

Abstract
Camptothecin (CPT), a potent antitumor drug, exhibits poor aqueous solubility and rapid conversion from the pharmacologically active lactone form to inactive carboxylate form at physiological pH. Solid dispersion of CPT in Soluplus®, an amphiphilic polymeric solubilizer, was prepared to increase the aqueous solubility of CPT and the resultant solid dispersion along with citric acid was formulated as hard gelatin capsules that were subsequently coated with Eudragit S100 polymer for colonic delivery. FTIR spectrum of the solid dispersion confirmed the presence of CPT. PXRD and DSC revealed the semicrystalline nature of solid dispersion. The solubility of the drug was found to increase ~40 times in the presence of Soluplus and ~75 times in solid dispersion. The capsules showed no drug release in 0.01 N HCl but released 86.4% drug in lactone form in phosphate buffer (pH 7.4) and the result appears to be due to citric acid-induced lowering of pH of buffer from 7.4 to 6.0. Thus the presence of citric acid in the formulation led to stabilization of the drug in its pharmacologically active lactone form. Cytotoxicity studies conducted with the formulation of solid dispersion with citric acid, utilizing cell cytotoxicity test (MTT test) on Caco-2 cells, confirmed cytotoxic nature of the formulation.
KEY WORDS: Caco-2, Camptothecin, colon targeting, Eudragit S100, solid dispersion, Soluplus
INTRODUCTION
The incidence rate of colorectal cancer, a major cause of mortality in the western world (1) is increasing. Colorectal cancer begins as benign adenomatous polyps, progresses to adenoma and further to an invasive cancer (2). Cancer confined within the walls of the colon (stages I and II) is amenable to surgery. If left untreated, the cancer may spread to lymph nodes (stage III) and may be treated by surgery in combination with adjuvant chemotherapy. Further spread of cancer leads to metastasis (stage IV), in which case the chances of survival may be improved by chemotherapy, though the disease in this stage is mostly incurable (3,4). For advanced stages of colorectal cancer, chemotherapy is based on the thymidylate-synthase inhibitor, 5-Fluorouracil (5-FU) (5). A combination of 5-FU and leucovorin, another cytotoxic agent, augments the cytotoxic effects by enhancing the binding affinity of the active fluorouracil-metabolite to thymidylate-synthase. However, the said combination administered in different treatment schedules did not increase the overall life expectancy of patients (6). Therefore there is a need for development of new chemotherapeutic drugs and drug delivery systems for the treatment of colon cancer.
Camptothecin (CPT), a modified monoterpene indole alkaloid, produced by Camptotheca acuminate, was first isolated by Wall and co-workers in 1966 (7). CPT has a pentacyclic ring structure (Fig. 1), having a pyrrolo (3,4-β)-quinoline moiety (A, B, and C), conjugated pyridone moiety (ring D) and one chiral center at position 20 within the α-hydroxy lactone ring with (S) configuration (E ring) (8). CPT has exhibited potent antitumor activity in many experimental tumor types (9). CPT exerts the antitumor effects by specific inhibition of eukaryotic DNA topoisomerase-I (topo-I), an enzyme playing pivotal roles in DNA replication, transcription, recombination, and repair. CPT exerts its cytotoxicity by creating DNA break by covalently trapping topo-I-DNA cleavable complex, thereby converting topo-I into a cellular poison (10). However, the full therapeutic potential of CPT, could not be exploited because of its poor aqueous solubility and rapid inactivation to the water soluble, pharmacologically inactive carboxylate form, due to lactone ring hydrolysis at physiological pH (pH > 7) as depicted in Fig. 1 (11,12). The carboxylate form binds to human serum albumin and hence inhibits its own cellular uptake (13). Further, the carboxylate form is responsible for the severe toxicity of the drug including hemorrhagic cystitis and myelotoxicity (14,15). Furthermore, studies have revealed that the lactone ring is essential for the passive diffusion of the drug in cancer cells and for interaction with topo-I (16). Different approaches have been utilized to alleviate the solubility and stability issues of CPT. As CPT is readily soluble in various lipids, while maintaining its biological activity, liposomal drug delivery system was designed to improve the drug cellular internalization and decrease in systemic toxicity (17). Microspheres of CPT in H-series of poly(D, L-lactide-co-glycolide) were designed to be implanted or injected directly at the tumor site (18). A prodrug of CPT was prepared by condensation of CPT with poly (ethylene glycol) 40 kDa dicarboxylic acid in the presence of diisopropylcarbodiimide and resultant ester when administered intraperitoneally, as an aqueous solution in P388-treated mice, demonstrated increase in life expectancies and cure rate (19). In vitro drug release study of complexes of CPT with alpha-cyclodextrin, beta-cyclodextrin, gamma-cyclodextrin, hydroxypropyl-beta-cyclodextrin, and randomly substituted dimethyl-beta-cyclodextrin, respectively, exhibited an increase in solubility and stability of CPT in 0.02 N HCl (20). However, all these approaches focus on increasing the solubility and/or stability of CPT, and literature is silent on any study conducted to deliver the improved drug formulation directly to colorectal cancerous tumors, by oral administration.
Fig. 1.
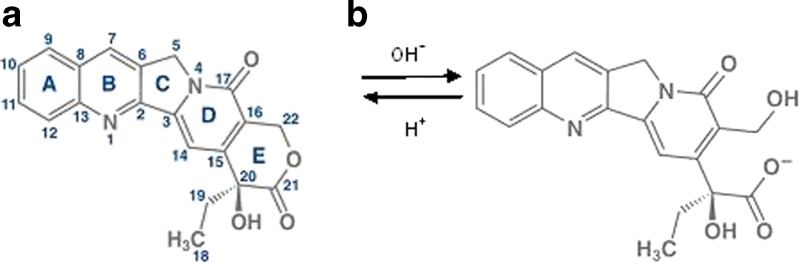
Chemical structure of a lactone and b carboxylate forms of Camptothecin
The aim of the present research was to increase the solubility and stability of CPT and to develop an oral formulation targeted to colon tumor cells in intact form, in order to reduce the side effects. In this study, attempts were made to prepare a solid dispersion of CPT in Solupus®, a polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol graft copolymer that keeps the drug molecularly dispersed in its matrix, potentially enhancing the aqueous solubility of the drug (21). A colon-targeted delivery system of the resultant solid dispersion was developed to achieve a high local concentration, and thus reduce the dose and undue side effects. The specific pH conditions of the colonic region were exploited to deliver the drug to the region, by developing a pH-dependent system, using Eudragit S100 (22). Eudragit S100 is an anionic polymer synthesized from methacrylic acid and methyl methacrylate which dissolves in the region of the digestive tract where pH is >7 (23,24). The system was designed in such a manner that an acidic microenvironment is developed in colon after the release of the drug from the system, to arrest the hydrolysis of the lactone ring of CPT. The cytotoxicity of CPT as well as its solid dispersion was also evaluated using Caco-2 cells.
MATERIALS AND METHODS
Materials
Camptothecin was purchased from Zhejiang Yixin Pharmaceutical Co. Ltd., Soluplus and Eudragit S100 was kindly donated by BASF and Evonic Industries (Mumbai, India), respectively. The following chemicals used in the studies were of analytical grade: ethanol anhydrous (Kemetyl A/S, Denmark), isobutanol, hydrochloric acid, potassioum dihydrogen phosphate, sodium hydroxide, triethylamine, and acetic acid (Merck K GaA, Darmstadt, Germany), MTT [3-(4,5-dimethylthiazol-2yl) 2,5-diphenyltetrazolium bromide], 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), bovine serum albumin, 2-(N-morpholino)ethanesulfonic acid (MES), sodium dodecyl sulfate (SDS), citric acid (anhydrous), all of Sigma Aldrich Labochemikalien GmbH, Hank's buffered salt solution (HBSS) ×10 (GIBCO batch 699997). Acetonitrile (Sigma) was of high-performance liquid chromatography (HPLC) grade.
HPLC Analysis of CPT
CPT was analyzed by HPLC as per the method described by Warner and Burke (25). This method is useful for simultaneous quantification of lactone and carboxylate forms of the drug. The HPLC system used was a Merck Hitachi Instrument, having a pump (L-7100), a Rheodyne injector fitted with a 100-μl sample loop (Cotati, California), an autosampler (L-7200), an interface (D-7000), and a fluorescence detector (L-7480). Separation was carried out using a “Phenomenex” C18 (4.6 × 150 mm) reverse phase column. Mobile phase used for the analysis contained 23 volumes of acetonitrile and 77 volumes of triethylammonium acetate (TEAA) buffer (1% v/v), pH 5.5. A sample volume of 20 μl was injected maintaining a flow rate of 1 ml/min at ambient temperature and CPT was detected employing excitation from 305 to 395 nm and emission from 430 to 470 nm.
Preparation of CPT Solid Dispersion
The ability of CPT and Soluplus to form a molecular dispersion was determined by solubility studies. The studies were carried out by incorporation of 2 g of CPT in 100 ml phosphate buffer pH 6.0 without Soluplus or in 5% solution of Soluplus in phosphate buffer pH 6.0 (100 ml) and placing in an incubator shaker at 37°C at 50 rpm. After 24 h, the suspensions were filtered through a 0.45-μm filter and the resultant solutions were analyzed for drug content by HPLC method as described above.
Based on the solubility experiment, solid dispersion of drug with Soluplus was prepared as follows: CPT (1 g) and Soluplus (3 g) were dissolved in ethanol (50 ml). Ethanol was evaporated on a rotary evaporator at 45°C and 70 mbar (BÜCHI, Switzerland. Rotavapor R-210).
Rotating evaporation was used to accelerate the solvent evaporation at low temperature. At high temperatures, the molecular mobility of drug and matrix remains high that may lead to phase separation (26). Total time consumed to evaporate the ethanol was 25 min.
Solubility of CPT in Solid Dispersion
The studies were carried out by placing 200 mg of pure CPT or 500 mg of solid dispersion (containing 125 mg CPT) in 50 ml phosphate buffer pH 6.0 each and placing in an incubator shaker at 37°C at 50 rpm. After 24 h, the suspensions were filtered through a 0.45 μm filter and the resultant solutions were analyzed for drug content by HPLC as described above.
Drug Content in Solid Dispersion
An accurately weighed solid dispersion of CPT equivalent to 20 mg of CPT was dispersed in 100 ml ethanol and subjected to centrifugation at 3,000 rpm for 10 min. The supernatant was withdrawn and diluted with ethanol to a concentration of ≈20 μg/ml. The resulting solution was analyzed for CPT content by HPLC as described above.
Determination of the Quantity of Citric Acid Required to Lower the pH from 7.4 to 6.0 of 100 ml Phosphate Buffer
It has been reported by researchers that the mean volume of colonic fluid varies between 13 ml (±12) in fasting state and 11 ml (±26) 1 h after meals. In no case, the minimum colonic fluid was less than 1 ml and more than 97 ml (27). Based on this, it was decided to design a system which can lower the pH of colonic fluid (100 ml) from 7.4 to 6.0 or less (pH of the colonic fluid was considered to be 7.4).
Citric acid (5 g) dissolved in 10 ml water was slowly added to 100 ml phosphate buffer (pH 7.4) (28). The volume of citric acid solution needed to lower the pH to 6.0 was noted and the amount of citric acid was calculated accordingly. The amount of citric acid was found to be 270 mg.
Preparation of Colonic Drug Delivery System
As reported, the dose of CPT was estimated to be 200 mg/day for an individual weighing 70 kg (19). Solid dispersion of CPT equivalent to 50 mg of CPT along with 270 mg of citric acid was filled into hard gelatin capsules (size 0). One hundred such capsules were filled using a hand-operated capsule filling machine. These filled capsules were spray coated with a 10% solution of Eudragit S100 in acetone in a coating pan (diameter 8 in., make Instacoat) at around 15 rpm with inlet and outlet temperature of 65°C and 42°C, respectively. The capsules were coated until a weight gain of 12% had been attained. Similarly coated capsules containing 50 mg of pure CPT with 270 mg citric acid each were also prepared to study the in vitro release profile.
On similar lines, coated capsules containing solid dispersion of CPT equivalent to 50 mg of CPT or 50 mg of pure CPT without citric acid were also prepared.
Characterization of CPT Solid Dispersion
Fourier Transform Infrared Spectroscopy
Fourier transform infrared (FTIR) spectra of CPT, Soluplus, and solid dispersion of CPT in Soluplus were recorded using a FTIR spectrophotometer (Bruker model Alpha) in the range of 4,000–500 cm−1 as neat sample.
X-ray Diffraction
X-ray diffractograms of CPT, Soluplus, and solid dispersion of CPT were recorded using an X-ray diffractometer (X'Pert Pro, PW 3050/PW 3071; Lelyweg, The Netherlands) using nickel-filtered CuKα radiation (λ = 1.540598 A°) generated at 40 kV and 30 mA and scanning rate of 2°/min over a 2θ range of 10°–80°.
Differential Scanning Calorimetry
Thermal analysis of CPT, Soluplus, and solid dispersion of CPT was performed using DSC821e system (Mettler Toledo). The sample was sealed in a crimped aluminum pan by application of the minimum possible pressure and heated at a rate of 10°C/min from 20°C to 300°C in a nitrogen atmosphere. An empty aluminum pan was utilized as the reference pan.
In Vitro Drug Release Profile
All the experiments for studying the in vitro release profile were conducted in triplicate. A Eudragit S100 coated capsule was placed in dissolution apparatus (USP type-2, make ERWEKA-DT 70) containing 100 ml of 0.01 N hydrochloric acid (HCl) (pH 2.0) as release medium maintained at 37°C and stirred for 2 h at 50 rpm. Height of the shaft was so adjusted that height of the lower surface of peddle remained 2.5 cm from the bottom of the dissolution vessel. A sinker was used to prevent the floating of the capsule in the release medium. A sample (5 ml) was withdrawn at the end of the experiment, filtered through a 0.45-μm filter and analyzed for drug content by HPLC as described above.
In similar experiments, dissolution of coated capsules containing solid dispersion of CPT or CPT parent bulk with or without citric acid was carried out in 100 ml of phosphate buffer (pH 7.4), keeping all other conditions same as above. Samples (5 ml) were withdrawn at regular intervals and each withdrawn sample was replaced with an equal volume of fresh release medium. The samples were filtered through a 0.45-μm filter and analyzed by HPLC as described previously.
Statistical Analysis
The in vitro drug release data from CPT and its solid dispersion were compared by statistical analysis using one-way analysis of variance (ANOVA). P < 0.05 was considered significant.
Cytotoxicity Study
In order to assess the antitumor activity of CPT and solid dispersion of CPT in acidic (pH 6.0) and mildly alkaline pH (pH 7.4), the MTT test was conducted on the Caco-2 cells as described by Anderberg and Artursson (29). The test is based on the principle that the enzyme dehydrogenase in the mitochondria of living cells converts the yellow MTT [3-(4, 5-dimethylthiazol-2yl) 2,5-diphenyltetrazolium bromide] to a blue-purple formazan crystal. If the cells are exposed to adverse treatment affecting their viability, the activity of the dehydrogenase enzyme is compromised.
Protocols for culturing Caco-2 cells were as previously described (30). Caco-2 cells of passage 40 were seeded onto 96-well Elisa plates (MicroWells, Nunk, Denmark). CPT alone and its solid dispersion with citric acid were tested in eight concentrations (0.05, 0.1, 0.2, 0.4, 1, 250, 500, and 1,000 μM/ml). HEPES (10 mM) was used as buffer pH 7.4 and MES (10 mM) was used as buffer pH 6.0. Cytotoxicity of CPT was tested at pH 7.4 in HEPES and at pH 6.0 in MES. Furthermore, cytotoxicity of solid dispersion with citric acid was tested in HEPES where the presence of citric acid lowered the pH from 7.4 to 6.0. All the solutions were prepared a day before the experiment in HBSS. SDS (concentration ranging from 0.05 to 5.0 mM/ml) was included as a positive control and blank HBSS served as a negative control. HBSS was placed in cell-free wells as an additional background control.
On the day of experiment, the cells were examined under a microscope to ensure that they were attached to the bottom of the wells. The medium was discarded and the test solutions along with the positive, negative, and background controls were transferred to the 96-well Elisa plates. All solutions were tested in triplicate. The tray was incubated at 37°C on a shaker table at 100 rpm (Edmund Bühler GmbH) for 4 h. Then, the wells were emptied and 100 μl of HBSS was added to all the wells along with 20 μl of MTT solution (5 mg/ml). The tray was wrapped in tin foil and incubated for 90 min at 37°C and 100 rpm (on shaker table). At this time, insoluble blue-purple formazan crystals were formed in viable cells. Subsequently 100 μl of solvent containing SDS (11% w/v), isobutanol (50% v/v), and 0.02 N hydrochloric acid (50% v/v) was added to all the wells and the tray was again wrapped in tin foil and set aside on the shaker table at 37°C overnight, to allow dissolution of the blue-purple crystals. On the following day, the absorbance was measured at 595 nm using a plate-reader (Labsystems Multiskan MS).
RESULTS AND DISCUSSION
Solubility Studies and Preparation of Solid Dispersion
Solubility of CPT alone and in the presence of Soluplus is depicted in Fig. 2. Solubility of CPT was 623 μg/100 ml at pH 6.0, while the solubility of CPT in presence of Soluplus was 26.65 mg/100 ml at the same pH. Thus, the solubility of CPT was increased almost 40 times in the presence of Soluplus, indicating excellent affinity between CPT and Soluplus to form a molecular dispersion. Soluplus is a polymeric solubilizer with an amphiphilic chemical structure, having large number of hydroxyl groups which make it a good solubilizer for poorly soluble drugs in aqueous media. As expected, Soluplus has been found to have substantial impact on the solubility of CPT.
Fig. 2.

Solubility of CPT alone, CPT in the presence of Soluplus, and solid dispersion of CPT in Soluplus in phosphate buffer pH 6.0
Solubility of CPT in Solid Dispersion
Solubility of CPT and solid dispersion is depicted in Fig. 2. Solubility of CPT was 623 μg/100 ml at pH 6.0, while the solubility of drug from solid dispersion was found to be 47.44 mg/100 ml at pH 6.0, indicating around 75 times increase in solubility.
Drug Content in Solid Dispersion
Drug content in solid dispersion was found to be 24.42% w/w.
Characterization of Solid Dispersion
FTIR Spectroscopy
FTIR spectra of CPT, Soluplus, and solid dispersion are shown in Fig. 3. FTIR spectrum of CPT showed –OH stretching at 3,425.7 cm−1, carbonyl stretching for cyclic ester (lactone) at 1,736 cm−1, C–C(=O)–O stretching at 1,148 cm−1, C=O stretching at 1,648 cm−1 for pyridone, C=C and C–N stretching for quinoline ring at 1,597, 1,572, and 1,436 cm−1. Peak at 767 cm−1 appears to be a contribution of four adjacent hydrogen bonds on hetero-aromatic nucleus. Spectrum of Soluplus, showed inter-molecularly hydrogen bonded –OH stretching in the 3,350–3,550 cm−1 range, ester carbonyl stretching at 1,729 cm−1, C=O stretching for tertiary amide at 1,616 cm−1, C–O stretching for ester at 1,233 and 1,109 cm−1 and CH3 bending at 1,436 cm−1. The FTIR spectrum of solid dispersion of CPT in Soluplus exhibited peaks at 1,734, 1,601, 1,436, 1,149, and 768 cm−1, suggesting presence of CPT in the solid dispersion. The –OH stretching, however, appeared at 3,424.7 cm−1 and became slightly broader, which could be due to an interaction with Soluplus. Similarly CPT peak of 1,597 cm−1 appeared as a principal peak at 1,601 cm−1 in the spectrum of solid dispersion, while CPT peaks of 1,648 and 1,572 cm−1 appeared as shoulders at 1,650 and 1,574 cm−1, which also suggest a possible interaction.
Fig. 3.

FTIR spectra of 1 CPT, 2 Soluplus, and 3 solid dispersion of CPT
Powder X-Ray Diffraction
The powder X-ray diffraction (XRD) pattern of CPT, Soluplus, and solid dispersion are shown in Fig. 4.The XRD indicated an amorphous structure of Soluplus while CPT was crystalline. The solid dispersion appears to be a mixture of crystalline and amorphous forms or semicrystalline in nature, as characteristic halo pattern for the amorphous form is also present along with the sharp peaks. XRD of CPT showed principal peaks at 25.59°, 17.70°, and 13.37° 2θ, while in solid dispersion, the said peaks appeared at 25.95°, 17.93°, and 13.33° 2θ, respectively, but the peak intensities were drastically reduced indicating marked reduction in crystallinity. The results suggest that the drug CPT is present as crystalline fraction in the solid dispersion and the crystallinity of the drug has been markedly reduced. The solid dispersion was stored at room temperature for 1 year and the sample was subjected to powder X-ray diffraction. The X-ray diffractogram indicated semicrystalline characteristic of drug in solid dispersion (Fig. 4), which suggests that storage at room temperature did not promote the crystallization of drug.
Fig. 4.

X-ray diffractograms of A CPT, B Soluplus, C solid dispersion of CPT in Soluplus, and D solid dispersion of CPT in Soluplus after 1 year storage at room temperature
Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) thermograms of the drug, polymer, and the solid dispersion are presented in Fig. 5. Pure CPT exhibited sharp melting endotherms at 259.07°C and 268.58°C, respectively, indicating the crystalline nature of the drug. However, in the thermogram of the solid dispersion of drug in Soluplus, the two melting endotherms of drug were replaced by a broad depressed endotherm having peak at 257.47°C. The physical mixture of drug and Soluplus (1:3) showed two endotherms at 260.74°C and 269.60°C, respectively. It appears that the replacement of the two melting endotherms of CPT by a broad depressed endotherm, in the DSC thermogram of CPT-Soluplus solid dispersion, where drug/polymer ratio was 1:3, was due to a dilution effect contributed by the amorphous polymer, as the solid dispersion contained 24.42% drug. The solid dispersion was stored at room temperature for 1 year and the sample was subjected to differential scanning calorimetry. The DSC thermogram showed a broad endotherm at 252.43°C (Fig. 5) which implies that storage at room temperature did not promote the crystallization of drug.
Fig. 5.

DSC thermograms of A Camptothecin, B Soluplus, C physical mixture of CPT and Soluplus in ratio 1:3, D solid dispersion of Camptothecin in Soluplus, E Camptothecin after 1 year storage at room temperatuere, and F solid dispersion of Camptothecin in Soluplus after 1 year storage at room temperature
In Vitro Drug Release
Exposure of coated capsule in 100 ml of 0.01 N HCl, as release medium, at 37°C for 2 h showed no drug release. Further, no breach of the coating of the capsule was observed indicating perfect enteric coating, which suggests the gastro-resistance of the formulation.
When the release medium was replaced with phosphate buffer (pH 7.4), the cumulative drug release (lactone form) was found to be 1.44% in case of CPT with citric acid as compared to 86.4% in case of solid dispersion of CPT with citric acid, which indicates the dissolution of Eudragit S100 coat at pH 7.4 and release of capsule contents. In the release medium no carboxylate form was detected. The presence of lactone form and absence of carboxylate form in the release medium could be attributed to the lowering of pH of release medium from 7.4 to 6 induced by citric acid. This in vitro release data, when compared statistically using one-way ANOVA, the extent of drug release was found to be significantly different (P < 0.05) between CPT (parent bulk) and solid dispersion of CPT.
But in case of CPT and its solid dispersion without citric acid, the cumulative drug release (carboxylate form) was 96.2% and 97.1%, respectively, in phosphate buffer (pH 7.4), indicating the dissolution of Eudragit S100 coat in pH 7.4. Lactone form of drug was found to be absent in the release medium, indicating the complete hydrolysis of lactone to the more soluble carboxylate form in release medium. As evident from Fig. 6, the cumulative drug release of carboxylate form in both cases, i.e., in case of CPT and its solid dispersion was not significantly different (P > 0.05).
Fig. 6.

In vitro release profile of CPT and solid dispersion of CPT in Soluplus; A CPT (lactone) in presence of citric acid; B Solid dispersion of CPT (lactone) in presence of citric acid; C CPT (carboxylate) in absence of citric acid; and D Solid dispersion of CPT (carboxylate) in absence of citric acid (values are mean±SE)
The results suggest that the Eudragit S100 coated capsules containing solid dispersion of CPT and citric acid, on oral ingestion, will disintegrate in the colon where the pH is between 7.0 and 8.0, because Eudragit S100 dissolves at pH > 7.0. On disruption of capsule shell, citric acid present in the capsule would lower the colonic fluid pH to 6.0 where solid dispersion of CPT would dissolve to release CPT in lactone form which would be subsequently available for uptake by tumor cells. Direct targeting of the drug in its absorbable form to colon would reduce the dose as well as systemic side effects.
Cytotoxicity Study
The cytotoxicity of CPT (in MES pH 6.0), solid dispersion of CPT along with citric acid (in HEPES, pH 6.0, due to presence of citric acid) and CPT (in HEPES pH 7.4) was assessed by recording color intensities (absorbance) of the wells. The percentage dehydrogenase activity was calculated using absorbance data and plotted against the concentration. Dehydrogenase activity of the cells in HBSS pH 7.4 varied from a minimum of 106% to a maximum of 130%, indicating the absence of toxicity of camptothecin to Caco-2 cells in the concentration range used (50 nM–1 mM/ml). This may be due to the hydrolysis of lactone ring of CPT to inactive carboxylate form (Figs. 7 and 8).
Fig. 7.

MTT test: percentage dehydrogenase activity Vs concentration of drug. A CPT at pH 7.4, B CPT at pH 6.0, C solid dispersion of CPT with citric acid
Fig. 8.

Photograph of 96-well tray depicting results of MTT test
The dehydrogenase activity of the cells with CPT at pH 6.0 and that of solid dispersion of CPT along with citric acid at pH 6.0 varied from 15% to 32% and 13% to 26%, respectively, indicating the cytotoxicity of CPT and its solid dispersion in acidic pH. This appears to be due to the presence of CPT with the intact lactone ring in acidic pH.
Therefore, CPT requires an acidic environment to prevent the hydrolysis of lactone ring and to retain its cytotoxic activity. CPT is taken up by tumor cells having acidic extracellular pH (31), which also favors the stability of the lactone ring, and hence the cytotoxicity of the drug.
The tumor suppressor protein p53 is required for induction of apoptosis in cells with damaged DNA (32). DNA strand break during replication, induces wild type p53 protein that may lead to arrest of cell cycle at G1–S transition (33,34). It has been reported that the p53 pathway is inactivated in the majority of human cancers by mutation of p53 (35). Also, in Caco2 cells a point mutation in the p53 gene in exon-6 (codon 204) leads to a change in the codon GAG (glu) to TAG (stop), which leads to termination of translation, leading to loss of protein expression (36). This suggests the existence of an alternate pathway other than the p53 pathway for the cytotoxicity of CPT in Caco-2 cells.
In addition to arrest of G1–S transition, arrest in G2 phase is also a cellular response to DNA damage (37). It has also been reported that p53 is not required for all forms of DNA-induced apoptosis and an important checkpoint for apoptopic induction exists at G2 (38). Moreover, the G2 checkpoint is critical at low levels of DNA damage and high CPT concentrations can arrest the cells in the S phase itself (39). Therefore the cytotoxicity of CPT observed in Caco2 cells may be attributed to apoptosis due to G2 and S checkpoints not involving p53 protein.
CONCLUSION
Solid dispersion of CPT in Soluplus was prepared to increase the aqueous solubility of CPT and the resultant solid dispersion along with citric acid was formulated as hard gelatin capsules that were subsequently coated with Eudragit S100 polymer for colonic delivery. The capsules showed no drug release in 0.01 N HCl but released 86.4% drug in lactone form in phosphate buffer (pH 7.4) and the result appears to be due to citric acid-induced lowering of pH of buffer from 7.4 to 6.0. CPT being in lactone form would be taken up by tumor cells in vivo. Direct targeting of the drug in its absorbable form to colon would reduce the dose as well as systemic side effects. Studies with Caco-2 cells confirmed the cytotoxicity of the formulation. Thus, the formulation of solid dispersion of CPT in Soluplus with citric acid could be beneficial in colon cancer. The authors believe that further research efforts are warranted to assess the potential of the developed delivery system targeted to release around colorectal tumors and eventually to ascertain the efficacy of the formulation in real-life situation.
REFERENCES
- 1.Giovannucci E. Modifiable risk factors for colon cancer. Gastroenterol Clin North Am. 2002;31:925–943. doi: 10.1016/S0889-8553(02)00057-2. [DOI] [PubMed] [Google Scholar]
- 2.Kinzler KW, Vogelstein B. Colorectal tumors. In: Vogelstein B, Kinzler KW, editors. The genetic basis of human cancer. 2. New York: McGraw-Hill; 2002. pp. 583–612. [Google Scholar]
- 3.Compton C, Hawk ET, Grochow L, Lee F, Ritter M, Niederhuber JE. Colon cancer. In: Abeloff MD, Armitage J, Niederhuber JE, Kastan MB, McKenna GW, editors. Abeloff's Clinical Oncology. Philadelphia: Churchill Livingstone; 2008. pp. 1477–1534. [Google Scholar]
- 4.Libutti SK, Saltz LB, Tepper JE. Colon cancer. In: DeVita VT Jr, Lawrence TS, Rosenberg SA, editors. DeVita, Hellman and Rosenberg's Cancer Principles and Practice of Oncology. Vol.1. Philadelphia: Lippincott Williams & Wilkins; 2008. pp. 1232–1284. [Google Scholar]
- 5.Moertel CG. Chemotherapy for colorectal cancer. N Eng J Med. 1994;330:1136–1142. doi: 10.1056/NEJM199404213301608. [DOI] [PubMed] [Google Scholar]
- 6.DiPiro JT. Pharmacotherapy (a pathophysiologic approach) 7. New York: McGraw-Hill Medical; 2008. [Google Scholar]
- 7.Wall ME, Wani MC, Cook CE, Palmer KH, Mc Phail AT, Sim GA. Plant antitumor agents: I. Isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminata. J Am Chem Soc. 1966;88:3888–3890. doi: 10.1021/ja00968a057. [DOI] [Google Scholar]
- 8.Camptothecin. In: Wikipedia; the free encyclopedia. Accessed 25 June 2010
- 9.Wang JC. DNA topoisomerases. Ann Rev Biochem. 1985;54:665–697. doi: 10.1146/annurev.bi.54.070185.003313. [DOI] [PubMed] [Google Scholar]
- 10.Chen AY, Liu LF. DNA topoisomerases, essential enzymes and lethal targets. Ann Rev Pharmacol Toxicol. 1994;94:194–218. doi: 10.1146/annurev.pa.34.040194.001203. [DOI] [PubMed] [Google Scholar]
- 11.Scott DO, Bindra DS, Stella VJ. Plasma pharmacokinetics of the lactone and carboxylate forms of 20(S)-camptothecin in anesthetized rats. Pharm Res. 1993;10:1451–1457. doi: 10.1023/A:1018919224450. [DOI] [PubMed] [Google Scholar]
- 12.Chourpa I, Millot JM, Stockalingum GD, Riou JF, Manfait M. Kinetics of lactone hydrolysis in antitumor drugs of camptothecin series as studied by fluorescence spectroscopy. Biochem Biophys Acta. 1998;1379:353–366. doi: 10.1016/S0304-4165(97)00115-3. [DOI] [PubMed] [Google Scholar]
- 13.Burke TG, Mi Z. Preferential binding of the carboxylate form of camptothecin by human serum albumin. Anal Biochem. 1993;212:285–287. doi: 10.1006/abio.1993.1325. [DOI] [PubMed] [Google Scholar]
- 14.Moertel CG, Schutt AJ, Reitemeier RJ, Hahn R. Phase II study of camptothecin (NSC 100880) in the treatment of advanced gastrointestinal cancer. Cancer Chemother Rep. 1972;56:95–99. [PubMed] [Google Scholar]
- 15.Muggia FM, Creaven PJ, Hansen HH, Cohen MH, Sealwry OS. Phase 1 clinical trials of weekly and daily treatment with camptothecin (NSC 100880) Cancer Chemother Rep. 1972;56:515–521. [PubMed] [Google Scholar]
- 16.Hertzberg RP, Caranfa MJ, Holden KG, Jakas DJ, Liu LF. DNA topoisomer, topoisomerase-targeting drugs. New York: Academy Press; 1994. [Google Scholar]
- 17.Daoud SS, Fetouh MI, Giovanella BC. Antitumor effect of liposomes-incorporated camptothecin in human malignant xenografts. Anti-cancer Drugs. 1995;6:83–93. doi: 10.1097/00001813-199502000-00010. [DOI] [PubMed] [Google Scholar]
- 18.Ertl B, Platzer P, Wirth M, Gabor F. Poly(D, L-lactic-co-glycolic acid) microspheres for sustained delivery and stabilization of camptothecin. J Controlled Rel. 1999;61:305–317. doi: 10.1016/S0168-3659(99)00122-4. [DOI] [PubMed] [Google Scholar]
- 19.Greenwald RB, Pendri A, Conover C, Gilbert C. Drug delivery systems. 2. Camptothecin 20–O–poly(ethylene glycol) ester transport forms. J Med Chem. 1996;39:1938–1940. doi: 10.1021/jm9600555. [DOI] [PubMed] [Google Scholar]
- 20.Kang J, Kumat V, Yang D, Chowdhury PR, Hohl RJ. Cyclodextrin complexation, influence on the solubility, stability, and cytotoxicity of camptothecin, an antineoplastic agent. Eur J Pharm Sci. 2002;15:163–170. doi: 10.1016/S0928-0987(01)00214-7. [DOI] [PubMed] [Google Scholar]
- 21.Crowley MM, Zhang F, Repka MA, Thumma S, Upadhye SB, Battu SK, et al. Pharmaceutical application of hot melt extrusion, part 1. Drug Dev Ind Pharm. 2007;33:909–926. doi: 10.1080/03639040701498759. [DOI] [PubMed] [Google Scholar]
- 22.Thakral NK, Ray AR, Majumdar DK. Eudragit S-100 entrapped chitosan microspheres of valdecoxib for colon cancer. J Mater Sci Mater Med. 2010;21(9):2691–2699. doi: 10.1007/s10856-010-4109-2. [DOI] [PubMed] [Google Scholar]
- 23.Li SP, Feld KM, Kowarski CR. Preparation and evaluation of Eudragit acrylic resin from controlled drug release of pseudoephedrine hydrochloride. Drug Dev Ind Pharm. 1991;17:1655–1683. doi: 10.3109/03639049109057314. [DOI] [Google Scholar]
- 24.Thakral NK, Ray AR, Bar-Shalom D, Eriksson AH, Majumdar DK. The quest for targeted delivery in colon cancer: mucoadhesive valdecoxib microspheres. Int J Nanomedicine. 2011;6:1057–1068. doi: 10.2147/IJN.S19561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warner DL, Burke TG. Simple and versatile high-performance liquid chromatographic method for the simultaneous quantitation of the lactone and carboxylate forms of camptothecin anticancer drugs. J Chromatogr. 1997;691:161–171. doi: 10.1016/S0378-4347(96)00426-4. [DOI] [PubMed] [Google Scholar]
- 26.Slade L, Levine H. A food polymer science approach to structure–property relationships in aqueous food systems: non-equilibrium behavior of carbohydrate-water system. In: Slade L, Levine H, editors. Water relationships in food. New York: Plenum Press; 1991. pp. 29–101. [DOI] [PubMed] [Google Scholar]
- 27.Schiller C, Fröhlich CP, Giessmann T, Siegmund W, Mönnikes H, Hosten N, et al. Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Aliment Pharmacol Ther. 2005;22:971–979. doi: 10.1111/j.1365-2036.2005.02683.x. [DOI] [PubMed] [Google Scholar]
- 28.Indian Pharmacopoeia The Indian Pharmacopoeia Commission, Ghaziabad, Ministry of Health & Family Welfare. Government India. 2010;1:559–560. [Google Scholar]
- 29.Anderberg EK, Artursson P. Epithelial transport of drugs in cell culture: VIII. Effects of the pharmaceutical surfactant excipient sodium dodecyl sulfate on cell membrane and tight junctional permeability in human intestinal epithelial (Caco-2) cells. J Pharm Sci. 1993;82:392–398. doi: 10.1002/jps.2600820412. [DOI] [PubMed] [Google Scholar]
- 30.Larsen M, Larsen BB, Frolund B, Nielsen CU. Transport of amino acids and GABA analogues via the human proton-coupled amino acid transporter, hPAT1, characterization of conditions for affinity and transport experiments in Caco-2 cells. Eur J Pharm Sci. 2008;35:86–95. doi: 10.1016/j.ejps.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 31.Wike-Hooley JL, van den Berg AP, van der Zee J, Reinhold HS. Human tumor pH and its variation. Eur J Cancer Clin Oncol. 1985;21:785–791. doi: 10.1016/0277-5379(85)90216-0. [DOI] [PubMed] [Google Scholar]
- 32.Hartwell L. Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell. 1993;71:543–546. doi: 10.1016/0092-8674(92)90586-2. [DOI] [PubMed] [Google Scholar]
- 33.Nelson WG, Kastan MB. DNA strand break, the DNA template alterations that trigger p53-dependant DNA damage response pathways. Mol Cell Biol. 1994;14:1815–1823. doi: 10.1128/mcb.14.3.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slichenmyer WJ, Nelson WG, Slebos RJ, Kastan MB. Loss of a p53-associated G1 checkpoint does not increase cell survival following DNA damage. Cancer Res. 1993;53:4164–4168. [PubMed] [Google Scholar]
- 35.Rodrigues NR, Rowan A, Smith ME, Kerr IB, Bodmer WF, Gannon JV, et al. p53 mutation in colorectal cancer. Proc Natl Acad Sci USA. 1990;87:7555–7559. doi: 10.1073/pnas.87.19.7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Bodmar WF. Analysis of p53 mutations and their expression in 56 colorectal cell lines. Proc Natl Acad Sci USA. 2006;103:976–981. doi: 10.1073/pnas.0510146103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konopa J. G2 block induced by DNA crosslinking agents and its possible consequences. Biochem Pharmacol. 1988;37:2303–2309. doi: 10.1016/0006-2952(88)90355-3. [DOI] [PubMed] [Google Scholar]
- 38.Han Z, Chatterjee D, Ming He D, Early J, Pantazis P, Wyche JH, et al. Evidence for a G2 checkpoint in p53-independent apoptosis induction by X-irradiation. Mol Cell Bio. 1995;15:5849–5857. doi: 10.1128/mcb.15.11.5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldwasser F, Shimizu T, Jackman J, Hoki Y, O'Connor PM, Kohn KW, et al. Correlations between S and G2 arrest and the cytotoxicity of camptothecin in human colon carcinoma cells. Cancer Res. 1996;56:4430–4437. [PubMed] [Google Scholar]