

Abstract
The intimate connection between coagulation and inflammation in the pathogenesis of vascular disease has moved more and more into focus of clinical research. This paper focuses on the essential components of this interplay in the settings of cardiovascular disease and acute coronary syndrome. Tissue factor, the main initiator of the extrinsic coagulation pathway, plays a central role via causing a proinflammatory response through activation of coagulation factors and thereby initiating coagulation and downstream cellular signalling pathways. Regarding activated clotting factors II, X, and VII, protease-activated receptors provide the molecular link between coagulation and inflammation. Hereby, PAR-1 displays deleterious as well as beneficial properties. Unravelling these interrelations may help developing new strategies to ameliorate the detrimental reciprocal aggravation of inflammation and coagulation.
1. Introduction
Systemic and local proinflammatory changes are in focus when investigating the pathophysiology of arteriosclerosis and acute coronary syndromes. In acute myocardial infarction (AMI), proinflammatory markers such as C-reactive protein (CRP), interleukins, or monocyte-chemoattractant protein (MCP)-1 are elevated [1–3] and their increase is of prognostic relevance for future cardiovascular events [4–6] and mortality [7–9]. Moreover, in healthy persons elevated proinflammatory markers are associated with an increase in cardiovascular risk [10–12]. Patients with increased circulating proinflammatory markers in AMI present with decreased myocardial salvage after coronary reperfusion therapy [13]. Similarly, in experimental studies, high levels of CRP deteriorate infarct size [14].
Sources of inflammatory response are vascular cells such as activated endothelial cells, which release proinflammatory cytokines such as interleukin (IL)-8 [15]. IL-8 is a CXC cytokine that acts as a chemoattractant and agonist for neutrophils, lymphocytes, and monocytes and is found in macrophage-rich atherosclerotic plaques [16]. Under flow conditions, IL-8 facilitates the arrest of monocytes on endothelium [16], which is necessary for migration into the intima in evolution of arteriosclerosis. Reperfusion injury after AMI as well as systemic inflammatory response syndrome can be associated to increased levels of IL-8 [17]. In experimental setting, murine IL-8 receptor knock-out mice display smaller arteriosclerotic lesions with less macrophages [18]. Apart from their contribution to arteriosclerosis, CXC cytokines are also produced by malignant cells and can promote tumor progression of a large variety of malignancies [19].
Besides IL-8, many other cytokines such as IL-6 take part in inflammatory responses by inducing B-cell differentiation, T-cell activation, and synthesis of acute phase proteins [20], but also contributing to proliferation of vascular smooth muscle cells (SMCs) [21]. Moreover TH1 activation was observed in acute coronary syndromes [22].
The pathogenesis of proinflammatory changes in acute coronary syndromes as well as the interplay between coagulation and inflammation is poorly understood and is subject to intense research. The mechanisms by which the coagulation system is altered by inflammatory interactions comprise enhanced synthesis and activation of coagulant proteins, decreased synthesis of anticoagulants, and suppression of fibrinolysis [23]. However, not only inflammation activates coagulation but coagulation in turn perpetuates inflammatory response [24]. Accordingly, increased levels of prothrombin fragment F1+2, fibrinopeptide A and D-Dimer, reflecting activation of the coagulation cascade, are also associated with an unfavorable outcome in acute coronary syndromes [25–27].
In this paper we focus on possible mechanisms of the interplay between coagulation and inflammation in acute coronary syndromes.
2. Tissue Factor
A strict separation between the intrinsic and extrinsic coagulation cascade surely fails to reflect physiologic conditions. Nevertheless, in the setting of arteriosclerosis and acute coronary syndromes the extrinsic pathway of coagulation is of particular significance.
Tissue Factor (TF) is the most important initiator of the extrinsic coagulation cascade. TF, a 47 kDa transmembrane glycoprotein and member of the class II cytokine receptor family, is the cofactor for the activated plasma clotting factor VII (FVIIa). The TF-FVIIa complex catalyzes the activation of factor X and IX, which leads to the generation of thrombin and thus finally of a fibrin clot. Under physiologic conditions TF is abundantly expressed only in the adventitia and is induced by several inflammatory mediators such as IL-6, IL-8, and MCP-1 [28, 29]. After vascular injury, TF is rapidly augmented in SMC of the media and accumulates in the SMC of the developing neointima [30]. Consequently, TF is highly expressed within atherosclerotic lesions and displays high procoagulant activity suggesting a role in determining plaque thrombogenicity [30]. In atherosclerotic carotid lesions disruption of plaques exposes TF-positive cells within the plaque to plasma clotting factors and initiates local thrombosis with subsequent occlusion of the vessel [31].
Furthermore, increased TF expression can be noticed on circulating monocytes and microparticles in acute coronary syndromes and may, thereby, contribute to activation of coagulation [32–34]. A soluble form of TF within the circulating blood may also support coronary thrombosis [35]. It has been shown that cytokines can induce expression of soluble TF [36], which on the other hand has been shown to accumulate in developing thrombi [37]. However, the clinical significance and individual contributions of microparticle-derived and soluble TF remain a matter of debate. Several studies have demonstrated increased levels of circulating TF in patients with unstable Angina pectoris (uAP) and acute myocardial infarction (AMI) [32, 38–45]. Therefore, it has long been speculated that, in cases with no plaque rupture or only fractional superficial erosion, thrombus formation may mainly depend on circulating levels of TF. Consistent with this idea, several studies suggest that the levels of circulating TF and other haemostatic biomarkers may correlate to adverse cardiovascular events and mortality in patients with acute coronary syndrome [46–48].
Stimulation of the TF-thrombin pathway does not only occur at the site of the plaque but also within the ischemic myocardium where activated coagulation factors may enhance inflammatory responses and increase infarct size [49]. TF contributes to inflammation, cell migration, and remodelling after vascular injury [50]. Furthermore, TF expression has been reported in a number of cancers, such as glioma, pancreatic cancer, non-small-cell lung cancer, colorectal cancer, ovarian cancer, prostate cancer, hepatocellular cancer, and breast cancer [51]. TF expression in tumors not only correlates with the incidence of thrombosis [52] but also promotes metastasis [53], tumor progression, and tumor angiogenesis [54].
TF-mediated intracellular signal transduction has not been completely elucidated so far. On one hand TF allows docking and activation of FVII and, therefore, promotes the generation of downstream coagulation factors and activation of protease-activated receptors (PARs) which themselves possibly induce intracellular signal transduction. On the other hand there is evidence for direct signalling through the cytoplasmic domain of TF following TF-FVIIa complex formation [55, 56].
3. Tissue Factor Pathway Inhibitor
The endogenous Kunitz-type inhibitor Tissue Factor Pathway Inhibitor-1 (TFPI) inhibits initiation of TF-induced blood coagulation and is mainly expressed on vascular endothelial cells. TFPI binds and inactivates FXa. The TFPI-FXa complex then binds and inactivates FVIIa. Increased levels of the TFPI-FXa complex may reflect both increased FXa generation and increased TFPI concentrations [57]. In addition to the full length TFPI most of the plasma TFPI circulates in truncated forms that are bound to plasma lipoproteins. These truncated forms lack their C-terminal domains and exhibit reduced affinity for vascular wall proteolysis. Additionally, it has been shown that endogenous proteases [58] and elastase released by neutrophils degrade TFPI, resulting in enhanced local coagulation that contributes to prevent pathogen dissemination during infection [59]. Conversely, infusion of a mutant TFPI protein resistant to proteolysis by elastase strongly impaired host defence against systemic infection.
4. Protease-Activated Receptors
Important players in the interaction between coagulation and inflammation are protease-activated receptors (PARs). PARs are G-protein coupled receptors that mediate various cellular reactions as cytokine release, expression of adhesion molecules, cell migration, or proliferation. Unlike other receptors, PARs are not activated by a soluble, external ligand. Proteases, such as activated coagulation factors, detach a defined part of the NH2-terminal chain of the receptor, thereby inducing a conformational change of the receptor. This change causes a self-activation by a “tethered ligand.” This activating sequence comprises only few amino acids. PARs can also be activated by synthetic peptides consisting of the sequence of amino acids representing the tethered ligand.
In contrast to other receptors, the activation of PARs by enzymatic cleavage is irreversible. After proteolytic activation the receptor must be internalized, degraded, and resynthesized. PARs are mainly expressed in vascular cells, but also in many different other cell types such as gastrointestinal and bronchial epithelial cells. Four different PARs are known: PAR-1, -3, and -4 show responsible for thrombin signaling whereas PAR-2 is activated by trypsin-like serine proteases, FVIIa, and matriptase but not by thrombin. PAR-1 and PAR-2 are expressed on smooth muscle cells and endothelial cells, whereas mainly PAR-1 is expressed on monocytes.
PAR-1 agonists or thrombin induce IL-8 and IL-6 in SMC, EC, and mononuclear cells (MNCs) [60] therefore emphasizing the role of PAR-1 in inflammatory processes in vascular cells and confirming data about PAR-1-mediated cytokine release in EC and monocytes [61]. In smooth muscle cells PAR-1 and PAR-2 agonists induce cytokine release to a similar extent which underlines the relevance of both PARs [60]. In addition to coagulation factors other serine proteases, for example, matriptase secreted by monocytes stimulate proinflammatory cytokine release in endothelial cells via PAR-2 activation [62]. Increased PAR-2 expression in atherosclerotic lesions suggests a role for this proinflammatory pathway (Figure 1) [63].
Figure 1.
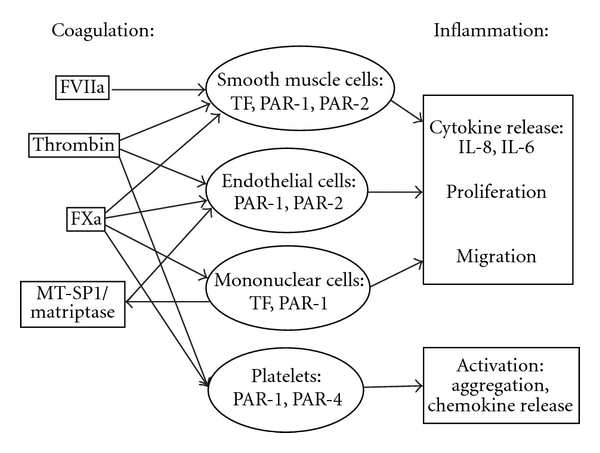
In addition, PARs can also be cleaved downstream of the tethered ligand, resulting in receptor inactivation by preventing further proteolytic activation [64]. PAR-1 signalling not only induces inflammatory responses but also causes antiapoptotic and vasculoprotective reactions [65]. Since the anticoagulant protease-activated protein C can activate PAR1 when in complex with the endothelial cell protein C receptor (EPCR), which may account for much of the protective effects conferred by activated protein C (APC) in severe sepsis [66]. Different contributions of these two pathways may prevail. First, APC acts via PAR-1 when attached to EPCR [65], resulting in cellular responses distinct from thrombin signalling [64] by a mechanism dependent in trans-activation of the sphingosine 1 phosphate receptor 1. In mouse models with strongly reduced EPCR expression or PAR-1 deficiency, the loss of EPCR/APC signalling via PAR-1 resulted in increased endotoxemia-induced lethality [67]. Concordantly, APC mutants have been shown to contribute to protective effects during sepsis by pathways independent from anticoagulant properties [67, 68]. The second pathway described is independent from EPCR. In this case, the availability of the integrin CD11b/CD18 has been shown to be crucial for PAR-1 mediated APC signaling on macrophages, thereby exhibiting anti-inflammatory effects and reducing endotoxin-induced lethality [69]. Thus the strength of PAR1 and PAR2 activation by thrombin, factor Xa, and activated protein C can either promote or protect against changes in vascular permeability depending on the status of the endothelium.
Platelet activation with subsequent thrombus generation plays a major role in the development of acute coronary syndromes. At low concentrations thrombin activates PAR-1 on platelets through a hirudin-like site and at high concentrations additional PAR-4. This induces shape change, P-selectin, and CD40L mobilization to the platelet membrane and promotes the release of platelet agonists ADP, thromboxane A2, chemokines, and growth factors [70] and, thereby, enhances proinflammatory changes. Thus, inhibition of PARs by thrombin of FXa inhibitors may prove beneficial in reducing not only thrombotic but also proinflammatory responses.
5. FXa
Binding of the serine protease FVII to TF results in generation of the coagulation protease FXa (FXa) and subsequently thrombin both known to induce cell signaling. FXa shows dose-dependent induction of intracellular calcium transients in endothelial cells that is active-site-dependent, and independent of thrombin [71]. Potential pathophysiological responses to FXa include stimulation of proliferation, production of proinflammatory cytokines, and prothrombotic TF [72].
Elevated TFPI-FXa and prothrombin fragments F1+2 plasma levels indicate activation of the coagulation cascade in acute coronary syndromes. Under physiological conditions an inverse relationship between TFPI-FXa and F1+2 suggests that TFPI-FXa regulates prothrombinase activity in vivo [73]. Under conditions associated with activation of the coagulation cascade, however, increased TFPI-Xa plasma levels occur [57, 74]. Activation of coagulation as measured by TFPI-FXa but not by F1+2 is associated with plasma concentrations of the proinflammatory cytokine IL-8 in acute coronary syndromes [60]. Furthermore, subsequent elevated IL-6 levels in the course of acute coronary syndromes are associated with initial TFPI-FXa concentrations [60]. These results argue for a proinflammatory role of FXa in acute coronary syndromes that is independent of thrombin. Although thrombin provokes similar proinflammatory effects as FXa in vitro the effects of thrombin may be diminished after heparin treatment in vivo. Several trials of unfractionated heparin (UFH) [75], direct thrombin inhibitors [76], and enoxaparin [77] have thus far failed to demonstrate mortality reductions in acute coronary syndromes. Yet, the OASIS-6 trial suggests a reduction in reinfarction and mortality without excess bleeding in patients not undergoing PCI [78]. Therapeutic inhibition of the proinflammatory effects of Factor Xa may, therefore, prove additional benefits as compared to thrombin inhibition in the clinical course in acute coronary syndromes. This is currently investigated in the ATLAS-ACS 2 TIMI 51 trial that is testing the hypothesis that anticoagulation with the oral factor Xa inhibitor rivaroxaban reduces cardiovascular death, MI, and stroke among patients with ACS treated with guideline-based therapies for ACS [79].
In vitro experiments revealed that FXa stimulates IL-8 and MCP-1 transcription in endothelial cells and mononuclear leukocytes [60]. Genetic studies and receptor desensitization experiments indicate that signaling by FXa is mediated by PAR-1 and PAR-2 [80, 81].
According to the expression of PAR-1 and PAR-2, PAR-1 and PAR-2 agonists induce IL-8 and MCP-1 release in endothelial cells, whereas, only PAR-1 agonists stimulated cytokine release in mononuclear cells [60].
6. FVIIa
Several studies suggest a signaling mechanism of the TF-FVIIa complex via PAR-2 [82]. In this model, TF-bound FVIIa proteolytically activates PAR-2 and, to a lesser extent, PAR-1, and thereby evokes intracellular signaling cascades [83].
In contrast to FXa, FVIIa does not elicit a proinflammatory response in endothelial cells or mononuclear cells [82]. The TF-FVIIa-PAR-2 signalling was only observed in SMC [82] since they express both TF and PAR-2 and both of them seem to be a prerequisite for FVIIa action. EC expressing PAR-2 but lacking TF will not permit FVIIa docking, whereas MNC displaying TF but only low PAR-2 expression, probably allow FVIIa binding but do not express sufficient PAR-2 molecules being subsequently activated. However, PAR-2 and TF are induced by cytokine stimulation [28, 84]. Thus, FVIIa may still have an important impact in atherosclerotic vessels [31] and acute coronary syndromes [29].
In recent studies [85], it has been demonstrated that FVII is synthesized by different cancer cells (liver, ovary, prostate, lung, gastric, thyroid, and breast). Considering that these tumor cells also synthesize TF it is conceivable that supraphysiologic concentrations of FVIIa after binding of FVII to TF occur. On tumor cells, the TF-FVIIa binary complex mediates activation of PAR-2 [86]. Therefore TF-FVIIa-PAR-2 interaction with subsequent cytokine release may be relevant within a tumor environment. TF/FVIIa/PAR2 signalling has been shown to promote proliferation and metastasis of tumor cells [87, 88]. Consistently, TF/FVIIa-specific upregulation of IL-8 expression in breast cancer cells has been shown to be mediated by PAR-2 and to increase cell migration [83]. Whether TF-FVIIa-PAR-2 interaction may also contribute to local thrombus formation and progression of atherosclerotic disease remains to be elucidated.
7. Conclusion
The interplay between coagulation and inflammation is a matter of intense research. Proinflammatory changes in acute coronary syndromes may substantially influence prognosis [6]. Experimental evidence suggests that this interplay may contribute to the development of vascular remodeling or support plaque disruption of the artery. However transfer of these data to the clinical settings remains controversial since additional optimized medical and interventional treatments interfere. Therefore, understanding the causes of inflammation facilitate the development of new therapeutic strategies. To analyse whether these new therapies translate to improved clinical outcome needs to be studied in appropriate clinical trials.
While inhibiting proinflammatory cytokines such as Tumor-Necrosis Factor-α (TNF) has been shown to effectively improve survival in several animal models of sepsis [89–91], anti-TNF therapy in septic humans failed to ameliorate or even worsened clinical outcome [92–95]. In chronic inflammatory diseases however, anti-inflammatory treatment has become clinical routine. For example, inhibition of IL-6 by the first anti-IL-6 antibody, tocilizumab, has been shown to completely block TF-dependent thrombin generation in experimental endotoxemia [23, 96], and tocilizumab will be of special future interest as it has been approved for rheumatoid arthritis.
Inhibiting coagulation could depict a more promising mechanism in fighting overwhelming inflammatory response. Experimental studies that have shown that anticoagulant treatment not only diminishes activation of coagulation but also inhibits inflammation, underline the cross-talk between activation of coagulation and cytokine release in vivo [49, 97, 98]. In acute inflammatory disorders such as severe sepsis, administration of recombinant APC significantly improves survival and long-term outcome. In future, APC mutants that lack anticoagulant properties but still enable sphingosine 1 phosphate receptor 1 dependent activation of PAR-1 will be of special clinical interest as they have been shown to reduce sepsis-induced in mice but do not predispose to bleeding complications.
So far, anti-inflammatory treatments displayed no explicit benefit in patients with acute coronary syndromes [99]. However, there is evidence that FXa inhibitors prove to be superior to thrombin inhibitors [100]. Particularly, treatment with low molecular weight heparins, that include additional anti-FXa activity as compared to unfractionated heparin, has been shown to decrease inflammatory changes in vitro and in vivo [101, 102].
Yet the question remains if and what anticoagulant therapies will prove beneficial to alter systemic inflammatory responses.
References
- 1.Aukrust P, Berge RK, Ueland T, et al. Interaction between chemokines and oxidative stress: possible pathogenic role in acute coronary syndromes. Journal of the American College of Cardiology. 2001;37(2):485–491. doi: 10.1016/s0735-1097(00)01110-4. [DOI] [PubMed] [Google Scholar]
- 2.Biasucci LM, Vitelli A, Liuzzo G, et al. Elevated levels of interleukin-6 in unstable angina. Circulation. 1996;94(5):874–877. doi: 10.1161/01.cir.94.5.874. [DOI] [PubMed] [Google Scholar]
- 3.Miyao Y, Yasue H, Ogawa H, et al. Elevated plasma interleukin-6 levels in patients with acute myocardial infarction. American Heart Journal. 1993;126(6):1299–1304. doi: 10.1016/0002-8703(93)90526-f. [DOI] [PubMed] [Google Scholar]
- 4.Haverkate F, Thompson SG, Pyke SD, Gallimore JR, Pepys MB, European Concerted Action on Thrombosis and Disabilities Angina Pectoris Study Group Production of C-reactive protein and risk of coronary events in stable and unstable angina. The Lancet. 1997;349(9050):462–466. doi: 10.1016/s0140-6736(96)07591-5. [DOI] [PubMed] [Google Scholar]
- 5.Lagrand WK, Visser CA, Hermens WT, et al. C-reactive protein as a cardiovascular risk factor more than an epiphenomenon? Circulation. 1999;100(1):96–102. doi: 10.1161/01.cir.100.1.96. [DOI] [PubMed] [Google Scholar]
- 6.Liuzzo G, Biasucci LM, Gallimore JR, et al. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. New England Journal of Medicine. 1994;331(7):417–424. doi: 10.1056/NEJM199408183310701. [DOI] [PubMed] [Google Scholar]
- 7.Lindahl B, Toss H, Siegbahn A, Venge P, Wallentin L. Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease. New England Journal of Medicine. 2000;343(16):1139–1147. doi: 10.1056/NEJM200010193431602. [DOI] [PubMed] [Google Scholar]
- 8.Lindmark E, Diderholm E, Wallentin L, Siegbahn A. Relationship between interleukin 6 and mortality in patients with unstable coronary artery disease: effects of an early invasive or noninvasive strategy. Journal of the American Medical Association. 2001;286(17):2107–2113. doi: 10.1001/jama.286.17.2107. [DOI] [PubMed] [Google Scholar]
- 9.Morrow DA, Rifai N, Antman EM, et al. C-reactive protein is a potent predictor of mortality independently of and in combination with troponin T in acute coronary syndromes: a TIMI 11A substudy. Journal of the American College of Cardiology. 1998;31(7):1460–1465. doi: 10.1016/s0735-1097(98)00136-3. [DOI] [PubMed] [Google Scholar]
- 10.Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. New England Journal of Medicine. 1997;336(14):973–979. doi: 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- 11.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. New England Journal of Medicine. 2000;342(12):836–843. doi: 10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- 12.Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101(15):1767–1772. doi: 10.1161/01.cir.101.15.1767. [DOI] [PubMed] [Google Scholar]
- 13.Dibra A, Mehilli J, Schwaiger M, et al. Predictive value of basal C-reactive protein levels for myocardial salvage in patients with acute myocardial infarction is dependent on the type of reperfusion treatment. European Heart Journal. 2003;24(12):1128–1133. doi: 10.1016/s0195-668x(03)00200-8. [DOI] [PubMed] [Google Scholar]
- 14.Barrett TD, Hennan JK, Marks RM, Lucchesi BR. C-reactive-protein-associated increase in myocardial infarct size after ischemia/reperfusion. Journal of Pharmacology and Experimental Therapeutics. 2002;303(3):1007–1013. doi: 10.1124/jpet.102.040600. [DOI] [PubMed] [Google Scholar]
- 15.Yamasaki Y, Matsuo Y, Matsuura N, et al. Transient increase of cytokine-induced neutrophil chemoattractant, a member of the interleukin-8 family, in ischemic brain areas after focal ischemia in rats. Stroke. 1995;26(2):318–323. doi: 10.1161/01.str.26.2.318. [DOI] [PubMed] [Google Scholar]
- 16.Gerszten RE, Garcia-Zepeda EA, Lim YC, et al. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 1999;398(6729):718–723. doi: 10.1038/19546. [DOI] [PubMed] [Google Scholar]
- 17.Dinarello CA, Gelfand JA, Wolff SM. Anticytokine strategies in the treatment of the systemic inflammatory response syndrome. Journal of the American Medical Association. 1993;269(14):1829–1835. [PubMed] [Google Scholar]
- 18.Boisvert WA, Santiago R, Curtiss LK, Terkeltaub RA. A leukocyte homologue of the IL-8 receptor CXCR-2 mediates the accumulation of macrophages in atherosclerotic lesions of LDL receptor- deficient mice. Journal of Clinical Investigation. 1998;101(2):353–363. doi: 10.1172/JCI1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leek RD, Landers R, Fox SB, Ng F, Harris AL, Lewis CE. Association of tumour necrosis factor alpha and its receptors with thymidine phosphorylase expression in invasive breast carcinoma. British Journal of Cancer. 1998;77(12):2246–2251. doi: 10.1038/bjc.1998.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yudkin JS, Kumari M, Humphries SE, Mohamed-Ali V. Inflammation, obesity, stress and coronary heart disease: is interleukin-6 the link? Atherosclerosis. 2000;148(2):209–214. doi: 10.1016/s0021-9150(99)00463-3. [DOI] [PubMed] [Google Scholar]
- 21.Ikeda U, Ikeda M, Oohara T, et al. Interleukin 6 stimulates growth of vascular smooth muscle cells in a PDGF-dependent manner. American Journal of Physiology—Heart and Circulatory Physiology. 1991;260(5, part 2):H1713–H1717. doi: 10.1152/ajpheart.1991.260.5.H1713. [DOI] [PubMed] [Google Scholar]
- 22.Steppich BA, Moog P, Matissek C, et al. Cytokine profiles and T cell function in acute coronary syndromes. Atherosclerosis. 2007;190(2):443–451. doi: 10.1016/j.atherosclerosis.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 23.Lipinski S, Bremer L, Lammers T, Thieme F, Schreiber S, Rosenstiel P. Coagulation and inflammation. Molecular insights and diagnostic implications. Hamostaseologie. 2011;31(2):94–104. doi: 10.5482/ha-1134. [DOI] [PubMed] [Google Scholar]
- 24.Ott I. Soluble tissue factor emerges from inflammation. Circulation Research. 2005;96(12):1217–1218. doi: 10.1161/01.RES.0000172745.09928.87. [DOI] [PubMed] [Google Scholar]
- 25.Ardissino D, Merlini PA, Gamba G, et al. Thrombin activity and early outcome in unstable angina pectoris. Circulation. 1996;93(9):1634–1639. doi: 10.1161/01.cir.93.9.1634. [DOI] [PubMed] [Google Scholar]
- 26.Ernofsson M, Strekerud F, Toss H, Abildgaard U, Wallentin L, Siegbahn A. Low-molecular weight heparin reduces the generation and activity of thrombin in unstable coronary artery disease. Thrombosis and Haemostasis. 1998;79(3):491–494. [PubMed] [Google Scholar]
- 27.Moss AJ, Goldstein RE, Marder VJ, et al. Thrombogenic factors and recurrent coronary events. Circulation. 1999;99(19):2517–2522. doi: 10.1161/01.cir.99.19.2517. [DOI] [PubMed] [Google Scholar]
- 28.Mackman N. Regulation of the tissue factor gene. The Journal of the Federation of American Societies for Experimental Biology. 1995;9(10):883–889. doi: 10.1096/fasebj.9.10.7615158. [DOI] [PubMed] [Google Scholar]
- 29.Neumann FJ, Ott I, Marx N, et al. Effect of human recombinant interleukin-6 and interleukin-8 on monocyte procoagulant activity. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17(12):3399–3405. doi: 10.1161/01.atv.17.12.3399. [DOI] [PubMed] [Google Scholar]
- 30.Hatakeyama K, Asada Y, Marutsuka K, Sato Y, Kamikubo Y, Sumiyoshi A. Localization and activity of tissue factor in human aortic atherosclerotic lesions. Atherosclerosis. 1997;133(2):213–219. doi: 10.1016/s0021-9150(97)00132-9. [DOI] [PubMed] [Google Scholar]
- 31.Wilcox JN, Smith KM, Schwartz SM, Gordon D. Localization of tissue factor in the normal vessel wall and in the atherosclerotic plaque. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(8):2839–2843. doi: 10.1073/pnas.86.8.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mallat Z, Benamer H, Hugel B, et al. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation. 2000;101(8):841–843. doi: 10.1161/01.cir.101.8.841. [DOI] [PubMed] [Google Scholar]
- 33.Ott I, Andrassy M, Zieglgänsberger D, Geith S, Schömig A, Neumann FJ. Regulation of monocyte procoagulant activity in acute myocardial infarction: role of tissue factor and tissue factor pathway inhibitor-1. Blood. 2001;97(12):3721–3726. doi: 10.1182/blood.v97.12.3721. [DOI] [PubMed] [Google Scholar]
- 34.Ott I, Neumann FJ, Kenngott S, Gawaz M, Schömig A. Procoagulant inflammatory responses of monocytes after direct balloon angioplasty in acute myocardial infarction. American Journal of Cardiology. 1998;82(8):938–942. doi: 10.1016/s0002-9149(98)00509-8. [DOI] [PubMed] [Google Scholar]
- 35.Bogdanov VY, Balasubramanian V, Hathcock J, Vele O, Lieb M, Nemerson Y. Alternatively spliced human tissue factor: a circulating, soluble, thrombogenic protein. Nature Medicine. 2003;9(4):458–462. doi: 10.1038/nm841. [DOI] [PubMed] [Google Scholar]
- 36.Szotowski B, Antoniak S, Poller W, Schultheiss HP, Rauch U. Procoagulant soluble tissue factor is released from endothelial cells in response to inflammatory cytokines. Circulation Research. 2005;96(12):1233–1239. doi: 10.1161/01.RES.0000171805.24799.fa. [DOI] [PubMed] [Google Scholar]
- 37.Giesen PL, Rauch U, Bohrmann B, et al. Blood-borne tissue factor: another view of thrombosis. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(5):2311–2315. doi: 10.1073/pnas.96.5.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Misumi K, Ogawa H, Yasue H, et al. Comparison of plasma tissue factor levels in unstable and stable angina pectoris. American Journal of Cardiology. 1998;81(1):22–26. doi: 10.1016/s0002-9149(97)00801-1. [DOI] [PubMed] [Google Scholar]
- 39.Soejima H, Ogawa H, Yasue H, Suefuji H, Kaikita K, Nishiyama K. Effects of imidapril therapy on endogenous fibrinolysis in patients with recent myocardial infarction. Clinical Cardiology. 1997;20(5):441–445. doi: 10.1002/clc.4960200507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Falciani M, Gori AM, Fedi S, et al. Elevated tissue factor and tissue factor pathway inhibitor circulating levels in ischaemic heart disease patients. Thrombosis and Haemostasis. 1998;79(3):495–499. [PubMed] [Google Scholar]
- 41.Sakai T, Inoue S, Takei M, et al. Activated inflammatory cells participate in thrombus size through tissue factor and plasminogen activator inhibitor-1 in acute coronary syndrome: immunohistochemical analysis. Thrombosis Research. 2011;127(5):443–449. doi: 10.1016/j.thromres.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 42.Bis J, Vojáček J, Dušek J, et al. Time-course of tissue factor plasma level in patients with acute coronary syndrome. Physiological Research. 2009;58(5):661–667. doi: 10.33549/physiolres.931521. [DOI] [PubMed] [Google Scholar]
- 43.Andrié RP, Bauriedel G, Braun P, Höpp HW, Nickenig G, Skowasch D. Increased expression of C-reactive protein and tissue factor in acute coronary syndrome lesions. Correlation with serum C-reactive protein, angioscopic findings, and modification by statins. Atherosclerosis. 2009;202(1):135–143. doi: 10.1016/j.atherosclerosis.2008.03.028. [DOI] [PubMed] [Google Scholar]
- 44.Malarstig A, Tenno T, Johnston N, et al. Genetic variations in the tissue factor gene are associated with clinical outcome in acute coronary syndrome and expression levels in human monocytes. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(12):2667–2672. doi: 10.1161/01.ATV.0000191637.48129.9b. [DOI] [PubMed] [Google Scholar]
- 45.Maly M, Vojacek J, Hrabos V, Kvasnicka J, Salaj P, Durdil V. Tissue factor, tissue factor pathway inhibitor and cytoadhesive molecules in patients with an acute coronary syndrome. Physiological Research. 2003;52(6):719–728. [PubMed] [Google Scholar]
- 46.Morange PE, Alessi MC, Juhan-Vague I. Relations between hemostatic variables, insulin resistance and inflammation. Hematology Journal. 2004;5(supplement 3):S15–S19. doi: 10.1038/sj.thj.6200415. [DOI] [PubMed] [Google Scholar]
- 47.Morange PE, Bickel C, Nicaud V, et al. Haemostatic factors and the risk of cardiovascular death in patients with coronary artery disease: the AtheroGene study. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(12):2793–2799. doi: 10.1161/01.ATV.0000249406.92992.0d. [DOI] [PubMed] [Google Scholar]
- 48.Steppich BA, Braun SL, Stein A, et al. Plasma TF activity predicts cardiovascular mortality in patients with acute myocardial infarction. Thrombosis Journal. 2009;7, article no. 11 doi: 10.1186/1477-9560-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Erlich JH, Boyle EM, Labriola J, et al. Inhibition of the tissue factor-thrombin pathway limits infarct size after myocardial ischemia-reperfusion injury by reducing inflammation. American Journal of Pathology. 2000;157(6):1849–1862. doi: 10.1016/S0002-9440(10)64824-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ott I, Michaelis C, Schuermann M, et al. Vascular remodeling in mice lacking the cytoplasmic domain of tissue factor. Circulation Research. 2005;97(3):293–298. doi: 10.1161/01.RES.0000177533.48483.12. [DOI] [PubMed] [Google Scholar]
- 51.Rickles FR, Hair GA, Zeff RA, Lee E, Bona RD. Tissue factor expression in human leukocytes and tumor cells. Thrombosis and Haemostasis. 1995;74(1):391–395. [PubMed] [Google Scholar]
- 52.White RH, Chew H, Wun T. Targeting patients for anticoagulant prophylaxis trials in patients with cancer: who is at highest risk? Thrombosis Research. 2007;120(supplement 2):S29–S40. doi: 10.1016/S0049-3848(07)70128-7. [DOI] [PubMed] [Google Scholar]
- 53.Mueller BM, Reisfeld RA, Edgington TS, Ruf W. Expression of tissue factor by melanoma cells promotes efficient hematogenous metastasis. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(24):11832–11836. doi: 10.1073/pnas.89.24.11832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu JL, May L, Lhotak V, et al. Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood. 2005;105(4):1734–1741. doi: 10.1182/blood-2004-05-2042. [DOI] [PubMed] [Google Scholar]
- 55.Ott I, Weigand B, Michl R, et al. Tissue factor cytoplasmic domain stimulates migration by activation of the GTPase Rac1 and the mitogen-activated protein kinase p38. Circulation. 2005;111(3):349–355. doi: 10.1161/01.CIR.0000153333.52294.42. [DOI] [PubMed] [Google Scholar]
- 56.Ettelaie C, Li C, Collier ME, et al. Differential functions of tissue factor in the trans-activation of cellular signalling pathways. Atherosclerosis. 2007;194(1):88–101. doi: 10.1016/j.atherosclerosis.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 57.Iversen N, Lindahl AK, Abildgaard U. Elevated plasma levels of the factor Xa-TFPI complex in cancer patients. Thrombosis Research. 2002;105(1):33–36. doi: 10.1016/s0049-3848(01)00404-2. [DOI] [PubMed] [Google Scholar]
- 58.Ott I, Malcouvier V, Schömig A, Neumann FJ. Proteolysis of tissue factor pathway inhibitor-1 by thrombolysis in acute myocardial infarction. Circulation. 2002;105(3):279–281. doi: 10.1161/hc0302.103591. [DOI] [PubMed] [Google Scholar]
- 59.Massberg S, Grahl L, von Bruehl ML, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nature Medicine. 2010;16(8):887–896. doi: 10.1038/nm.2184. [DOI] [PubMed] [Google Scholar]
- 60.Busch G, Seitz I, Steppich B, et al. Coagulation factor Xa stimulates interleukin-8 release in endothelial cells and mononuclear leukocytes: implications in acute myocardial infarction. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(2):461–466. doi: 10.1161/01.ATV.0000151279.35780.2d. [DOI] [PubMed] [Google Scholar]
- 61.Schaffner A, Rhyn P, Schoedon G, Schaer DJ. Regulated expression of platelet factor 4 in human monocytes—role of PARs as a quantitatively important monocyte activation pathway. Journal of Leukocyte Biology. 2005;78(1):202–209. doi: 10.1189/jlb.0105024. [DOI] [PubMed] [Google Scholar]
- 62.Seitz I, Hess S, Schulz H, et al. Membrane-type serine protease-1/matriptase induces interleukin-6 and -8 in endothelial cells by activation of protease-activated receptor-2: potential implications in atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(4):769–775. doi: 10.1161/01.ATV.0000258862.61067.14. [DOI] [PubMed] [Google Scholar]
- 63.Napoli C, de Nigris F, Wallace JL, et al. Evidence that protease activated receptor 2 expression is enhanced in human coronary atherosclerotic lesions. Journal of Clinical Pathology. 2004;57(5):513–516. doi: 10.1136/jcp.2003.015156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van der Poll T, Boer JD, Levi M. The effect of inflammation on coagulation and vice versa. Current Opinion in Infectious Diseases. 2011;24(3):273–278. doi: 10.1097/QCO.0b013e328344c078. [DOI] [PubMed] [Google Scholar]
- 65.Danese S, Vetrano S, Zhang L, Poplis VA, Castellino FJ. The protein C pathway in tissue inflammation and injury: pathogenic role and therapeutic implications. Blood. 2009;115(6):1121–1131. doi: 10.1182/blood-2009-09-201616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ruf W, Dorfleutner A, Riewald M. Specificity of coagulation factor signaling. Journal of Thrombosis and Haemostasis. 2003;1(7):1495–1503. doi: 10.1046/j.1538-7836.2003.00300.x. [DOI] [PubMed] [Google Scholar]
- 67.Niessen F, Furlan-Freguia C, Fernández JA, et al. Endogenous EPCR/aPC-PARI signaling prevents inflammation-induced vascular leakage and lethality. Blood. 2009;113(12):2859–2866. doi: 10.1182/blood-2008-12-192385. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 68.Kerschen EJ, Fernandez JA, Cooley BC, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant-activated protein C. Journal of Experimental Medicine. 2007;204(10):2439–2448. doi: 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cao C, Gao Y, Li Y, Antalis TM, Castellino FJ, Zhang L. The efficacy of activated protein C in murine endotoxemia is dependent on integrin CD11b. Journal of Clinical Investigation. 2010;120(6):1971–1980. doi: 10.1172/JCI40380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brass LF. Thrombin and platelet activation. Chest. 2003;124(supplement 3):18S–25S. doi: 10.1378/chest.124.3_suppl.18s. [DOI] [PubMed] [Google Scholar]
- 71.Senden NH, Jeunhomme TM, Heemskerk JW, et al. Factor Xa induces cytokine production and expression of adhesion molecules by human umbilical vein endothelial cells. Journal of Immunology. 1998;161(8):4318–4324. [PubMed] [Google Scholar]
- 72.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacological Reviews. 2001;53(2):245–282. [PubMed] [Google Scholar]
- 73.Gouin-Thibault I, Dewar L, Craven S, Kulczycky M, Wun TC, Ofosu FA. Probable regulation of factor VIIa-tissue factor and prothrombinase by factor Xa-TFPI and TFPI in vivo. British Journal of Haematology. 1996;95(4):738–746. doi: 10.1046/j.1365-2141.1996.d01-1963.x. [DOI] [PubMed] [Google Scholar]
- 74.Ohkura N, Soe G, Kohno I, et al. Monoclonal antibody specific for tissue factor pathway inhibitor-factor Xa complex: its characterization and application to plasmas from patients with disseminated intravascular coagulation and pre-disseminated intravascular coagulation. Blood Coagulation and Fibrinolysis. 1999;10(6):309–319. [PubMed] [Google Scholar]
- 75.Collins R, Peto R, Baigent C, Sleight P. Aspirin, heparin, and fibrinolytic therapy in suspected acute myocardial infarction. New England Journal of Medicine. 1997;336(12):847–860. doi: 10.1056/NEJM199703203361207. [DOI] [PubMed] [Google Scholar]
- 76.The Direct Thrombin Inhibitor Trialists’ Collaborative Group. Direct thrombin inhibitors in acute coronary syndromes: principal results of a meta-analysis based on individual patients’ data. The Lancet. 2002;359(9303):294–302. doi: 10.1016/S0140-6736(02)07495-0. [DOI] [PubMed] [Google Scholar]
- 77.The Assessment of the Safety and Efficacy of a New Thrombolytic Regimen (ASSENT)-3 Investigators. Efficacy and safety of tenecteplase in combination with enoxaparin, abciximab, or unfractionated heparin: the ASSENT-3 randomised trial in acute myocardial infarction. The Lancet. 2001;358(9282):605–613. doi: 10.1016/S0140-6736(01)05775-0. [DOI] [PubMed] [Google Scholar]
- 78.Yusuf S, Mehta SR, Chrolavicius S, et al. Effects of fondaparinux on mortality and reinfarction in patients with acute ST-segment elevation myocardial infarction: the OASIS-6 randomized trial. The Journal of the American Medical Association. 2006;295(13):1519–1530. doi: 10.1001/jama.295.13.joc60038. [DOI] [PubMed] [Google Scholar]
- 79.Gibson CM, Mega JL, Burton P, et al. Rationale and design of the Anti-Xa therapy to lower cardiovascular events in addition to standard therapy in subjects with acute coronary syndrome-thrombolysis in myocardial infarction 51 (ATLAS-ACS 2 TIMI 51) trial: a randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of rivaroxaban in subjects with acute coronary syndrome. American Heart Journal. 2011;161(5):815–821.e6. doi: 10.1016/j.ahj.2011.01.026. [DOI] [PubMed] [Google Scholar]
- 80.Camerer E, Kataoka H, Kahn M, Lease K, Coughlin SR. Genetic evidence that protease-activated receptors mediate factor Xa signaling in endothelial cells. Journal of Biological Chemistry. 2002;277(18):16081–16087. doi: 10.1074/jbc.M108555200. [DOI] [PubMed] [Google Scholar]
- 81.Riewald M, Kravchenko VV, Petrovan RJ, et al. Gene induction by coagulation factor Xa is mediated by activation of protease-activated receptor 1. Blood. 2001;97(10):3109–3116. doi: 10.1182/blood.v97.10.3109. [DOI] [PubMed] [Google Scholar]
- 82.Demetz G, Seitz I, Stein A, et al. Tissue factor-factor VIIa complex induces cytokine expression in coronary artery smooth muscle cells. Atherosclerosis. 2010;212(2):466–471. doi: 10.1016/j.atherosclerosis.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 83.Hjortoe GM, Petersen LC, Albrektsen T, et al. Tissue factor-factor VIIa-specific up-regulation of IL-8 expression in MDA-MB-231 cells is mediated by PAR-2 and results in increased cell migration. Blood. 2004;103(8):3029–3037. doi: 10.1182/blood-2003-10-3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moons AH, Levi M, Peters RJ. Tissue factor and coronary artery disease. Cardiovascular Research. 2002;53(2):313–325. doi: 10.1016/s0008-6363(01)00452-7. [DOI] [PubMed] [Google Scholar]
- 85.Koizume S, Jin MS, Miyagi E, et al. Activation of cancer cell migration and invasion by ectopic synthesis of coagulation factor VII. Cancer Research. 2006;66(19):9453–9460. doi: 10.1158/0008-5472.CAN-06-1803. [DOI] [PubMed] [Google Scholar]
- 86.Schaffner F, Ruf W. Tissue factor and PAR2 signaling in the tumor microenvironment. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29(12):1999–2004. doi: 10.1161/ATVBAHA.108.177428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gessler F, Voss V, Dützmann S, Seifert V, Gerlach R, Kögel D. Inhibition of tissue factor/protease-activated receptor-2 signaling limits proliferation, migration and invasion of malignant glioma cells. Neuroscience. 2010;165(4):1312–1322. doi: 10.1016/j.neuroscience.2009.11.049. [DOI] [PubMed] [Google Scholar]
- 88.Zerbib P, Grimonprez A, Corseaux D, et al. Inhibition of tissue factor-factor VIIa proteolytic activity blunts hepatic metastasis in colorectal cancer. Journal of Surgical Research. 2009;153(2):239–245. doi: 10.1016/j.jss.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 89.Hinshaw LB, Emerson TE, Jr., Chang AC, Duerr M, Peer G, Fournel M. Study of septic shock in the non-human primate: relationship of pathophysiological response to therapy with anti-TNF antibody. Circulatory Shock. 1994;44(4):221–229. [PubMed] [Google Scholar]
- 90.Hinshaw LB, Emerson TE, Jr., Taylor FB, Jr., et al. Lethal Staphylococcus aureus-induced shock in primates: prevention of death with anti-TNF antibody. Journal of Trauma. 1992;33(4):568–573. [PubMed] [Google Scholar]
- 91.Hinshaw LB, Tekamp-Olson P, Chang AC, et al. Survival of primates in LD100 septic shock following therapy with antibody to tumor necrosis factor (TNF alpha) Circulatory Shock. 1990;30(3):279–292. [PubMed] [Google Scholar]
- 92.Clark MA, Plank LD, Connolly AB, et al. Effect of a chimeric antibody to tumor necrosis factor-alpha on cytokine and physiologic responses in patients with severe sepsis—a randomized, clinical trial. Critical Care Medicine. 1998;26(10):1650–1659. doi: 10.1097/00003246-199810000-00016. [DOI] [PubMed] [Google Scholar]
- 93.Favalli EG, Desiati F, Atzeni F, et al. Serious infections during anti-TNFα treatment in rheumatoid arthritis patients. Autoimmunity Reviews. 2009;8(3):266–273. doi: 10.1016/j.autrev.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 94.Goode S, Tierney G, Deighton C. Life threatening intra-abdominal sepsis in patients on anti-TNF-α therapy. Gut. 2006;55(4):590–591. doi: 10.1136/gut.2005.085449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ho GT, Mowat A, Potts L, et al. Efficacy and complications of adalimumab treatment for medically-refractory Crohn’s disease: analysis of nationwide experience in Scotland (2004–2008) Alimentary Pharmacology and Therapeutics. 2009;29(5):527–534. doi: 10.1111/j.1365-2036.2008.03919.x. [DOI] [PubMed] [Google Scholar]
- 96.Levi M, van der Poll T, Büller HR. Bidirectional relation between inflammation and coagulation. Circulation. 2004;109(22):2698–2704. doi: 10.1161/01.CIR.0000131660.51520.9A. [DOI] [PubMed] [Google Scholar]
- 97.Carr C, Bild GS, Chang AC, et al. Recombinant E. coli-derived tissue factor pathway inhibitor reduces coagulopathic and lethal effects in the baboon gram-negative model of septic shock. Circulatory Shock. 1994;44(3):126–137. [PubMed] [Google Scholar]
- 98.Hisama N, Yamaguchi Y, Okajima K, et al. Anticoagulant pretreatment attenuates production of cytokine-induced neutrophil chemoattractant following ischemia-reperfusion of rat liver. Digestive Diseases and Sciences. 1996;41(7):1481–1486. doi: 10.1007/BF02088576. [DOI] [PubMed] [Google Scholar]
- 99.Giugliano GR, Giugliano RP, Gibson CM, Kuntz RE. Meta-analysis of corticosteroid treatment in acute myocardial infarction. American Journal of Cardiology. 2003;91(9):1055–1059. doi: 10.1016/s0002-9149(03)00148-6. [DOI] [PubMed] [Google Scholar]
- 100.Antman EM, McCabe CH, Gurfinkel EP, et al. Enoxaparin prevents death and cardiac ischemic events in unstable angina/non-Q-wave myocardial infarction. Results of the thrombolysis in myocardial infarction (TIMI) 11B trial. Circulation. 1999;100(15):1593–1601. doi: 10.1161/01.cir.100.15.1593. [DOI] [PubMed] [Google Scholar]
- 101.Manduteanu I, Voinea M, Capraru M, Dragomir E, Simionescu M. A novel attribute of enoxaparin: inhibition of monocyte adhesion to endothelial cells by a mechanism involving cell adhesion molecules. Pharmacology. 2002;65(1):32–37. doi: 10.1159/000056183. [DOI] [PubMed] [Google Scholar]
- 102.Montalescot G, Bal-dit-Sollier C, Chibedi D, et al. Comparison of effects on markers of blood cell activation of enoxaparin, dalteparin, and unfractionated heparin in patients with unstable angina pectoris or non-ST-segment elevation acute myocardial infarction (the ARMADA study) American Journal of Cardiology. 2003;91(8):925–930. doi: 10.1016/s0002-9149(03)00105-x. [DOI] [PubMed] [Google Scholar]