

Abstract
Post translational modification (PTM) and proteolytic processing of proteins contributes to regulation of their stability, intracellular localization and interactions with other proteins, and to direct enhancement or repression of their activity. Both PTM and proteolysis are dynamic; levels and rates change in response to changes in the cellular environment. Tissue excision, post mortem interval and subsequent methods of tissue processing for protein analysis unavoidably alter the cellular environment and therefore features of protein profiles. To aid in understanding the time frame and protein specificity of these changes and the biological and technical contributions to them, we have compared features of protein profiles in mouse hippocampus and cortex following three methods of tissue handling: immediate lysate preparation, and rapid heating to 95°C and standard snap freezing in liquid nitrogen, prior to lysate preparation. In spite of the very short time frames involved, we observe protein-specific differences in levels of phosphorylation, general differences in patterns of sumoylation, and specific differences in levels of proteolytic cleavage of calcineurin and the neurotrophin receptor, TRKA. These differences vary with brain region and with post excision time to processing, and highlight the challenges inherent in accurately profiling the in vivo proteome.
Keywords: Post translational modification, phosphorylation, sumoylation, proteolytic processing, protein lysates, in vivo proteome
1. Introduction
Protein activity is commonly regulated by post translational modification (PTM). Signaling cascades, protein stability and protein interactions are known to be influenced by phosphorylation, sumoylation, ubiquitination, methylation, and other modifications (Deribe et al., 2010). In addition, for some neurologically critical proteins such as APP and nerve growth factor receptors, activities are also regulated by site specific cleavage (Thinakaran and Koo, 2008; Diaz-Rodriguez et al., 1999). Investigating in vivo cellular regulatory mechanisms, and recognizing abnormalities associated with disease, requires accurate evaluation of protein activity and therefore measurements of PTM and proteolysis. There are, however, significant biological and technical challenges to such assessments, and because these differ with handling and processing, they are affected by the source of the tissue samples. For human tissues, obtained from biopsies and from autopsies, timing of harvest and initial handling of samples largely cannot be controlled, while for laboratory animals, typically mouse or rat, euthanasia and tissue procurement can in principle be well controlled. In both cases, however, biological challenges arise because PTMs are dynamic and reversible and can change rapidly in response to changes in the cellular and environmental conditions that occur following tissue excision and during post mortem interval. Technical challenges arise because proteolytic cleavage rates and PTMs also change during tissue processing where proteases and enzymes responsible for protein modifications are released from cellular compartments or complexes where they normally are sequestered from, or regulated in access to, their substrates. For both biological and technical reasons, therefore, preserving protein profiles requires inactivation of associated enzymes. Laboratory protocols for processing tissues for protein measurement typically focus on phosphorylation, and rely on rapid tissue dissection, carrying out processing steps on ice, and including phosphatase inhibitors in homogenization buffers. Concerns that this may be inadequate for preserving phosphorylation have led to the introduction of alternative procedures. Focused microwave irradiation to the brain, using specialized instruments, was developed as a method to simultaneously kill a rat or mouse and inactivate all enzymes by rapidly heating the brain to high temperatures, thus preserving the protein phosphorylation profile at the instant of death (Hossain et al., 1994; O'Callaghan and Sriram, 2004). Because this can be used only for rodent brain tissue, an instrument with broader utility was recently introduced, the Stabilizor T1, which uses heat and pressure under vacuum to inactivate enzymes in fresh or frozen dissected tissues (Svensson et al., 2009).
Here we describe several features of protein profiles in lysates prepared from mouse brain regions that were treated in the Stabilizor T1. We compare them with results obtained from the standard laboratory protocol of snap freezing of tissues in liquid nitrogen prior to processing and from immediately processing tissues. We show that rapid heating of whole brains to 95°C followed by dissection vs. rapid brain dissection on ice followed by transfer to liquid nitrogen, or immediate processing, results in differences in the levels of protein-specific phosphorylation, in patterns of sumoylation, and in cleavage products of calcineurin and the nerve growth factor receptor, TRKA. Variations in these differences are seen between cortex and hippocampus, however, heat stabilization largely preserves the initial features against further changes associated with room temperature incubation. These data emphasize the challenges inherent in determining in vivo protein features and therefore in studying the mechanisms of their regulation. They also emphasize the need for consistent methods of fresh tissue sample handling, in particular when the goal is to assess abnormalities associated with a disease state or genetic variability.
2. Materials and methods
2.1. Mice and tissue processing
Male C57BL6/J mice, three per group, approximately two months of age were used. Animals were housed together in standard environmental conditions under a 12h dark/light cycle and allowed to free access to food and water. Animals were treated in accordance with guidelines provided by the National Institutes of Health and protocols have been approved by the Animal Care and Use Committee of the University of Colorado Denver.
All mice were euthanized by cervical dislocation and, for all three protocols, brains were removed from the skull within 30 seconds. For the heat stabilization protocol (ST tissues), whole brains were thermally denatured in the Stabilizor T1 (Denator, AB) following the manufacturer's directions. Briefly, following excision, whole brains were immediately placed in a new teflon sample cassette and inserted into the Stabilizor T1 that had been preheated to 95°C. Evacuation of air from the sample chamber and sample heating were completed within ∼50 seconds. Stabilized brains were frozen in liquid nitrogen and stored at -80°C until lysate preparation. For the standard frozen tissue protocol (frozen, FZ, tissues), the brain was held on ice and hippocampus and cortex were dissected within 90 seconds; tissues were then immediately snap frozen in liquid nitrogen and stored at -80°C until lysate preparation. For the fresh lysate preparation protocol (fresh, FSH, tissues), brains were similarly dissected. Hippocampus and cortex were then cut in half; one half was immediately added to lysis buffer and lysates prepared as described below; the second half was held at room temperature for 30 minutes prior to lysate preparation. Steps, temperature and time for each protocol are compared in Table 1.
Table 1. Protocol comparisons, steps and time frames, post euthanasia to lysis.
ST, heat stabilized; FZ, frozen; FSH, fresh. RT, room temperature incubation. N/A, not applicable.
| Protocol | Remove brain |
Dissect | Heat | Freeze | Thaw | Lyse, 0′ RT |
Lyse, +30′ RT |
|---|---|---|---|---|---|---|---|
| ST | 30″ | N/A | 50″, 95° | Yes | Yes, dissect | ½ | ½ |
| FZ | 30″ | 90″, ice | N/A | Yes | No | ½ | ½ |
| FSH | 30″ | 90″, ice | N/A | N/A | N/A | ½ | ½ |
2.3. Preparation of protein lysates
ST whole brains were thawed and dissected into hippocampus and cortex prior to lysate preparation. For each ST, FZ and FSH sample, each brain region was cut into two pieces, and one piece was processed immediately (0 minutes room temperature, RT), while the other piece was held at room temperature for 30 minutes (30 min RT) (Table 1). Whole cell lysates from all samples were prepared in IEF buffer (8M urea, 4% CHAPS, 50 mM Tris) (Svensson et al., 2009) with the addition of protease and phosphatase inhibitors. Tissues were homogenized by sonication (three bursts of five seconds duration in the Branson Sonic Power Co U.S.A). Protein concentrations were determined using the 660 nM Protein Assay (Pierce); yields from ST, FZ and FSH tissues were equivalent.
2.4. Western blots
Twenty μg of protein lysates per lane were separated at 170 V, constant voltage for 2 ½ hours on SDS-PAGE gels; 10% (w/v) polyacrylamide gels were used for all antibodies, with additional 8% gels for TrkA and 12% gels for SUMO3. Following electrophoresis, proteins were transferred to PVDF membranes at 250 mA constant current for 1 ½ hours. Membranes were blocked with 5% (w/v) Bovine Serum Albumin (BSA) in TBST (Tris-buffered saline, 1% Tween20), followed by overnight incubation at 4°C with primary antibody. Sources of primary antibody were as follows: pERK1/2(Tyr204), pELK1(Ser383) (Santa Cruz Biotechnology Inc., Santa Cruz, CA); pCREB(Ser133), pCAMKII(Thr286) (PhosphoSolutions, CO, USA); pJNK(Thr183/Tyr185), pAKT(Ser473), pRSK(Ser380), pGSK3β(Ser9), tRSK, pan-calcineurin A (Cell Signaling Technology, MA, USA), pGJA1(Ser368)(Connexin 43) (Abnova, Taiwan); phosphoserine, SUMO3 (Invitrogen, CA, USA; cat#700186), TRKA (Epitomics, Inc, CA, USA). Detection of bound primary antibodies was performed with alkaline-phosphatase-conjugated goat anti-rabbit (Invitrogen, CA, USA) or goat anti-mouse (Cell Signaling Technology, MA, USA) secondary antibodies. Signals were detected with CDP-Star Chemiluminescence reagent; imaging and quantitation were carried out using the Diana III CCD camera and Aida software (Raytest, Inc, Germany). All membranes were stripped in 0.2M Glycine pH 2.5/0.05% Tween, at 70°C for 20 minutes, and re-probed with actin antibody (Sigma).
2.5. Statistical analysis
For each antibody, signals were normalized to actin; average values from replicate experiments were used in statistical analysis using the unpaired Student t-test (GraphPad Prizm). P< 0.05 was considered statistically significant.
3. Results
3.1. Phosphorylation patterns at initial time points
We first compared results in heat stabilized (ST) tissues with those obtained by the protocol most commonly used in laboratory rodent experiments, snap freezing in liquid nitrogen (FZ). In Figure 1, we show patterns of phospho-serine in lysates from ST and FZ cortex. There are no obvious differences in overall band patterns or intensities, either between ST and FZ samples or among samples within the same treatment group. Figure 2 shows results of Western blot analysis for twelve phospho-proteins in hippocampal lysates prepared from the same brains. Levels of pELK, pERK1, pERK2 and pCREB appear similar in ST and FZ. In contrast, levels of pJNK1 and pJNK2/3 appear slightly higher in FZ tissues, and levels of pCAMKIIα, pCAMKIIβ, pGSK3β, pAKT, pRSK and the Connexin 43 protein, pGJA1, are dramatically higher in FZ tissues compared with ST. These visual interpretations are confirmed by quantitation after normalization to actin signals (Figure 3; Table 1a). Results were similar for cortex tissues (Figures 4a and 5; Table 2a).
Figure 1. Patterns of phospho-serine appear similar in frozen and heat stabilized cortex.
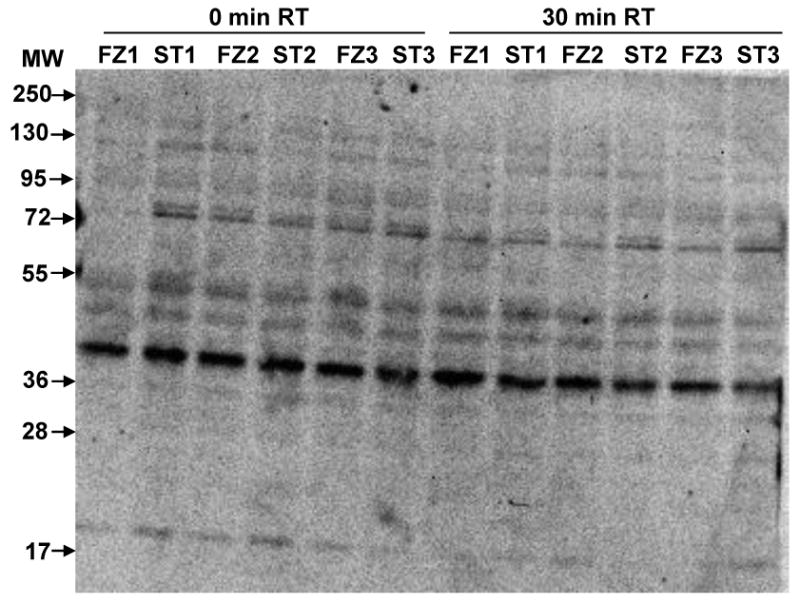
Lysates were prepared after snap freezing in liquid nitrogen (FZ1-FZ3) or heat stabilization (ST1-ST3). Immediately prior to lysate preparation, brain regions were cut in half; one half was immediately placed in IEF buffer and processed (0 minutes room temperature (0 min RT)) and the other half was held for 30 minutes at room temperature (30 min RT) followed by buffer addition and processing. Gel membrane was screened with an antibody to phospho-serine.
Figure 2. Hippocampal levels of specific phospho-proteins differ between frozen and heat stabilized tissues and between 0 and 30 minutes room temperature incubation.
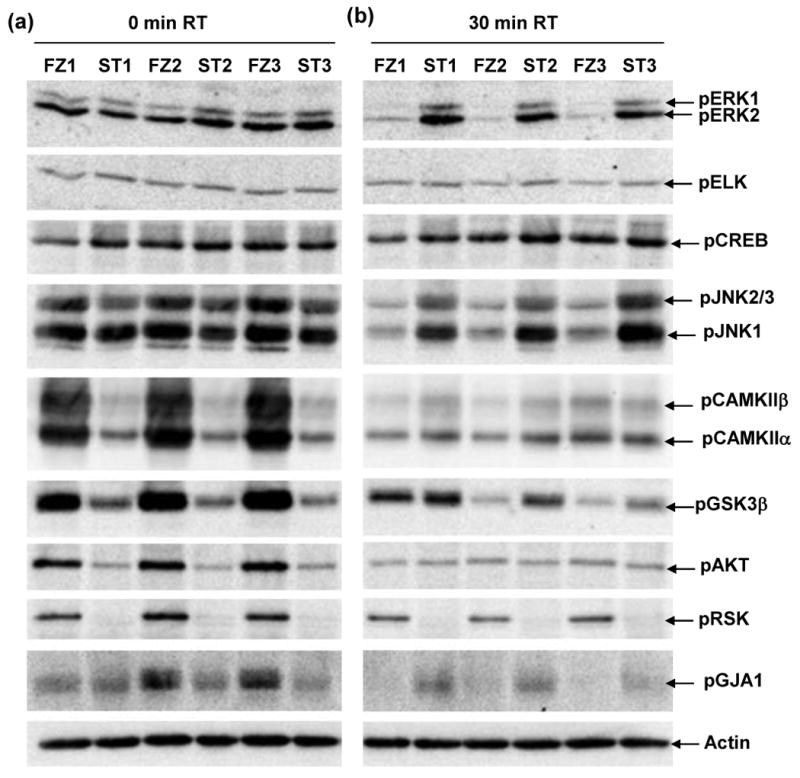
Lysates were prepared from hippocampus from the same frozen (FZ1-FZ3) and stabilized (ST1-ST3) brains as in Figure 1. Gel membranes were screened with antibodies to the indicated proteins.
Figure 3. Quantitation of phospho-proteins in hippocampal lysates shows that heat stabilized levels remain unchanged while frozen tissue levels are decreased with room temperature incubation.

Signals from replicate Western blots as in Figure 2 were normalized to actin. Histograms represent average values for each group of samples relative to 100% for frozen tissues at 0 min RT (white bars); black bars, stabilized tissues 0′ RT; grey bars, frozen tissues 30′ RT; striped bars, stabilized tissues 30′ RT. Errors bars are the standard error of the mean. Statistical significance was determined by the unpaired Student's t-test. *p<0.05; **p <0.001:***p <0.0001.
Figure 4. Cortex levels of specific phospho-proteins differ between frozen and heat stabilized tissues and between 0 and 30 minutes room temperature incubation.
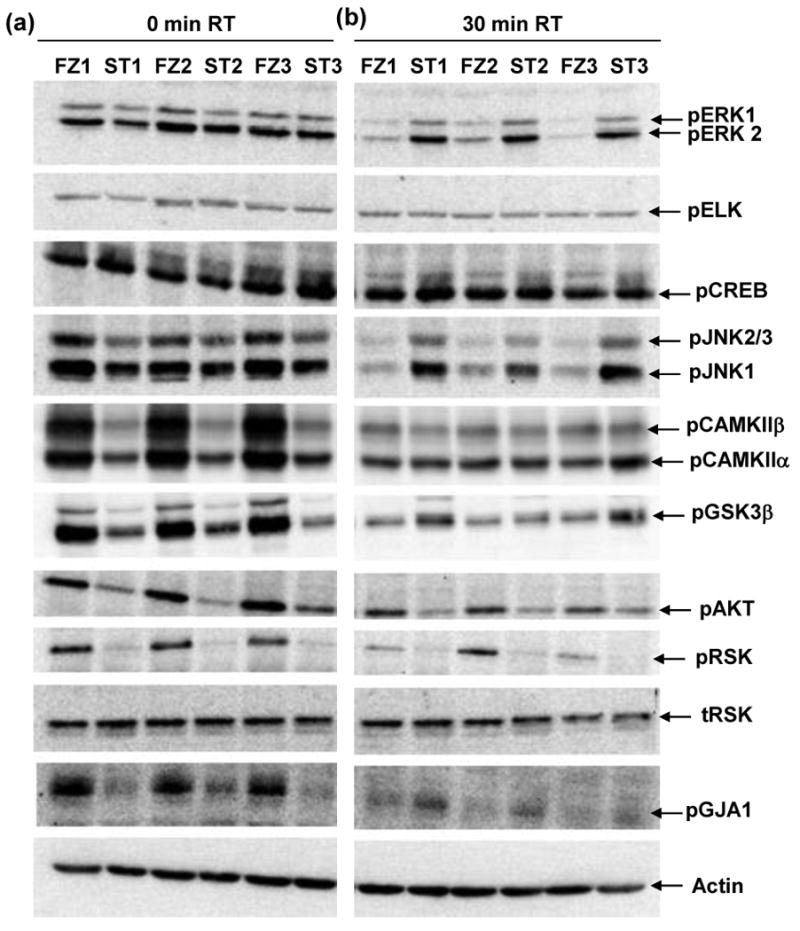
Lysates are those used in Figure 1. Gel membranes were screened with antibodies to the indicated proteins as in Figure 2.
Figure 5. Unchanged levels of phospho-proteins in cortical lysates from heat stabilized tissue and decreased levels in frozen tissue with room temperature incubation.

Signals from replicate Western blots as in Figure 4 were normalized to actin. Histograms represent average values for each group of samples relative to 100% for frozen tissues at 0 min RT (white bars); black bars, stabilized tissues 0′ RT; grey bars, frozen tissues 30′ RT; striped bars, stabilized tissues 30′ RT. Errors bars are the standard error of the mean. Statistical significance was determined by the unpaired Student's t-test. *p<0.05; **p <0.001:***p <0.0001.
Table 2. Levels of phosphorylation at 0 minutes room temperature incubation differ between heat stabilized and frozen or fresh tissues.
Values are compared to 100% in snap frozen (FZ) hippocampus (Hp) and cortex. a) ST, heat stabilized; b) FSH, fresh. *, p<0.05; **, p<0.01, ***, p<0.001.
| (a) ST | (b) FSH | |||
|---|---|---|---|---|
| Protein | Hp | Cortex | Hp | Cortex |
| pERK1 | 133% | 90% | 102% | 88% |
| pERK2 | 130 | 92% | 116% | 90% |
| pELK | 121% | 93% | 95% | 91% |
| pCREB | 116% | 98% | 119%* | 123%* |
| pJNK1 | 73% *** | 71%* | 107% | 93%* |
| pJNK2/3 | 72%* | 73%* | 103% | 93% |
| pCAMKIIα | 35%*** | 49%** | 104% | 108% |
| pCAMKIIβ | 28%*** | 33%*** | 104% | 92% |
| pGSK3β | 27%*** | 33%** | 123% | 115% |
| pAKT | 25%*** | 36%* | 144%* | 130% |
| pRSK | 27%** | 31%* | 125% | 147%* |
| pGJA1 | 63%* | 47%** | 69%* | 90% |
| tRSK | ND | 95% | ND | 114% |
Similar Western analyses were carried out with freshly processed (FSH) tissues. As shown in Table 1b, results are highly similar to those obtained with FZ tissues. Phospho-CREB levels are slightly higher in hippocampus and cortex, while there are region-specific marginal differences in pJNK1, pAKT, pRSK and pGJA1.
3.2. Phosphorylation patterns following incubation at room temperature
Although protein-specific phosphorylation levels at initial time points were frequently lower in ST than FZ tissues, heat stabilization was effective, as seen by comparing phosphorylation levels in tissues that were held at room temperature for 30 minutes prior to lysate preparation (Figures 2b and 4b). The loss of signal in FZ tissues for the majority of phospho-proteins is visually striking. Data in Figure 3 and Table 3a show that, for all twelve proteins, the reductions were significant, and that, for the majority, the losses were greater than 50%. In ST tissues, however, for the majority of proteins, phosphorylation levels were statistically unchanged from initial values (Table 3b; Figures 3 and 5).
Table 3. Levels of phosphorylation are largely unchanged in heat stabilized tissues but significantly decreased in frozen and fresh tissues after 30 minutes at room temperature.
All comparisons are relative to 100% for that method at 0 minutes. a) Frozen, b)Stabilized, c) Fresh. *, p<0.05; **, p<0.01, ***, p<0.001.
| (a) FZ | (b) ST | (c) FSH | ||||
|---|---|---|---|---|---|---|
| Protein | Hp | Cortex | Hp | Cortex | Hp | Cortex |
| pERK1 | 25%** | 40%** | 98% | 109% | 52%* | 48%* |
| pERK2 | 19%*** | 23%*** | 105% | 112% | 48%* | 46%* |
| pELK | 69% | 75%* | 85% | 91% | 83% | 91% |
| pCREB | 62%* | 62%** | 87% | 86% | 61%*** | 60%*** |
| pJNK1 | 24%*** | 22%*** | 104% | 70%* | 100% | 64%*** |
| pJNK2/3 | 25%*** | 25%*** | 90% | 67%* | 93% | 60%** |
| pCAMKIIα | 25%*** | 44%** | 93% | 97% | 38%** | 35%*** |
| pCAMKIIβ | 16%*** | 34%*** | 87% | 86% | 29%** | 30%*** |
| pGSK3β | 14%*** | 16%*** | 90% | 83% | 53%* | 43%** |
| pAKT | 19%** | 38%* | 75%* | 54%* | 37%*** | 42%** |
| pRSK | 48%* | 58% | 44%* | 29%** | 53%** | 44%** |
| pGJA1 | 18%** | 30%** | 79% | 99% | 45%** | 36%** |
| tRSK | ND | 59%** | ND | 73% | ND | 80%* |
Data in Table 3 also illustrate protein-specific and brain region-specific differences in stability. In frozen tissues, phosphorylation of ELK, CREB and RSK were the least labile with room temperature incubation, decreasing by <40%-50% of initial levels in both hippocampus and cortex. All other phosphorylations decreased to <25% in hippocampus and <40% in cortex, with the most labile being phosphorylation of GSK3B, which was reduced to <20% in both hippocampus and cortex. Stabilized tissues also showed region-specific effects, although reductions were few and less dramatic compared with FZ tissues. Levels of pRSK and pAKT were decreased in both hippocampus and cortex, while decreases in pJNK1 and pJNK2/3 were seen only in cortex. Notably, in both hippocampus and cortex, levels of pGSK3B, extremely labile without stabilization, were completely preserved.
Phosphorylation in FSH tissues shows the same overall patterns of lability as FZ, although pELK and pJNK1/2/3 levels are modestly better preserved.
3.3. Sumoylation patterns
Sumoylation is reversible and known to regulate activities and intracellular localizations of a number of proteins and to function in concert with phosphorylation (Seeler and Dejean 2003; Manza et al., 2004; Yang et al., 2003). Figure 6a shows patterns of SUMO3 modification in lysates from cortex. As expected from prior reports, free SUMO3 monomers, dimers and trimers are observed in addition to a complex pattern of higher molecular weight bands (>100 kD) indicative of SUMO3 ligated to target proteins. The intensities of the SUMO3 monomer and the higher molecular weight bands appear similar in FZ and ST tissues at 0′ RT. However careful inspection shows two differences: in FZ vs. ST tissues, the SUMO3 trimer band at ∼50 kD is slightly more intense and there is a faint smear in the size range of 70-95 kD. After 30′ RT incubation, both these signals, in particular the SUMO3 trimer band, are increased in the FZ tissues. Furthermore, quantitation of the SUMO3 monomer band relative to actin shows that it too is increased in FZ vs. ST tissues (data not shown). These data suggest that, like phosphorylation, sumoylations is labile in excised tissues.
Figure 6. Decreased levels of sumoylation and increased proteolytic processing of calcineurin and TRKA in lysates from frozen vs. stabilized cortex.
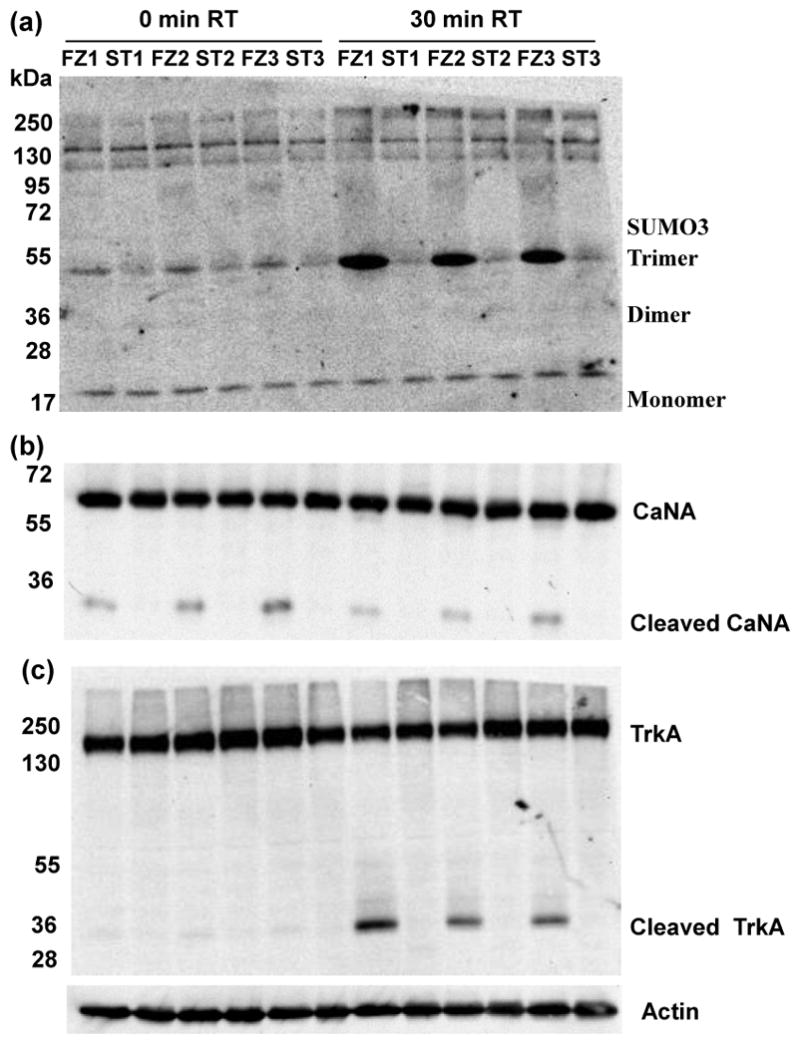
Samples are the same as in Figure 4. (a) Free SUMO3 monomers, dimers and trimers are indicated. Higher molecular weight bands are SUMO-modified proteins. A faint smear between 72 and 95 kD is visible on the frozen (FZ) samples. (b) Calcineurin catalytic subunit, CaNA major signal at ∼60 is indicated. A smaller fragment at ∼30 kD is seen only in FZ samples. (c) TRKA signal is indicated at ∼140 kD; a TRKA-specific fragment at ∼35 kD is seen only in FZ samples.
3.4. Proteolytic processing
Lee et al., (2007) have shown that, under conditions of increased oxidative stress, the protein phosphatase, calcineurin, specifically the catalytic subunit, CaNA, is subject to cleavage by a lysosomal protease of unknown identity. Cleavage is site specific and releases a 28 kD band, destroying phosphatase activity. In Figure 6b, western analysis shows that in FZ tissues there is a significant amount of CaNA cleavage even at 0 minutes. In contrast, in ST tissues, the cleavage product is almost undetectable even at 30 minutes. This cleavage could be a technical artifact, due to rapid loss of the integrity of intracellular compartments during FZ tissue processing and consequent release of proteases to the cytosol, or it could be due to a biologically controlled response to ischemia, one that programs inactivation of some protein modification enzymes. In a separate experiment, we examined levels of the neurotrophin receptor, TRKA, as an example of a large transmembrane protein that is also subject to proteolytic processing (Diaz-Rodriguez et al 1999). In Figure 6c, the large TRKA 140 kD band appears intact in FZ and ST cortex at 0 minutes. After 30 minutes, however, in FZ tissue, a strong band is visible at ∼30 kD. Closer inspection shows that this band was present, although extremely faint, even at 0 minutes. Consistent with the appearance of the smaller band, the intensity of the intact 140 kD band is visually decreased in the FZ tissue. Together, these data suggest that, similar to CaNA, TRKA is being specifically cleaved. Further experiments are required to determine if this cleavage is due to the same metalloproteases which produced a band described as ∼40 kD (Diaz-Rodriguez et al 1999) or a technical artifact due to disruption of cellular compartments.
4. Discussion
These experiments directly compared the effects of heat stabilization using the Stabilizor T1 vs. snap freezing and fresh processing for the preservation of protein features in mouse brain tissue. PTMs in general are dynamic and reversible, and changes in phosphorylation and sumoylation are expected to occur in brain as a result of ischemia and hypoxia following death as cells alter the activity levels and targets of their endogenous phosphatases, kinases and sumo ligases and proteases. In all three protocols, the time between sacrifice and tissue preservation (by heat, freezing or lysis) was short (Table 1). Common to all protocols is the ∼30 seconds required to remove the brain from the skull. Completion of whole brain heat stabilization required a further 50 seconds, during which time the brain was being heated to 95°. Dissection of brain regions in the other two protocols, whether to freezing in liquid nitrogen or to lysis buffer for fresh processing, required 90 seconds, during which time the brains were held on ice. Given differences in time and temperature, it is therefore reasonable that cellular and enzymatic responses will differ during processing and result in different endpoint protein profiles. Notably, none of these protocols, not even FSH, can be claimed to faithfully reflect the in vivo state for every protein. One general interpretation, for example, is that phosphorylation rapidly increases in FZ and FSH tissues in the initial two minutes post euthanasia due to the time required for dissection (on ice) and an imbalance in enzyme activities that favors kinases over phosphatases. These changes are either different (favoring phosphatases over kinases) or more limited in stabilized tissues, due to the shorter time frame and higher temperatures, giving rise to the significantly lower levels of specific phosphorylation at time 0 in stabilized vs. non-stabilized tissues shown in Table 2. It is not possible to distinguish between these two scenarios. During a 30 minute room temperature incubation, in non-stabilized tissues, enzyme activity levels continue to change. In stabilized tissues, however, enzymes are largely inactivated, and further phosphorylation profile changes are very limited. Consistent with the latter observation, Svensson et al (2009), in evaluation of the Stabilizor T1, demonstrated residual phosphatase activity of approximately 10% relative to non-stabilized tissue in the presence of phosphatase inhibitors. While this seems negligible, if it is tissue and protein-specific, it could give rise to the decreases in phosphorylation levels of RSK, AKT and JNK listed in Table 3b.
Analysis of protein feature preservation in other studies (Svensson et al 2008; O'Callahan and Sriram 2004) has focused on phosphorylation profiles. Here, we have shown that sumoylation profiles also change with post mortem interval and tissue handling. This is not inconsistent with the observations of dramatic changes in sumolyation profiles in response to oxidative stress (Manza et al., 2004). We also show apparent site-specific processing of a subunit of the protein phosphatase, calcineurin, and of the nerve growth factor receptor, TRKA. It remains to be determined if these are biologically programmed responses to post mortem intervals and temperature changes, and indeed, the same cleavages as reported under other conditions (Lee et al 2007; Diaz-Rodriguez et al 199), or if one or both are technical artifacts due to release of normally sequestered proteases.
Time delays between death (and attendant cell molecular responses to ischemia and hypoxia) and protein preservation are unavoidable. The data presented here make clear that unambiguous determination of in vivo levels of protein modification and processing is a challenge and that different protocols for tissue handling and protein preparation will result in significantly different protein profiles. In evaluating the relative efficacy of these protocols, three recent observations from analysis of human and mouse tissues are relevant. First, Ferrer et al (2007) showed that tissue samples from human autopsies stored at 0°C had higher levels of specific phosphorylation that remained stable for longer time periods (several hours to several days), in comparison with tissues stored at 4°C. Second, Espina et al (2008), in analysis of eleven different normal or tumor tissue biopsy samples, showed that kinases remained active, and contrary to an expected rapid loss of phosphorylation at room temperature, for most protein phosphorylation examined, levels increased significantly over tens of minutes before decreasing after a few hours. Lastly, Snyder et al (2007) showed that treatment of mice with common general anesthetics altered phosphorylation of numerous neurologically relevant proteins, among them ERK2, DARRP32 and the NR1 subunit of the NMDA receptor, in a brain region-specific fashion. Notably, they used focused microwave irradiation to euthanize the mice; given the potential for inducing stress responses in mice in application of this method, use of preliminary anesthesia might be attractive, but clearly would add to variation in the protein profiles.
Heat stabilization appears to rapidly and effectively inactivate a broad range of protein modifying and processing enzymes. However, there are additional practical considerations to its use. We noted protein-specific differences between hippocampus and cortex in responses to heat stabilization, and therefore additional differences may exist in similarly treated non-brain tissues. Multiple tissues from a single animal cannot be stabilized within the same time frame. The alternative is rapid freezing of these tissues in liquid nitrogen, to be followed at a later time by stabilization. A second consideration is that heat stabilization, whether with the Stabilizor T1 or focused microwaves, in denaturing proteins, destroys intracellular structures (although NOT tissue morphology); while this does not affect tissue dissection, either manually as used here or by laser capture microdissection, it precludes subcellular fractionation, thus limiting some types of studies. Lastly, the short time delays, <2 minutes, used here are practical only for laboratory animals. For analysis of human samples, collected at biopsies or autopsies, time delays will be longer, variable and uncontrollable. Rather than reliable measurement of in vivo levels, a more reasonable goal will be consistent timing and treatment of tissue samples to generate consistent post mortem or post excision alterations. Maintenance of samples on ice during delays required by pathology followed by stabilization should provide more consistent results. It must be recognized that post mortem and post excision delays may have altered some patterns of disease vs. normal differences. This may be of particular note if the disease phenotype includes mutation or variation in protein modification or processing enzymes. These abnormalities could in theory be obscured or exacerbated by time frame and method of protein lysate preparation.
Acknowledgments
The authors thank Alberto Costa and Melissa Stasko for technical assistance in providing mouse tissues. This work was supported by the Fondation Jerome Lejeune, the National Institutes of Health (HD056235), the Anna and John J Sie Foundation and the University of Colorado School of Medicine. Denator AB provided the Stabilizor T1 instrument and funding for purchase of antibodies; they did not participate in planning of experiments, analysis of results or writing of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Deribe YL, Pawson T, Dikic I. Post-translational modifications in signal integration. Nat Struct Mol Biol. 2010;17:666–672. doi: 10.1038/nsmb.1842. [DOI] [PubMed] [Google Scholar]
- Díaz-Rodríguez E, Cabrera N, Esparís-Ogando A, Montero JC, Pandiella A. Cleavage of the TrkA neurotrophin receptor by multiple metalloproteases generates signalling-competent truncated forms. Eur J Neurosci. 1999;11:1421–1430. doi: 10.1046/j.1460-9568.1999.00552.x. [DOI] [PubMed] [Google Scholar]
- Espina V, Edmiston KH, Heiby M, Pierobon M, Sciro M, Merritt B, Banks S, Deng J, VanMeter AJ, Geho DH, Pastore L, Sennesh J, Petricoin EF, 3rd, Liotta LA. A portrait of tissue phosphoprotein stability in the clinical tissue procurement process. Mol Cell Proteomics. 2008;7:1998–2018. doi: 10.1074/mcp.M700596-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Santpere G, Arzberger T, Bell J, Blanco R, Boluda S, Budka H, Carmona M, Giaccone G, Krebs B, Limido L, Parchi P, Puig B, Strammiello R, Strobel T, Kretzschmar H. Brain protein preservation largely depends on the postmortem staorage temperature: Implications for study of proteins in human neurologic diseases and management of brain banks: A brainNet Europe study. J Neuropathol Exp Neurol. 2007;66:35–46. doi: 10.1097/nen.0b013e31802c3e7d. [DOI] [PubMed] [Google Scholar]
- Lee JE, Kim H, Jang H, Cho EJ, Youn HD. Hydrogen peroxide triggers the proteolytic cleavage and the inactivation of calcineurin. J Neurochem. 2007;100:1703–1712. doi: 10.1111/j.1471-4159.2006.04340.x. [DOI] [PubMed] [Google Scholar]
- Manza LL, Codreanu SG, Stamer SL, Smith DL, Wells KS, Roberts RL, Liebler DC. Global shifts in protein sumoylation in response to electrophile and oxidative stress. Chem Res Toxicol. 2004;17:1706–15. doi: 10.1021/tx049767l. [DOI] [PubMed] [Google Scholar]
- O'Callaghan JP, Sriram K. Focused Microwave irradiation of the brain preserves in vivio protein phosphorylation: comparison with other methods of sacrifice and analysis of multiple phosphorylations. J Neurosci Methods. 2004;135:159–168. doi: 10.1016/j.jneumeth.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Seeler JS, Dejean A. Nuclear and unclear functions of SUMO. Nat Rev Mol Cell Biol. 2003;4:690–699. doi: 10.1038/nrm1200. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Galdi S, Hendrick JP, Hemming HC., Jr General anesthetics selectively modulate glutamatergic and dopaminergic signaling via site-specific phosphorylation in vivo. Neuropharmacol. 2007;53:619–630. doi: 10.1016/j.neuropharm.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Svensson M, Boren M, Sköld K, Fälth M, Sjögren B, Andersson M, Svenningsson P, Andren PE. Heat stabilization of the tissue proteome: a new technology for improved proteomics. J Proteome Res. 2009;8:974–81. doi: 10.1021/pr8006446. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Jaffray E, Hay RT, Sharrocks AD. Dynamic interplay of the SUMO and ERK pathways in regulating Elk-1 transcriptional activity. Mol Cell. 2003;12:63–74. doi: 10.1016/s1097-2765(03)00265-x. [DOI] [PubMed] [Google Scholar]