2.60–5.41] | 2.72 [1.78–4.16] |
*Odds ratios and their 95% confidence intervals.
†Adjusted odds ratios were derived from multiple logistic regression analysis including age at diagnosis, gender, race/ethnicity, marital status, smoking status, alcohol use, ABL, and missing teeth.
The potential association between chronic inflammation and HNSCC is further supported by two recent case control studies suggesting a beneficial effect of nonsteroidal anti-inflammatory drugs (NSAID) against HNSCC [50, 51]. In a hospital-based case control study among 529 patients with HNSCC and 529 control subjects matched by age, sex, and smoking status, Aspirin use was associated with a 25% reduction in the risk of HNSCC (OR = 0.75; 95% CI = 0.58–0.96). Risk reduction was observed across all primary tumor sites, with cancers of the oral cavity and oropharynx exhibiting greater risk reduction [50]. In another more recent hospital-based case-control study among 71 incident HNSCC cases and 71 healthy controls, daily NSAID use was associated with 86% reduction in HNSCC risk (OR = 0.14; 95% CI = 0.04–0.54) after adjusting for educational level and marital status [51].
The biological mechanism of the association between chronic inflammation and cancer has been described extensively but is also evolving continuously since both inflammation and cancer are complex processes under the control of many driving forces [15–18]. Bacteria and their products, including endotoxins, enzymes, and metabolic by-products, may directly induce genetic and epigenetic changes in surrounding epithelial cells [52–54]. They also increase the production of carcinogenic acetaldehydes [55–57] and nitrosamines [58, 59]. However, the bulk of the available evidence supports an indirect association through stimulation of inflammation. Host cells, including neutrophils, macrophages, monocytes, lymphocytes, fibroblasts, and epithelial cells, respond to bacteria by generating (1) cytokines, chemokines, prostaglandins, growth factors, and other signals that provide an environment for cell survival, proliferation, migration, angiogenesis, and inhibition of apoptosis [60]. This environment helps epithelial cells to accumulate mutations, and drives these mutant epithelial cells to proliferate and migrate and gives them a growth advantage; (2) reactive oxygen species (hydrogen peroxide and oxy radicals), reactive nitrogen species (nitric oxides), reactive lipids and metabolites (malondialdehyde, 4-hydroxy-2-nonenal), and matrix metalloproteinases (MMPs) which can act as endogenous mutagens. Numerous in vivo and in vitro studies have confirmed the associations of several genes and proteins involved in different stages of inflammation with carcinogenesis [61–71]. In addition to its independent association with HNSCC, chronic inflammation may also act synergistically with other carcinogens to increase the risk of HNSCC. For example, breaks in the mucosal barrier due to chronic inflammation may lead to enhanced penetration of other carcinogens such as tobacco, alcohol, and dietary metabolites [72, 73].
3. Chronic Inflammation and Oral HPV Infection
The steady increase in the incidence of oropharyngeal cancers over the last four decades has been mainly attributed to oral HPV infection which has been accepted as an etiological factor for a subset of HNSCC [10–14]. HPV is a commonly transmitted virus but the majority of the infections are cleared rapidly by the immune system. Rather than its mere presence at one time point, persistence of the virus seems to be the critical factor for the development of HPV-related diseases [74, 75]. Therefore, targeting factors associated not only with the acquisition but also with the persistence of oral HPV infection will contribute to both prevention and treatment.
HPV is a small DNA virus with a specific tropism for squamous epithelia. More than 120 different HPV types have been isolated to date. Low-risk HPVs, including HPV-6 and HPV-11, induce benign hyperproliferations of the epithelium such as papillomas or warts which rarely progress to cancer [76]. High-risk oncogenic types such as HPV-16 and HPV-18 are associated with squamous cell carcinoma. HPV-16 and HPV-18 are capable of transforming epithelial cells. This transforming potential is largely a result of the function of two viral oncoproteins, E6 and E7, which inactivate two tumor suppressor proteins, p53 and pRb, respectively. Expression of E6 and E7 results in cellular proliferation, loss of cell cycle regulation, impaired cellular differentiation, increased frequency of mutations, and chromosomal instability [76, 77].
Population-based studies estimate that the prevalence of HPV DNA in normal oral mucosa is 5–10% [14, 76]. In contrast with cervical cancer, in which HPV is present 100% of the time, HPV is present at a smaller and variable percentage in HNSCC, depending on the subsite and the geographic region studied. In a review of 60 studies, the prevalence of HPV DNA was 23.5% in oral cavity SCC, 35.6% in oropharyngeal SCC, and 24.0% in laryngeal SCC [78]. HPV-16 accounted for a larger majority of HPV-positive oropharyngeal SCC (86.7%) compared with HPV-positive oral cavity SCC (68.2%) and HPV-positive laryngeal SCC (69.2%). Conversely, HPV-18 was rare in HPV-positive oropharyngeal SCC (2.8%) compared with HPV-positive oral cavity SCC (34.1%) and HPV-positive laryngeal SCC (17.0%). Aside from HPV-16 and HPV-18, other oncogenic HPV DNAs were rarely detected in HNSCC [78]. In a multi-national study conducted by the International Agency for Research on Cancer (IARC), only 18% of oropharyngeal tumors were HPV-positive [4]. In the majority of recent studies, >50% of oropharyngeal SCC contained the HPV DNA [10–14]. Probability of HPV DNA being detected in the oral mucosa increases with increasing degree of dysplasia. Overall, HPV DNA is 2- to 3-times more likely to be detected in premalignant lesions and about 5-times more likely to be detected in HNSCC compared to normal oral mucosa [79].
HPV-positive HNSCCs have a unique risk factor profile. These tumors are more common in younger patients, have a male predominance, are often staged higher, yet have a survival advantage. These patients are less likely to have used tobacco and alcohol excessively [14]. Pathologically, they are more likely to be basaloid type (poorly differentiated) and lack keratinization. The p16 protein, also known as cyclin-dependent kinase 4 (CDK4) inhibitor, is upregulated in HPV-positive HNSCC, and may serve as a surrogate marker for HPV transcriptional activity. Molecularly, these tumors expresses oncoproteins E6 and E7 demonstrate less chromosomal disruption than HPV-negative HNSCC. Tumor suppressor genes p53 and pRb are usually intact, but the expressed proteins are inactivated by the viral oncoproteins E6 and E7, respectively. In contrast, HPV-negative HNSCC, which is usually initiated by smoking and alcohol, shows considerable chromosomal aberration, including mutation of the p53 gene in 50% of cases [80–82].
Although the natural history of cervical HPV infection is well established, the mechanism of transmission for oral HPV infection, which has a distinct immunologic environment, is largely unknown. There is increasing evidence that oral HPV transmission may be related to sexual history, and particularly oral sexual behavior. Case-control studies have shown associations of oral HPV infection with the number of oral and genital sexual partners, age at first of oral-genital contact, frequency of sexual contacts, and history of genital warts [83–85]. Vertical transmission of oral HPV infection through maternal milk is also possible [86]. HPV infection at each anatomic site is localized and concordance of oral and genital HPV infection seems to be low [14] implying the importance of local oral factors. Research evaluating the role of local oral factors on natural history of oral HPV infection is lacking and is critical to develop effective prevention strategies.
Few epidemiological studies suggest that chronic inflammation of the cervix increases the risks of HPV infection and cervical cancer [87–90]. In addition, molecular studies have shown that inflammatory cytokines, including IL-1, IL-6, and TNF-α, modulate proliferation of HPV and expression of its oncogenes E6 and E7 in cervical epithelial cells [91–96]. Supporting these findings, an in vitro study has shown that a nonsteroidal anti-inflammatory drug strongly induced degradation of the HPV oncoproteins causing growth arrest and apoptosis in cervical carcinoma cells in a concentration-dependent manner [97]. Continuous expression of the HPV oncoproteins has also been shown to be critical for the maintenance of malignant phenotype in HPV-positive oropharyngeal cancers [98]. The oral cavity and cervix are lined with similar types of mucosa, and the same HPV types cause cervical and head and neck cancers. In a recent hospital-based case-control study with 30 incident primary base of tongue SCC, a history of chronic periodontitis was associated with tumor HPV status after adjustment for age at diagnosis, gender, race/ethnicity, alcohol use, smoking status, ABL, and number of missing teeth (OR = 3.96, 95% CI = 1.18–13.36) (Table 4) [99].
Table 4.
Odds Ratios for Tumor HPV Status Associated with Periodontal Variables.
| Crude OR | Adjusted† OR | |
|---|---|---|
| [95% CI]* | [95% CI] | |
| Alveolar Bone Loss (per millimeter) |
2.86 [1.03–7.98] | 3.96 [1.18– 13.36] |
| Missing Teeth (per tooth) |
0.94 [0.86–1.02] | 0.95 [0.74–1.21] |
*Odds ratios and 95% confidence intervals.
†Adjusted odds ratios were derived from multiple logistic regression model including age at diagnosis, gender, race/ethnicity, alcohol use, smoking status, ABL and number of missing teeth.
An association between chronic inflammation and HPV infection is biologically plausible. HPV infects basal cells of the epithelium exclusively, and it usually gains access through microabrasions. In addition, replication of the virus is closely associated with basal cell proliferation [74, 75]. Mucosal injury and consequent breaks within the oral mucosa mediated by inflammatory cytokines may facilitate the acquisition as well as the persistence of oral HPV infection [70]. In the presence of chronic inflammation, basal cell proliferation is also increased leading to higher viral load in saliva as well as higher risk of transmission [75]. The observed association between periodontitis and oral HPV infection may be alternatively explained by the direct effects of bacteria [100]. Prospective cohort studies have shown that concurrent infection with Chlamydia trachomatis increased the persistence of cervical HPV infection and the risk of cervical cancer [101, 102]. Periodontal bacteria, derived from periodontal pockets, are extensively present in the oral mucosa [103, 104]. However, whether the association between periodontitis and oral HPV infection is through direct effects of bacteria or through stimulation of inflammation is yet to be determined. It is likely that both mechanisms are involved but the bulk of the current evidence suggests that the periodontal bacteria travel from affected tissues to distant sites via saliva and bloodstream and cause tissue injury through inflammatory reactions [105–109].
4. Implications for Prevention and Treatment
The potential role of chronic inflammation in the etiology of HNSCC is summarized in Figure 1. Briefly, chronic inflammation may increase the risk of HNSCC both independently and by facilitating the acquisition and persistence of oral HPV infection.
Figure 1.
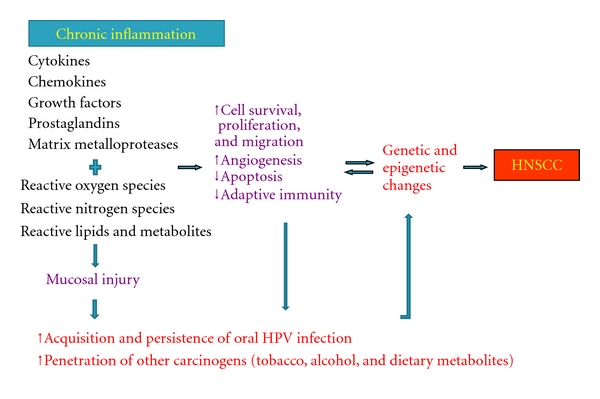
Model for the role of chronic inflammation in the etiology of head and neck squamous cell carcinoma (HNSCC).
Understanding the interactions among risk factors may be the most important step towards effective prevention and treatment of HNSCC. It is well established that carcinogenesis is a multifactorial process and the presence of a single risk factor is usually not sufficient to cause cancer. Yet, most studies have focused on independent effects of specific risk factors [6, 7]. Recently, two separate entities, “tobacco-related” and “HPV-related” HNSCC, have been suggested [80–82]. However, this classification overlooks the fact that most HPV-positive HNSCC patients also have a history of tobacco use. It is more likely that the interaction between different combinations of risk factors in each individual determines the disease outcome or its clinical presentation. For example, in our study population consisting of newly diagnosed base of tongue cancer patients, 83.5% had a smoking history and 63.3% had HPV-positive tumors. The majority (71.4%) of patients with HPV-positive tumors also had a smoking history [99]. The results further suggested that smokers with chronic periodontitis were more likely to have HPV-positive tumors, and smokers without periodontitis history were more likely to have HPV-negative tumors [99]. Smoking is a strong risk factor for chronic periodontitis [21]. Although validation with further studies is needed, these observations suggest that only a subset of smokers who develop chronic inflammatory diseases (perhaps long-term, moderate to heavy smokers) are at increased risk for oral HPV infection.
Saliva provides a means of interaction between different carcinogens. It also provides a means of transport from one surface to another promoting disease at distant sites. Salivary enzymes also metabolize tobacco, alcohol, and dietary components into carcinogens [110]. This may contribute to the field cancerization in the head and neck region. The fact that the majority of HNSCC occurs at saliva-draining areas supports this hypothesis [111]. While periodontitis is localized to the structures around teeth, periodontal pocket contents, including bacteria, viruses, inflammatory cytokines, enzymes, and toxins are continuously shed into saliva [22, 23, 112]. On the other hand, saliva has no access into periodontal pockets. Therefore, periodontal pockets are probably a source of inflammatory and microbial markers in saliva rather than being the actual sites of carcinogenesis.
It is critical to understand the characteristics of the dental biofilm to effectively treat periodontitis. Biofilms are sessile communities of bacteria embedded in a polymeric matrix irreversibly attached to a surface. The bacteria in the biofilm differ profoundly from their free-floating counterparts and are resistant to antimicrobials as well to host defense [113]. Thus, effective treatment of periodontitis is based on mechanical removal of the biofilm. In the initial stages of the disease, a thorough professional dental cleaning and maintenance of a good personal oral hygiene, by brushing and flossing, is usually sufficient to reverse the disease process. In more advanced stages, however, the biofilm or calculus in periodontal pockets is inaccessible by personal oral hygiene practices. In these cases, surgical reduction of the periodontal pockets and/or periodic professional cleanings may be necessary to control the disease [19, 113].
If prospective studies confirm that chronic local inflammation is in fact a significant player in the natural histories of oral HPV infection and HNSCC, this may have important implications, not only for prevention, but also for treatment. Chronic local inflammation, such as periodontitis, is easy to detect and may represent a clinical high-risk profile for both oral HPV infection and HNSCC. Subjects with sources of chronic inflammation in the oral cavity may be screened for oral HPV infection and HNSCC for early diagnosis.
5. Conclusion
Chronic local inflammation, both independently and by facilitating oral HPV infection, may be an important player in the etiology of HNSCC.
Acknowledgments
This paper was supported by Grants R03CA119262 from the National Cancer Institute and T32-DE07034 from the National Institute of Dental and Craniofacial Research.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: A Cancer Journal for Clinicians. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Hayat MJ, Howlader N, Reichman ME, Edwards BK. Cancer statistics, trends, and multiple primary cancer analyses from the surveillance, epidemiology, and end results (SEER) program. Oncologist. 2007;12(1):20–37. doi: 10.1634/theoncologist.12-1-20. [DOI] [PubMed] [Google Scholar]
- 3.Ernster JA, Sciotto CG, O’Brien MM, et al. Rising incidence of oropharyngeal cancer and the role of oncogenic human papilloma virus. Laryngoscope. 2007;117(12):2115–2128. doi: 10.1097/MLG.0b013e31813e5fbb. [DOI] [PubMed] [Google Scholar]
- 4.Herrero R, Castellsagué X, Pawlita M, et al. Human papillomavirus and oral cancer: the international agency for research on cancer multicenter study. Journal of the National Cancer Institute. 2003;95(23):1772–1783. doi: 10.1093/jnci/djg107. [DOI] [PubMed] [Google Scholar]
- 5.Hammarstedt L, Lindquist D, Dahlstrand H, et al. Human papillomavirus as a risk factor for the increase in incidence of tonsillar cancer. International Journal of Cancer. 2006;119(11):2620–2623. doi: 10.1002/ijc.22177. [DOI] [PubMed] [Google Scholar]
- 6.Scully C. Oral cancer aetiopathogenesis; past, present and future aspects. Medicina Oral, Patologia Oral y Cirugia Bucal. 2011;16(3):306–311. doi: 10.4317/medoral.16.e306. [DOI] [PubMed] [Google Scholar]
- 7.Scully C, Bagan JV. Recent advances in oral oncology 2008; squamous cell carcinoma imaging, treatment, prognostication and treatment outcomes. Oral Oncology. 2009;45(6):e25–e30. doi: 10.1016/j.oraloncology.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 8.Kelloff GJ, Lippman SM, Dannenberg AJ, et al. Progress in chemoprevention drug development: the promise of molecular biomarkers for prevention of intraepithelial neoplasia and cancer—a plan to move forward. Clinical Cancer Research. 2006;12(12):3661–3697. doi: 10.1158/1078-0432.CCR-06-1104. [DOI] [PubMed] [Google Scholar]
- 9.Choe MS, Zhang X, Shin HJC, Shin DM, Zhuo C. Interaction between epidermal growth factor receptor- and cyclooxygenase 2-mediated pathways and its implications for the chemoprevention of head and neck cancer. Molecular Cancer Therapeutics. 2005;4(9):1448–1455. doi: 10.1158/1535-7163.MCT-04-0251. [DOI] [PubMed] [Google Scholar]
- 10.Gillison ML, Koch WM, Capone RB, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. Journal of the National Cancer Institute. 2000;92(9):709–720. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- 11.Pintos J, Black MJ, Sadeghi N, et al. Human papillomavirus infection and oral cancer: a case-control study in Montreal, Canada. Oral Oncology. 2008;44(3):242–250. doi: 10.1016/j.oraloncology.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 12.D’Souza G, Kreimer AR, Viscidi R, et al. Case-control study of human papillomavirus and oropharyngeal cancer. New England Journal of Medicine. 2007;356(19):1944–1956. doi: 10.1056/NEJMoa065497. [DOI] [PubMed] [Google Scholar]
- 13.Joura EA, Leodolter S, Hernandez-Avila M, et al. Efficacy of a quadrivalent prophylactic human papillomavirus (types 6, 11, 16, and 18) L1 virus-like-particle vaccine against high-grade vulval and vaginal lesions: a combined analysis of three randomised clinical trials. The Lancet. 2007;369(9574):1693–1702. doi: 10.1016/S0140-6736(07)60777-6. [DOI] [PubMed] [Google Scholar]
- 14.Adelstein DJ, Ridge JA, Gillison ML, et al. Head and neck squamous cell cancer and the human papillomavirus: summary of a National Cancer Institute State of the Science Meeting, November 9-10, 2008, Washington, D.C. Head & neck. 2009;31(11):1393–1422. doi: 10.1002/hed.21269. [DOI] [PubMed] [Google Scholar]
- 15.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. Journal of Clinical Investigation. 2007;117(5):1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 17.Culig Z. Cytokine disbalance in common human cancers. Biochimica et Biophysica Acta. 2011;1813(2):308–314. doi: 10.1016/j.bbamcr.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 18.Zhu Z, Zhong S, Shen Z. Targeting the inflammatory pathways to enhance chemotherapy of cancer. Cancer Biology and Therapy. 2011;12(2):95–105. doi: 10.4161/cbt.12.2.15952. [DOI] [PubMed] [Google Scholar]
- 19.Loesche WJ, Grossman NS. Periodontal disease as a specific, albeit chronic, infection: diagnosis and treatment. Clinical Microbiology Reviews. 2001;14(4):727–752. doi: 10.1128/CMR.14.4.727-752.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Armitage GC. Periodontal diagnoses and classification of periodontal diseases. Periodontology 2000. 2004;34:9–21. doi: 10.1046/j.0906-6713.2002.003421.x. [DOI] [PubMed] [Google Scholar]
- 21.Burt B. Academy Report. Epidemiology of Periodontal Diseases. Journal of Periodontology. 2005;76:1406–1419. doi: 10.1902/jop.2005.76.8.1406. [DOI] [PubMed] [Google Scholar]
- 22.Scannapieco FA, Ng P, Hovey K, Hausmann E, Hutson A, Wactawski-Wende J. Salivary biomarkers associated with alveolar bone loss. Annals of the New York Academy of Sciences. 2007;1098:496–497. doi: 10.1196/annals.1384.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giannobile WV, Beikler T, Kinney JS, Ramseier CA, Morelli T, Wong DT. Saliva as a diagnostic tool for periodontal disease: current state and future directions. Periodontology 2000. 2009;50(1):52–64. doi: 10.1111/j.1600-0757.2008.00288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scannapieco FA, Wang B, Shiau HJ. Oral bacteria and respiratory infection: effects on respiratory pathogen adhesion and epithelial cell proinflammatory cytokine production. Annals of Periodontology. 2001;6(1):78–86. doi: 10.1902/annals.2001.6.1.78. [DOI] [PubMed] [Google Scholar]
- 25.Genco RJ, Trevisan M, Wu T, et al. Periodontal disease and risk of coronary heart disease [2] (multiple letters) Journal of the American Medical Association. 2001;285(1):40–41. doi: 10.1001/jama.285.1.40. [DOI] [PubMed] [Google Scholar]
- 26.Nichols TC, Fischer TH, Deliargyris EN, Baldwin AS., Jr Role of nuclear factor-κB (NF-κB) in inflammation, periodontitis, and atherogenesis. Annals of Periodontology. 2001;6(1):20–29. doi: 10.1902/annals.2001.6.1.20. [DOI] [PubMed] [Google Scholar]
- 27.Shultis WA, Weil EJ, Looker HC, et al. Effect of periodontitis on overt nephropathy and end-stage renal disease in type 2 diabetes. Diabetes Care. 2007;30(2):306–311. doi: 10.2337/dc06-1184. [DOI] [PubMed] [Google Scholar]
- 28.Mealey BL, Rose LF. Diabetes mellitus and inflammatory periodontal diseases. Current Opinion in Endocrinology, Diabetes and Obesity. 2008;15(2):135–141. doi: 10.1097/MED.0b013e3282f824b7. [DOI] [PubMed] [Google Scholar]
- 29.Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ. Identification of periodontal pathogens in atheromatous plaques. Journal of Periodontology. 2000;71(10):1554–1560. doi: 10.1902/jop.2000.71.10.1554. [DOI] [PubMed] [Google Scholar]
- 30.Dorn BR, Dunn WA, Progulske-Fox A. Invasion of human coronary artery cells by periodontal pathogens. Infection and Immunity. 1999;67(11):5792–5798. doi: 10.1128/iai.67.11.5792-5798.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruma M, Boggess K, Moss K, et al. Maternal periodontal disease, systemic inflammation, and risk for preeclampsia. American Journal of Obstetrics and Gynecology. 2008;198(4):389–e1. doi: 10.1016/j.ajog.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 32.López NJ, Da Silva I, Ipinza J, Gutiérrez J. Periodontal therapy reduces the rate of preterm low birth weight in women with pregnancy-associated gingivitis. Journal of Periodontology. 2005;76(11):2144–2153. doi: 10.1902/jop.2005.76.11-S.2144. [DOI] [PubMed] [Google Scholar]
- 33.D’Aiuto F, Parkar M, Andreou G, et al. Periodontitis and systemic inflammation: control of the local infection is associated with a reduction in serum inflammatory markers. Journal of Dental Research. 2004;83(2):156–160. doi: 10.1177/154405910408300214. [DOI] [PubMed] [Google Scholar]
- 34.Radnai M, Pal A, Novak T, Urban E, Eller J, Gorzo I. Benefits of periodontal therapy when preterm birth threatens. Journal of Dental Research. 2009;88(3):280–284. doi: 10.1177/0022034508330229. [DOI] [PubMed] [Google Scholar]
- 35.Graham S, Dayal H, Rohrer T. Dentition, diet, tobacco, and alcohol in the epidemiology of oral cancer. Journal of the National Cancer Institute. 1977;59(6):1611–1618. doi: 10.1093/jnci/59.6.1611. [DOI] [PubMed] [Google Scholar]
- 36.Zheng T, Boyle P, Hu H, et al. Dentition, oral hygiene, and risk of oral cancer: a case-control study in Beijing, People’s Republic of China. Cancer Causes and Control. 1990;1(3):235–241. doi: 10.1007/BF00117475. [DOI] [PubMed] [Google Scholar]
- 37.Winn DM, Blot WJ, McLaughlin JK, et al. Mouthwash use and oral conditions in the risk of oral and pharyngeal cancer. Cancer Research. 1991;51(11):3044–3047. [PubMed] [Google Scholar]
- 38.Marshall JR, Graham S, Haughey BP, et al. Smoking, alcohol, dentition and diet in the epidemiology of oral cancer. European Journal of Cancer Part B. 1992;28(1):9–15. doi: 10.1016/0964-1955(92)90005-l. [DOI] [PubMed] [Google Scholar]
- 39.Bundgaard T, Wildt J, Frydenberg M, Elbrond O, Nielsen JE. Case-control study of squamous cell cancer of the oral cavity in Denmark. Cancer Causes and Control. 1995;6(1):57–67. doi: 10.1007/BF00051681. [DOI] [PubMed] [Google Scholar]
- 40.Velly AM, Franco EL, Schlecht N, et al. Relationship between dental factors and risk of upper aerodigestive tract cancer. Oral Oncology. 1998;34(4):284–291. [PubMed] [Google Scholar]
- 41.Moreno-López LA, Esparza-Gómez GC, González-Navarro A, Cerero-Lapiedra R, González-Hernández MJ, Domínguez-Rojas V. Risk of oral cancer associated with tobacco smoking, alcohol consumption and oral hygiene: a case-control study in Madrid, Spain. Oral Oncology. 2000;36(2):170–174. doi: 10.1016/s1368-8375(99)00084-6. [DOI] [PubMed] [Google Scholar]
- 42.Talamini R, Vaccarella S, Barbone F, et al. Oral hygiene, dentition, sexual habits and risk of oral cancer. British Journal of Cancer. 2000;83(9):1238–1242. doi: 10.1054/bjoc.2000.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balaram P, Sridhar H, Rajkumar T, et al. Oral cancer in Southern India: the influence of smoking, drinking, paan-chewing and oral hygiene. International Journal of Cancer. 2002;98(3):440–445. doi: 10.1002/ijc.10200. [DOI] [PubMed] [Google Scholar]
- 44.Lissowska J, Pilarska A, Pilarski P, et al. Smoking, alcohol, diet, dentition and sexual practices in the epidemiology of oral cancer in Poland. European Journal of Cancer Prevention. 2003;12(1):25–33. doi: 10.1097/00008469-200302000-00005. [DOI] [PubMed] [Google Scholar]
- 45.Rosenquist K, Wennerberg J, Schildt EB, Bladström A, Göran Hansson B, Andersson G. Oral status, oral infections and some lifestyle factors as risk factors for oral and oropharyngeal squamous cell carcinoma. A population-based case-control study in southern Sweden. Acta Oto-Laryngologica. 2005;125(12):1327–1336. doi: 10.1080/00016480510012273. [DOI] [PubMed] [Google Scholar]
- 46.Guha N, Boffetta P, Wünsch Filho V, et al. Oral health and risk of squamous cell carcinoma of the head and neck and esophagus: results of two multicentric case-control studies. American Journal of Epidemiology. 2007;166(10):1159–1173. doi: 10.1093/aje/kwm193. [DOI] [PubMed] [Google Scholar]
- 47.Tezal M, Grossi SG, Genco RJ. Is periodontitis associated with oral neoplasms? Journal of Periodontology. 2005;76(3):406–410. doi: 10.1902/jop.2005.76.3.406. [DOI] [PubMed] [Google Scholar]
- 48.Tezal M, Sullivan MA, Reid ME, et al. Chronic periodontitis and the risk of tongue cancer. Archives of Otolaryngology—Head and Neck Surgery. 2007;133(5):450–454. doi: 10.1001/archotol.133.5.450. [DOI] [PubMed] [Google Scholar]
- 49.Tezal M, Sullivan MA, Hyland A, et al. Chronic periodontitis and the incidence of head and neck squamous cell carcinoma. Cancer Epidemiology Biomarkers and Prevention. 2009;18(9):2406–2412. doi: 10.1158/1055-9965.EPI-09-0334. [DOI] [PubMed] [Google Scholar]
- 50.Jayaprakash V, Rigual NR, Moysich KB, et al. Chemoprevention of head and neck cancer with aspirin: a case-control study. Archives of Otolaryngology—Head and Neck Surgery. 2006;132(11):1231–1236. doi: 10.1001/archotol.132.11.1231. [DOI] [PubMed] [Google Scholar]
- 51.Ahmadi N, Goldman R, Seillier-Moiseiwitsch F, Noone AM, Kosti O, Davidson BJ. Decreased risk of squamous cell carcinoma of the head and neck in users of nonsteroidal anti-inflammatory drugs. International Journal of Otolaryngology. 2010;2010:8 pages. doi: 10.1155/2010/424161. Article ID 424161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lax AJ, Thomas W. How bacteria could cause cancer: one step at a time. Trends in Microbiology. 2002;10(6):293–299. doi: 10.1016/s0966-842x(02)02360-0. [DOI] [PubMed] [Google Scholar]
- 53.Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124(4):823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 54.Huycke MM, Gaskin HR. Commensal bacteria, redox stress, and colorectal cancer: mechanisms and models. Experimental Biology and Medicine. 2004;229(7):586–597. doi: 10.1177/153537020422900702. [DOI] [PubMed] [Google Scholar]
- 55.Homann N, Tillonen J, Rintamäki H, Salaspuro M, Lindqvist C, Meurman JH. Poor dental status increases acetaldehyde production from ethanol in saliva: a possible link to increased oral cancer risk among heavy drinkers. Oral Oncology. 2001;37(2):153–158. doi: 10.1016/s1368-8375(00)00076-2. [DOI] [PubMed] [Google Scholar]
- 56.Visapää JP, Götte K, Benesova M, et al. Increased cancer risk in heavy drinkers with the alcohol dehydrogenase 1C∗1 allele, possibly due to salivary acetaldehyde. Gut. 2004;53(6):871–876. doi: 10.1136/gut.2003.018994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salaspuro MP, Mezey E. Acetaldehyde, microbes, and cancer of the digestive tract. Critical Reviews in Clinical Laboratory Sciences. 2003;40(2):183–208. doi: 10.1080/713609333. [DOI] [PubMed] [Google Scholar]
- 58.Shapiro KB, Hotchkiss JH, Roe DA. Quantitative relationship between oral nitrate-reducing activity and the endogenous formation of N-nitrosoamino acids in humans. Food and Chemical Toxicology. 1991;29(11):751–755. doi: 10.1016/0278-6915(91)90183-8. [DOI] [PubMed] [Google Scholar]
- 59.Mirvish SS. Role of N-nitroso compounds (NOC) and N-nitrosation in etiology of gastric, esophageal, nasopharyngeal and bladder cancer and contribution to cancer of known exposures to NOC. Cancer Letters. 1995;93(1):17–48. doi: 10.1016/0304-3835(95)03786-V. [DOI] [PubMed] [Google Scholar]
- 60.Rüegg C. Leukocytes, inflammation, and angiogenesis in cancer: fatal attractions. Journal of Leukocyte Biology. 2006;80(4):682–684. doi: 10.1189/jlb.0606394. [DOI] [PubMed] [Google Scholar]
- 61.Asting AG, Carén H, Andersson M, Lönnroth C, Lagerstedt K, Lundholm K. COX-2 gene expression in colon cancer tissue related to regulating factors and promoter methylation status. BMC Cancer. 2011;11:p. 238. doi: 10.1186/1471-2407-11-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Annals of the Rheumatic Diseases. 2011;70(supplement 1):i104–i108. doi: 10.1136/ard.2010.140145. [DOI] [PubMed] [Google Scholar]
- 63.Tatemichi M, Ogura T, Esumi H. Impact of inducible nitric oxide synthase gene on tumor progression. European Journal of Cancer Prevention. 2009;18(1):1–8. doi: 10.1097/CEJ.0b013e3282f75f29. [DOI] [PubMed] [Google Scholar]
- 64.Allen C, Duffy S, Teknos T, et al. Nuclear factor-κB-related serum factors as longitudinal biomarkers of response and survival in advanced oropharyngeal carcinoma. Clinical Cancer Research. 2007;13(11):3182–3190. doi: 10.1158/1078-0432.CCR-06-3047. [DOI] [PubMed] [Google Scholar]
- 65.Greten FR, Eckmann L, Greten TF, et al. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118(3):285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 66.Wang D, Wang H, Shi Q, et al. Prostaglandin E2 promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor δ . Cancer Cell. 2004;6(3):285–295. doi: 10.1016/j.ccr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 67.Stetler-Stevenson WG. The tumor microenvironment: regulation by MMP-independent effects of tissue inhibitor of metalloproteinases-2. Cancer and Metastasis Reviews. 2008;27(1):57–66. doi: 10.1007/s10555-007-9105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leaner VD, Donninger H, Birrer MJ. Transcription factors as targets for cancer therapy: AP-1 a potential therapeutic target. Current Cancer Therapy Reviews. 2007;3(1):1–6. [Google Scholar]
- 69.Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6(5):447–458. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 70.Dauer DJ, Ferraro B, Song L, et al. Stat3 regulates genes common to both wound healing and cancer. Oncogene. 2005;24(21):3397–3408. doi: 10.1038/sj.onc.1208469. [DOI] [PubMed] [Google Scholar]
- 71.Cassatella MA, Huber V, Calzetti F, et al. Interferon-activated neutrophils store a TNF-related apoptosis-inducing ligand (TRAIL/Apo-2 ligand) intracellular pool that is readily mobilizable following exposure to proinflammatory mediators. Journal of Leukocyte Biology. 2006;79(1):123–132. doi: 10.1189/jlb.0805431. [DOI] [PubMed] [Google Scholar]
- 72.Williams DA. Inflammatory cytokines and mucosal injury. Journal of the National Cancer Institute. Monographs. 2001;(29):26–30. doi: 10.1093/oxfordjournals.jncimonographs.a003435. [DOI] [PubMed] [Google Scholar]
- 73.Pöllänen MT, Salonen JI, Uitto VJ. Structure and function of the tooth-epithelial interface in health and disease. Periodontology 2000. 2003;31:12–31. doi: 10.1034/j.1600-0757.2003.03102.x. [DOI] [PubMed] [Google Scholar]
- 74.Huh WK. Human papillomavirus infection a concise review of natural history. Obstetrics and Gynecology. 2009;114(1):139–143. doi: 10.1097/AOG.0b013e3181ab6878. [DOI] [PubMed] [Google Scholar]
- 75.Stubenrauch F, Laimins LA. Human papillomavirus life cycle: active and latent phases. Seminars in Cancer Biology. 1999;9(6):379–386. doi: 10.1006/scbi.1999.0141. [DOI] [PubMed] [Google Scholar]
- 76.Zhang ZY, Sdek P, Cao J, Chen WT. Human papillomavirus type 16 and 18 DNA in oral squamous cell carcinoma and normal mucosa. International Journal of Oral and Maxillofacial Surgery. 2004;33(1):71–74. doi: 10.1054/ijom.2002.0443. [DOI] [PubMed] [Google Scholar]
- 77.Ha PK, Califano JA. The role of human papillomavirus in oral carcinogenesis. Critical Reviews in Oral Biology and Medicine. 2004;15(4):188–196. doi: 10.1177/154411130401500402. [DOI] [PubMed] [Google Scholar]
- 78.Kreimer AR, Clifford GM, Boyle P, Franceschi S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systemic review. Cancer Epidemiology Biomarkers and Prevention. 2005;14(2):467–475. doi: 10.1158/1055-9965.EPI-04-0551. [DOI] [PubMed] [Google Scholar]
- 79.Miller CS, Johnstone BM. Human papillomavirus as a risk factor for oral squamous cell carcinoma: a meta-analysis, 1982–1997. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics. 2001;91(6):622–635. doi: 10.1067/moe.2001.115392. [DOI] [PubMed] [Google Scholar]
- 80.Gillison ML, D’Souza G, Westra W, et al. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. Journal of the National Cancer Institute. 2008;100(6):407–420. doi: 10.1093/jnci/djn025. [DOI] [PubMed] [Google Scholar]
- 81.Klussmann JP, Weissenborn SJ, Wieland U, et al. Human papillomavirus-positive tonsillar carcinomas: a different tumor entity? Medical Microbiology and Immunology. 2003;192(3):129–132. doi: 10.1007/s00430-002-0126-1. [DOI] [PubMed] [Google Scholar]
- 82.Charfi L, Jouffroy T, Cremoux PD, et al. Two types of squamous cell carcinoma of the palatine tonsil characterized by distinct etiology, molecular features and outcome. Cancer Letters. 2008;260(1-2):72–78. doi: 10.1016/j.canlet.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 83.Ragin C, Edwards R, Larkins-Pettigrew M, et al. Oral HPV infection and sexuality: a cross-sectional study in women. International Journal of Molecular Sciences. 2011;12(6):3928–3940. doi: 10.3390/ijms12063928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schwartz SM, Daling JR, Doody DR, et al. Oral cancer risk in relation to sexual history and evidence of human papillomavirus infection. Journal of the National Cancer Institute. 1998;90(21):1626–1636. doi: 10.1093/jnci/90.21.1626. [DOI] [PubMed] [Google Scholar]
- 85.Kreimer AR, Alberg AJ, Daniel R, et al. Oral Human Papillomavirus Infection in Adults Is Associated with Sexual Behavior and HIV Serostatus. Journal of Infectious Diseases. 2004;189(4):686–698. doi: 10.1086/381504. [DOI] [PubMed] [Google Scholar]
- 86.Yoshida K, Furumoto H, Abe A, et al. The possibility of vertical transmission of human papillomavirus through maternal milk. Journal of Obstetrics and Gynaecology. 2011;31(6):503–506. doi: 10.3109/01443615.2011.570814. [DOI] [PubMed] [Google Scholar]
- 87.Castle PE, Hillier SL, Rabe LK, et al. An association of cervical inflammation with high-grade cervical neoplasia in women infected with oncogenic human papillomavirus (HPV) Cancer Epidemiology Biomarkers and Prevention. 2001;10(10):1021–1027. [PubMed] [Google Scholar]
- 88.Altuglu I, Terek MC, Ozacar T, Ozsaran AA, Bilgiç A. The prevalence of human papilloma virus DNA in women with mucopurulent endocervicitis. European Journal of Gynaecological Oncology. 2002;23(2):166–168. [PubMed] [Google Scholar]
- 89.Prokopakis P, Sourvinos G, Koumantaki Y, Koumantakis E, Spandidos DA. K-ras mutations and HPV infection in cervicitis and intraepithelial neoplasias of the cervix. Oncology Reports. 2002;9(1):129–133. [PubMed] [Google Scholar]
- 90.Parashari A, Singh V, Gupta MM, et al. Significance of inflammatory cervical smears. APMIS : Acta Pathologica, Microbiologica, et Immunologica Scandinavica. 1995;103(1–6):273–278. doi: 10.1111/j.1699-0463.1995.tb01106.x. [DOI] [PubMed] [Google Scholar]
- 91.Gaiotti D, Chung J, Iglesias M, et al. Tumor necrosis factor-α promotes human papillomavirus (HPV) E6/E7 RNA expression and cyclin-dependent kinase activity in HPV-immortalized keratinocytes by a ras-dependent pathway. Molecular Carcinogenesis. 2000;27(2):97–109. doi: 10.1002/(sici)1098-2744(200002)27:2<97::aid-mc5>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 92.Woodworth CD, Simpson S. Comparative lymphokine secretion by cultured normal human cervical keratinocytes, papillomavirus-immortalized, and carcinoma cell lines. American Journal of Pathology. 1993;142(5):1544–1555. [PMC free article] [PubMed] [Google Scholar]
- 93.Woodworth CD, Notario V, DiPaolo JA. Transforming growth factors beta 1 and 2 transcriptionally regulate human papillomavirus (HPV) type 16 early gene expression in HPV-immortalized human genital epithelial cells. Journal of Virology. 1990;64(10):4767–4775. doi: 10.1128/jvi.64.10.4767-4775.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Woodworth CD, Lichti U, Simpson S, Evans CH, DiPaolo JA. Leukoregulin and γ-interferon inhibit human papillomavirus type 16 gene transcription in human papillomavirus-immortalized human cervical cells. Cancer Research. 1992;52(2):456–463. [PubMed] [Google Scholar]
- 95.Woodworth CD, McMullin E, Iglesias M, Plowman GD. Interleukin 1α and tumor necrosis factor α stimulate autocrine amphiregulin expression and proliferation of human papillomavirus- immortalized and carcinoma-derived cervical epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(7):2840–2844. doi: 10.1073/pnas.92.7.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Iglesias M, Plowman GD, Woodworth CD. Interleukin-6 and interleukin-6 soluble receptor regulate proliferation of normal, human papillomavirus-immortalized, and carcinoma-derived cervical cells in vitro. American Journal of Pathology. 1995;146(4):944–952. [PMC free article] [PubMed] [Google Scholar]
- 97.Karl T, Seibert N, Stöhr M, Osswald H, Rösl F, Finzer P. Sulindac induces specific degradation of the HPV oncoprotein E7 and causes growth arrest and apoptosis in cervical carcinoma cells. Cancer Letters. 2007;245(1-2):103–111. doi: 10.1016/j.canlet.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 98.Rampias T, Sasaki C, Weinberger P, Psyrri A. E6 and E7 gene silencing and transformed phenotype of human papillomavirus 16-positive oropharyngeal cancer cells. Journal of the National Cancer Institute. 2009;101(6):412–423. doi: 10.1093/jnci/djp017. [DOI] [PubMed] [Google Scholar]
- 99.Tezal M, Nasca MS, Stoler DL, et al. Chronic periodontitis-human papillomavirus synergy in base of tongue cancers. Archives of Otolaryngology—Head and Neck Surgery. 2009;135(4):391–396. doi: 10.1001/archoto.2009.6. [DOI] [PubMed] [Google Scholar]
- 100.Khan SA, Wingard JR. Infection and mucosal injury in cancer treatment. Journal of the National Cancer Institute. Monographs. 2001;(29):31–36. doi: 10.1093/oxfordjournals.jncimonographs.a003437. [DOI] [PubMed] [Google Scholar]
- 101.Samoff E, Koumans EH, Markowitz LE, et al. Association of Chlamydia trachomatis with persistence of high-risk types of human papillomavirus in a cohort of female adolescents. American Journal of Epidemiology. 2005;162(7):668–675. doi: 10.1093/aje/kwi262. [DOI] [PubMed] [Google Scholar]
- 102.Madeleine MM, Anttila T, Schwartz SM, et al. Risk of cervical cancer associated with Chlamydia trachomatis antibodies by histology, HPV type and HPV cofactors. International Journal of Cancer. 2007;120(3):650–655. doi: 10.1002/ijc.22325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rudney JD, Chen R, Sedgewick GJ. Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis, and Tannerella forsythensis are components of a polymicrobial intracellular flora within human buccal cells. Journal of Dental Research. 2005;84(1):59–63. doi: 10.1177/154405910508400110. [DOI] [PubMed] [Google Scholar]
- 104.Yilmaz O. The chronicles of porphyromonas gingivalis: the microbium, the human oral epithelium and their interplay. Microbiology. 2008;154(10):2897–2903. doi: 10.1099/mic.0.2008/021220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Scannapieco FA, Wang B, Shiau HJ. Oral bacteria and respiratory infection: effects on respiratory pathogen adhesion and epithelial cell proinflammatory cytokine production. Annals of Periodontology. 2001;6(1):78–86. doi: 10.1902/annals.2001.6.1.78. [DOI] [PubMed] [Google Scholar]
- 106.Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ. Identification of periodontal pathogens in atheromatous plaques. Journal of Periodontology. 2000;71(10):1554–1560. doi: 10.1902/jop.2000.71.10.1554. [DOI] [PubMed] [Google Scholar]
- 107.Dorn BR, Dunn WA, Progulske-Fox A. Invasion of human coronary artery cells by periodontal pathogens. Infection and Immunity. 1999;67(11):5792–5798. doi: 10.1128/iai.67.11.5792-5798.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ruma M, Boggess K, Moss K, et al. Maternal periodontal disease, systemic inflammation, and risk for preeclampsia. American Journal of Obstetrics and Gynecology. 2008;198(4):389–e1. doi: 10.1016/j.ajog.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 109.Iacopino AM. Periodontitis and diabetes interrelationships: role of inflammation. Annals of Periodontology. 2001;6(1):125–137. doi: 10.1902/annals.2001.6.1.125. [DOI] [PubMed] [Google Scholar]
- 110.Reznick AZ, Hershkovich O, Nagler RM. Saliva-a pivotal player in the pathogenesis of oropharyngeal cancer. British Journal of Cancer. 2004;91(1):111–118. doi: 10.1038/sj.bjc.6601869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cawson RA. Cawson’s Essentials of Oral Pathology and Oral Medicine. 8th edition. New York, NY, USA: Elsevier Limited; 2008. Oral cancer; p. 280. [Google Scholar]
- 112.Hormia M, Wilberg J, Ruokonen H, Syrjanen S. Marginal periodontium as a potential reservoir of human pappilomavirus in oral mucosa. Journal of Periodontology. 2005;76(3):358–363. doi: 10.1902/jop.2005.76.3.358. [DOI] [PubMed] [Google Scholar]
- 113.Socransky SS, Haffajee AD. Dental biofilms: difficult therapeutic targets. Periodontology 2000. 2002;28(1):12–55. doi: 10.1034/j.1600-0757.2002.280102.x. [DOI] [PubMed] [Google Scholar]