

Abstract
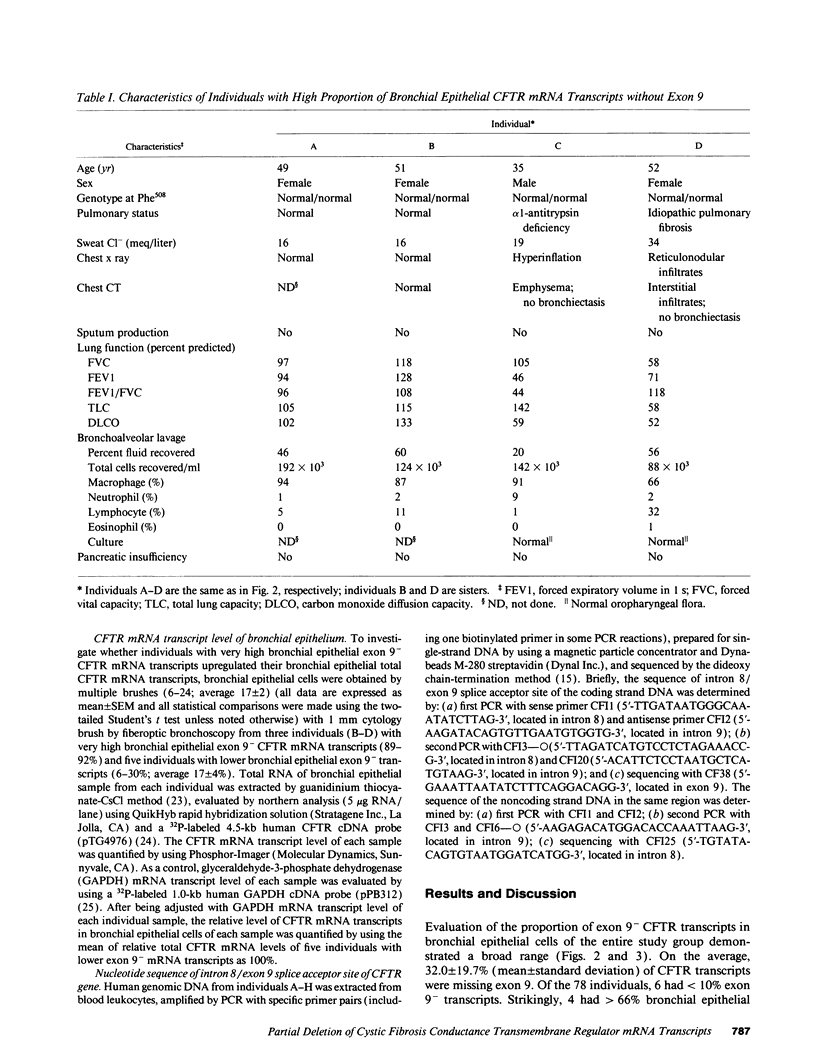
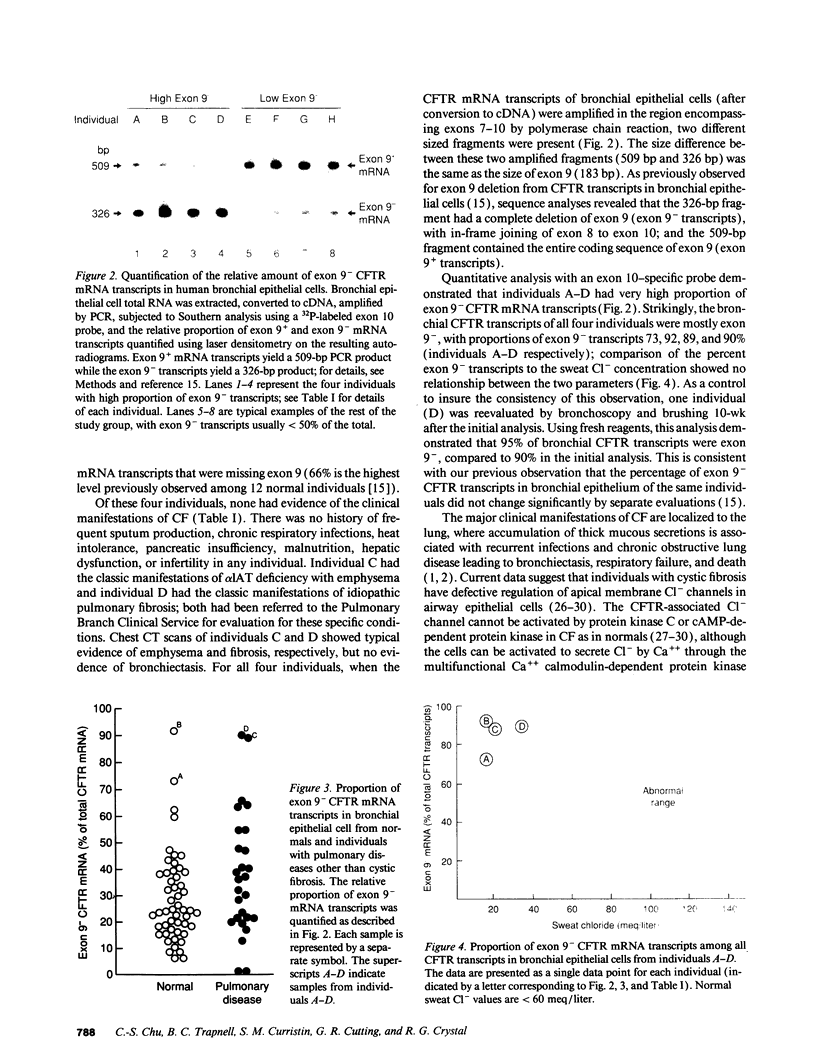
Cystic fibrosis (CF) is a recessive hereditary disorder, requiring both parental cystic fibrosis conductance transmembrane regulator (CFTR) genes to carry mutations for clinical disease to manifest, i.e., only 50% of normal CFTR gene expression is required to maintain a normal phenotype. To help define the minimum amount of normal CFTR gene expression necessary to maintain normalcy, we have capitalized on our prior observation (Chu, C.-S., B. C. Trapnell, J. J. Murtagh, Jr., J. Moss, W. Dalemans, S. Jallat, A. Mercenier, A. Pavirani, J.-P. Lecocq, G. R. Cutting, et al. 1991. EMBO [Eur. Mol. Biol. Organ] J. 10:1355-1363) that normal individuals can have up to 66% of bronchial CFTR mRNA transcripts that are missing exon 9, a region representing 21% of the sequence coding for the critical nucleotide (ATP)-binding fold 1 (NBF1) of the predicted CFTR protein. The study population included 78 individuals with no prior diagnosis of CF. Evaluation of bronchial epithelial cells (obtained by bronchoscopy) revealed that exon 9 was variably deleted in all individuals. Remarkably, there were four individuals, all greater than or equal to 35 yr, in whom bronchial epithelial cells exhibited 73, 89, 90, and 92% CFTR transcripts with inframe deletion of exon 9, respectively, despite normal sweat Cl- and no clinical manifestation of CF. In the context that only 8% or less of bronchial CFTR transcripts need exon 9 to maintain normal airway function, these observations strongly suggest that either exon 9 is not necessary for CFTR structure and/or function or that only a very small fraction of bronchial epithelial cells need to express normal CFTR mRNA transcripts with exon 9 to perform the function of CFTR sufficient to maintain a normal phenotype in vivo.
Full text
PDF




Images in this article


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Anderson M. P., Gregory R. J., Thompson S., Souza D. W., Paul S., Mulligan R. C., Smith A. E., Welsh M. J. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991 Jul 12;253(5016):202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- Anderson M. P., Rich D. P., Gregory R. J., Smith A. E., Welsh M. J. Generation of cAMP-activated chloride currents by expression of CFTR. Science. 1991 Feb 8;251(4994):679–682. doi: 10.1126/science.1704151. [DOI] [PubMed] [Google Scholar]
- Brantly M. L., Paul L. D., Miller B. H., Falk R. T., Wu M., Crystal R. G. Clinical features and history of the destructive lung disease associated with alpha-1-antitrypsin deficiency of adults with pulmonary symptoms. Am Rev Respir Dis. 1988 Aug;138(2):327–336. doi: 10.1164/ajrccm/138.2.327. [DOI] [PubMed] [Google Scholar]
- Brantly M. L., Wittes J. T., Vogelmeier C. F., Hubbard R. C., Fells G. A., Crystal R. G. Use of a highly purified alpha 1-antitrypsin standard to establish ranges for the common normal and deficient alpha 1-antitrypsin phenotypes. Chest. 1991 Sep;100(3):703–708. doi: 10.1378/chest.100.3.703. [DOI] [PubMed] [Google Scholar]
- Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., Smith A. E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990 Nov 16;63(4):827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Cheng S. H., Rich D. P., Marshall J., Gregory R. J., Welsh M. J., Smith A. E. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell. 1991 Sep 6;66(5):1027–1036. doi: 10.1016/0092-8674(91)90446-6. [DOI] [PubMed] [Google Scholar]
- Chirgwin J. M., Przybyla A. E., MacDonald R. J., Rutter W. J. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979 Nov 27;18(24):5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- Chu C. S., Trapnell B. C., Murtagh J. J., Jr, Moss J., Dalemans W., Jallat S., Mercenier A., Pavirani A., Lecocq J. P., Cutting G. R. Variable deletion of exon 9 coding sequences in cystic fibrosis transmembrane conductance regulator gene mRNA transcripts in normal bronchial epithelium. EMBO J. 1991 Jun;10(6):1355–1363. doi: 10.1002/j.1460-2075.1991.tb07655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal R. G., Fulmer J. D., Roberts W. C., Moss M. L., Line B. R., Reynolds H. Y. Idiopathic pulmonary fibrosis. Clinical, histologic, radiographic, physiologic, scintigraphic, cytologic, and biochemical aspects. Ann Intern Med. 1976 Dec;85(6):769–788. doi: 10.7326/0003-4819-85-6-769. [DOI] [PubMed] [Google Scholar]
- Cuppens H., Marynen P., De Boeck C., De Baets F., Eggermont E., Van den Berghe H., Cassiman J. J. A child, homozygous for a stop codon in exon 11, shows milder cystic fibrosis symptoms than her heterozygous nephew. J Med Genet. 1990 Nov;27(11):717–719. doi: 10.1136/jmg.27.11.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutting G. R., Kasch L. M., Rosenstein B. J., Tsui L. C., Kazazian H. H., Jr, Antonarakis S. E. Two patients with cystic fibrosis, nonsense mutations in each cystic fibrosis gene, and mild pulmonary disease. N Engl J Med. 1990 Dec 13;323(24):1685–1689. doi: 10.1056/NEJM199012133232407. [DOI] [PubMed] [Google Scholar]
- Cutting G. R., Kasch L. M., Rosenstein B. J., Zielenski J., Tsui L. C., Antonarakis S. E., Kazazian H. H., Jr A cluster of cystic fibrosis mutations in the first nucleotide-binding fold of the cystic fibrosis conductance regulator protein. Nature. 1990 Jul 26;346(6282):366–369. doi: 10.1038/346366a0. [DOI] [PubMed] [Google Scholar]
- Davies K. Cystic fibrosis. Complementary endeavours. Nature. 1990 Nov 8;348(6297):110–111. doi: 10.1038/348110a0. [DOI] [PubMed] [Google Scholar]
- Davis P. B., Del Rio S., Muntz J. A., Dieckman L. Sweat chloride concentration in adults with pulmonary diseases. Am Rev Respir Dis. 1983 Jul;128(1):34–37. doi: 10.1164/arrd.1983.128.1.34. [DOI] [PubMed] [Google Scholar]
- Ercolani L., Florence B., Denaro M., Alexander M. Isolation and complete sequence of a functional human glyceraldehyde-3-phosphate dehydrogenase gene. J Biol Chem. 1988 Oct 25;263(30):15335–15341. [PubMed] [Google Scholar]
- Frizzell R. A., Rechkemmer G., Shoemaker R. L. Altered regulation of airway epithelial cell chloride channels in cystic fibrosis. Science. 1986 Aug 1;233(4763):558–560. doi: 10.1126/science.2425436. [DOI] [PubMed] [Google Scholar]
- Fulmer J. D., Roberts W. C., von Gal E. R., Grystal R. G. Small airways in idiopathic pulmonary fibrosis. Comparison of morphologic and physiologic observations. J Clin Invest. 1977 Sep;60(3):595–610. doi: 10.1172/JCI108811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory R. J., Rich D. P., Cheng S. H., Souza D. W., Paul S., Manavalan P., Anderson M. P., Welsh M. J., Smith A. E. Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Mol Cell Biol. 1991 Aug;11(8):3886–3893. doi: 10.1128/mcb.11.8.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang T. C., Lu L., Zeitlin P. L., Gruenert D. C., Huganir R., Guggino W. B. Cl- channels in CF: lack of activation by protein kinase C and cAMP-dependent protein kinase. Science. 1989 Jun 16;244(4910):1351–1353. doi: 10.1126/science.2472005. [DOI] [PubMed] [Google Scholar]
- Hyde S. C., Emsley P., Hartshorn M. J., Mimmack M. M., Gileadi U., Pearce S. R., Gallagher M. P., Gill D. R., Hubbard R. E., Higgins C. F. Structural model of ATP-binding proteins associated with cystic fibrosis, multidrug resistance and bacterial transport. Nature. 1990 Jul 26;346(6282):362–365. doi: 10.1038/346362a0. [DOI] [PubMed] [Google Scholar]
- Kartner N., Hanrahan J. W., Jensen T. J., Naismith A. L., Sun S. Z., Ackerley C. A., Reyes E. F., Tsui L. C., Rommens J. M., Bear C. E. Expression of the cystic fibrosis gene in non-epithelial invertebrate cells produces a regulated anion conductance. Cell. 1991 Feb 22;64(4):681–691. doi: 10.1016/0092-8674(91)90498-n. [DOI] [PubMed] [Google Scholar]
- Kerem B. S., Zielenski J., Markiewicz D., Bozon D., Gazit E., Yahav J., Kennedy D., Riordan J. R., Collins F. S., Rommens J. M. Identification of mutations in regions corresponding to the two putative nucleotide (ATP)-binding folds of the cystic fibrosis gene. Proc Natl Acad Sci U S A. 1990 Nov;87(21):8447–8451. doi: 10.1073/pnas.87.21.8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerem B., Rommens J. M., Buchanan J. A., Markiewicz D., Cox T. K., Chakravarti A., Buchwald M., Tsui L. C. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989 Sep 8;245(4922):1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- Li M., McCann J. D., Anderson M. P., Clancy J. P., Liedtke C. M., Nairn A. C., Greengard P., Welsch M. J. Regulation of chloride channels by protein kinase C in normal and cystic fibrosis airway epithelia. Science. 1989 Jun 16;244(4910):1353–1356. doi: 10.1126/science.2472006. [DOI] [PubMed] [Google Scholar]
- Li M., McCann J. D., Liedtke C. M., Nairn A. C., Greengard P., Welsh M. J. Cyclic AMP-dependent protein kinase opens chloride channels in normal but not cystic fibrosis airway epithelium. Nature. 1988 Jan 28;331(6154):358–360. doi: 10.1038/331358a0. [DOI] [PubMed] [Google Scholar]
- Rich D. P., Gregory R. J., Anderson M. P., Manavalan P., Smith A. E., Welsh M. J. Effect of deleting the R domain on CFTR-generated chloride channels. Science. 1991 Jul 12;253(5016):205–207. doi: 10.1126/science.1712985. [DOI] [PubMed] [Google Scholar]
- Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J. L. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989 Sep 8;245(4922):1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rommens J. M., Iannuzzi M. C., Kerem B., Drumm M. L., Melmer G., Dean M., Rozmahel R., Cole J. L., Kennedy D., Hidaka N. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989 Sep 8;245(4922):1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- Saiki R. K., Gelfand D. H., Stoffel S., Scharf S. J., Higuchi R., Horn G. T., Mullis K. B., Erlich H. A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988 Jan 29;239(4839):487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- Saltini C., Hance A. J., Ferrans V. J., Basset F., Bitterman P. B., Crystal R. G. Accurate quantification of cells recovered by bronchoalveolar lavage. Am Rev Respir Dis. 1984 Oct;130(4):650–658. doi: 10.1164/arrd.1984.130.4.650. [DOI] [PubMed] [Google Scholar]
- Trapnell B. C., Chu C. S., Paakko P. K., Banks T. C., Yoshimura K., Ferrans V. J., Chernick M. S., Crystal R. G. Expression of the cystic fibrosis transmembrane conductance regulator gene in the respiratory tract of normal individuals and individuals with cystic fibrosis. Proc Natl Acad Sci U S A. 1991 Aug 1;88(15):6565–6569. doi: 10.1073/pnas.88.15.6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell B. C., Zeitlin P. L., Chu C. S., Yoshimura K., Nakamura H., Guggino W. B., Bargon J., Banks T. C., Dalemans W., Pavirani A. Down-regulation of cystic fibrosis gene mRNA transcript levels and induction of the cystic fibrosis chloride secretory phenotype in epithelial cells by phorbol ester. J Biol Chem. 1991 Jun 5;266(16):10319–10323. [PubMed] [Google Scholar]
- Wagner J. A., Cozens A. L., Schulman H., Gruenert D. C., Stryer L., Gardner P. Activation of chloride channels in normal and cystic fibrosis airway epithelial cells by multifunctional calcium/calmodulin-dependent protein kinase. Nature. 1991 Feb 28;349(6312):793–796. doi: 10.1038/349793a0. [DOI] [PubMed] [Google Scholar]
- Walker J. E., Saraste M., Runswick M. J., Gay N. J. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1(8):945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh M. J. Abnormal regulation of ion channels in cystic fibrosis epithelia. FASEB J. 1990 Jul;4(10):2718–2725. doi: 10.1096/fasebj.4.10.1695593. [DOI] [PubMed] [Google Scholar]
- Welsh M. J., Fick R. B. Cystic fibrosis. J Clin Invest. 1987 Dec;80(6):1523–1526. doi: 10.1172/JCI113237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura K., Nakamura H., Trapnell B. C., Dalemans W., Pavirani A., Lecocq J. P., Crystal R. G. The cystic fibrosis gene has a "housekeeping"-type promoter and is expressed at low levels in cells of epithelial origin. J Biol Chem. 1991 May 15;266(14):9140–9144. [PubMed] [Google Scholar]
- Zielenski J., Rozmahel R., Bozon D., Kerem B., Grzelczak Z., Riordan J. R., Rommens J., Tsui L. C. Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics. 1991 May;10(1):214–228. doi: 10.1016/0888-7543(91)90503-7. [DOI] [PubMed] [Google Scholar]