

Abstract
The present study aims to prepare carvedilol (CAR) nanosuspensions using the anti-solvent precipitation–ultrasonication technique to improve its dissolution rate and oral bioavailability. Alpha-tocopherol succinate (VES) was first used as a co-stabilizer to enhance the stability of the nanosuspensions. The effects of the process parameters on particle size of the nanosuspensions were investigated. The optimal values of the precipitation temperature, power inputs, and the time length of ultrasonication were selected as 10°C, 400 W, and 15 min, respectively. Response surface methodology based on central composite design was utilized to evaluate the formulation factors that affect the size of nanosuspensions, i.e., the concentration of CAR and VES in the organic solution, and the level of sodium dodecyl sulfate in the anti-solvent phase, respectively. The optimized formulation showed a mean size of 212 ± 12 nm and a zeta potential of −42 ± 3 mV. Scanning electron microscopy revealed that the nanosuspensions were flaky-shaped. Powder X-ray diffraction and differential scanning calorimetry analysis confirmed that the nanoparticles were in the amorphous state. Fourier transform infrared analysis demonstrated that the reaction between CAR and VES is probably due to hydrogen bonding. The nanosuspension was physically stable at 25°C for 1 week, which allows it to be further processing such as drying. The dissolution rate of the nanosuspensions was markedly enhanced by reducing the size. The in vivo test demonstrated that the Cmax and AUC0–36 values of nanosuspensions were approximately 3.3- and 2.9-fold greater than that of the commercial tablets, respectively.
KEY WORDS: alpha-tocopherol succinate, anti-solvent precipitation, carvedilol, nanosuspensions, oral bioavailability
INTRODUCTION
Solubility and gastrointestinal permeability are the major factors that determine the bioavailability and absorption of a drug(1). However, a major portion of drugs on the market have poor aqueous solubility, a situation that is estimated to be even more striking in the future(2). Over the past decades, nanosuspensions have been widely used to tackle problems associated with poor solubility and low bioavailability. A reduction of particle size would lead to a dramatic increase in the dissolution rate of a drug, which in turn could result in substantial increases in oral bioavailability.
Techniques used to produce drug nanosuspensions can be categorized into two major classes: top-down and bottom-up technologies(3). Top-down approaches mainly comprise media milling and high-pressure homogenization(4). Although these techniques are widely used, their limitations include requiring a long preparation time, difficulty in achieving a narrow size distribution, high energy input, and contamination, which diverts greater attention toward the bottom-up process(3,5). Bottom-up approaches exhibit tremendous potential with respect to improving bioavailability by obtaining smaller particle sizes and amorphous drug particles(5). The anti-solvent precipitation process is one of the most promising techniques to prepare nanoparticles, which is cost-effective, rapid to perform, and suited for scaling up(6,7). In this method, the drug is first dissolved in a water-miscible organic solvent, and is then rapidly mixed with an aqueous stabilizer solution. The technique has been employed for the preparation of micro/nanosuspensions, such as itraconazole(6), spironolactone(8), and megestrol acetate(9). However, inhibiting the growth of freshly precipitated particles driven by Ostwald ripening is still a problem. The prolonged stabilization can only be achieved by immediate drying.
Recently, ultrasound has received much attention and has been proved to be an effective method for controlling the nucleation and crystallization process(10,11). It has been successfully employed to produce stable nanosuspensions.
In the present work, alpha-tocopherol succinate (VES) is introduced into the organic phase as a co-stabilizer during the anti-solvent precipitation process to inhibit the growth of precipitated nanoparticles for the first time. VES is a succinyl ester of alpha-tocopherol (Fig. 1). It contains an open (non-esterified) carboxylic acid group and possesses some amphiphilic characters. VES has reportedly been successfully used to stabilize a docetaxel-loaded intravenous emulsion(12). However, it has not been used to enhance the stability of nanosuspensions.
Fig. 1.

Chemical structures of a alpha-tocopherol and b alpha-tocopherol succinate
Carvedilol (CAR) is a non-selective β-blocking agent that also displays α1-adrenergic antagonism, resulting in a blood pressure-reducing action through vasodilatation(13). The chemical structure is shown in Fig. 2. As a Biopharmaceutics Classification System class II drug, CAR is well absorbed after oral administration, but is nearly insoluble in water. Due to the slow dissolution rate in the intestinal tract and a significant degree of first-pass metabolism, the oral bioavailability of CAR in humans is only 25% to 35%, and it is variable(14). Formulation strategies for overcoming the dissolution-related problems of CAR include the use of lipophilic solutions(15), conversion of the drug to a salt(16), formation of inclusion complex with cyclodextrin(17), the use of a self-emulsifying system(18), and preparation of solid dispersions with porous silica(19). The present work aims to prepare stable CAR nanosuspensions using the anti-solvent precipitation–ultrasonication method to improve the drug dissolution rate and oral bioavailability. VES was used as a co-stabilizer to enhance the stability of nanosuspensions for the first time. The effects of the process parameters on the particle size of the nanosuspension were investigated. A three-factor central composite design (CCD) was used to optimize the formulation, as well as to understand the effect of the variables. The optimal formulation was verified, and the physicochemical characteristics of the nanosuspensions were investigated in detail. Finally, in vitro and in vivo tests were carried out to compare the dissolution rate and oral bioavailability of the nanoparticles with that of the commercial tablet.
Fig. 2.

Chemical structure of carvedilol
MATERIALS AND METHODS
Materials
CAR was purchased from Shandong Qilu Pharmaceutical Co., Ltd. (Shandong, China). Alpha-tocopherol succinate (VES) was purchased from Jiangsu Xixin Vitamin Co., Ltd. (Jiangsu, China). Sodium dodecyl sulfate (SDS) was obtained from TianJin Bodi Chemical Holding Co., Ltd. (Tianjin, China). Propranolol was purchased from Changzhou Yabang Pharmaceutical Co., Ltd. (Jiangsu, China). Diethyl ether was obtained from Shandong Yuwang Industrial Co., Ltd. (Shandong, China). All other chemicals and reagents used were of analytical grade or better.
Preparation of CAR Nanosuspensions
CAR nanosuspensions were prepared through the anti-solvent precipitation–ultrasonication method(10). Briefly, CAR and VES were dissolved completely in acetone to prepare the organic phase and the solution was then passed through a 0.45-μm filter (Shanghai Huan’ao Trading Company, Shanghai, China) to remove the possible impurities. Meanwhile, the anti-solvent phase was prepared by dispersing stabilizer SDS in distilled water. At a fixed temperature, 1 ml of organic solution was quickly injected by syringe into 50 ml of anti-solvent using a B25 high shear homogenizer (BRT Equipment Technology Co. Ltd., Shanghai, China) at 10,000 rpm for 1 min. Immediately, drug particles precipitated from the anti-solvent, the samples were treated with an Ultrasonic Processor (20–25 kHz, Ningbo Scientz Biotechnology Co. Ltd., China) at different ultrasonic power inputs for different time lengths subsequently. The period of ultrasound burst was set to 3 s with a pause of 3 s between two ultrasound bursts. During the process, the temperature was controlled at 4–8°C using an ice–water bath. Then, the nanosuspensions were kept under vacuum at room temperature for 24 h to remove the acetone.
For long-term stability of the final product, the freshly prepared nanosuspensions were freeze-dried with cryoprotectant (i.e., maltose) at the concentration of 3% w/v. The nanosuspensions were pre-frozen in the refrigerator at −75°C for 12 h and subsequently freeze-dried in a FD-1C-50 freeze-drier (Boyikang Laboratory Instruments Co. Ltd., China) at −25°C for 12 h, followed by a secondary drying phase at 20°C for 4 h. Lyophilized preparation without cryoprotectant was also prepared as a control in this work.
Experimental Design and Statistical Analysis
In the present study, response surface methodology based on CCD(20) was utilized to evaluate the formulation factors that affect the mean particle size (Y) of nanosuspensions, i.e., the concentration of CAR (X1) and VES (X2) in the organic solution, and the level of SDS in the anti-solvent phase (X3), respectively. The experimental range of each variable was selected based on the results of preliminary experiments. The precipitation temperature, time, and power input of the ultrasonication was fixed at 10°C, 15 min, and 400 W, respectively. Table I shows the independent factors and their design levels in the present study. The experiments were designed using Design-Expert® software. To reduce systematic errors, the experiments were completely randomized.
Table I.
Independent Variables and Their Levels Investigated in the Central Composite Design
| Variables | Symbols | Range and levels | ||||
|---|---|---|---|---|---|---|
| −1.682 | −1 | 0 | 1 | 1.682 | ||
| CAR (mg/mL) | X 1 | 200 | 241 | 300 | 359 | 400 |
| VES (mg/mL) | X 2 | 120 | 144 | 180 | 216 | 240 |
| SDS (%) | X 3 | 0.20 | 0.32 | 0.50 | 0.68 | 0.80 |
A second-order polynomial model Eq. 1 was fitted to the response using the Design-Expert® software.
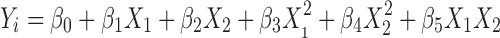 |
1 |
Where Yi represents the predicted response, X represents the independent variable, and β represents the coefficient. The three-dimensional (3D) response surface graphs were plotted using Origin 8.0 software according to the equation.
Particle Size Analysis and Zeta Potential Measurement
Particle size and size distribution of nanosuspensions were determined by laser diffraction using a Coulter LS 230 Analyzer (Beckman-Coulter Co. Ltd., USA) at room temperature. The measurements were performed with polarization intensity differential scattering technology included. The particle size was expressed as a volume distribution. The particle size distributions were evaluated by SPAN, defined as
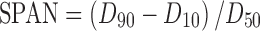 |
2 |
D10, D50, and D90 represent 10%, 50%, and 90% of the cumulative particle size distribution at the given size. The SPAN is a dimensionless number which illustrates whether or not the spread of the distribution is narrow or wide. A small SPAN indicates a narrow size distribution(21). The zeta potential was measured using a Malvern Zeta Sizer (Malvern Instruments, UK).
Scanning Electron Microscopy
The surface morphology of CAR nanosuspensions were visualized with a field emission scanning electron microscope (SUPRA 35, ZEISS, German). Before observation, the samples were fixed onto metal stubs using double-sided sticky tape previously secured onto aluminum stubs and then coated with gold under a vacuum.
Powder X-Ray Diffraction Analysis
The crystalline state of CAR in different samples was confirmed with a powder X-ray diffractometer (D/max 2500, Rigaku, Japan) using CuKα radiation. The obtained data were typically collected between 3° and 45° at a scan rate of 0.04°.
Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) analysis was performed using a DSC 1 calorimeter (Mettler Toledo, Schwerzenbach, Switzerland). Analysis was performed under a nitrogen purge (20 ml/min). The samples (about 3 mg) were weighed accurately, placed in aluminum pans, and then sealed with a pinhole-pierced cover. Heating curves were recorded at a scan rate of 10°C/min from 25°C to 250°C, and an empty pan was used as reference.
Fourier Transform Infrared Spectrometry
The infrared spectra of the samples were obtained using an IFS-55 Fourier transform infrared spectrometry (FTIR) spectrometer (Bruker, Germany). The measurements were performed over the range of 400–4,000 cm−1. Powder samples were milled with KBr to form a very fine powder. This powder was then compressed into a thin pellet for analysis.
Short-Term Physical Stability
The aqueous CAR nanosuspensions were stored in a closed glass vial at 25°C for up to 1 week. At the predetermined time intervals, aliquots were taken and subjected to particle size analysis as described above. The changes in appearance, particle size, SPAN value, and drug concentration were recorded. The concentration of CAR was determined by HPLC (Pump model: Shimadzu LC-20A, Japan) method(22). The analytical column was Diamonsil C18 (200 × 4.6 mm, 5 μm) (Dikma, USA). The HPLC mobile phase was a mixture of methanol, NaH2PO4/Na2HPO4 (12.3:1 M ratio) at 33 mM total concentration in water and glacial acetic acid (60:40:0.3, v/v/v), at a flow rate of 1.0 ml/min and the detection was performed at 242 nm using UV–VIS detector (Model: Shimadzu SPD-M20A, Japan).
In Vitro Dissolution Studies
An in vitro dissolution test was conducted in a dissolution apparatus (ZRS-6G, TiandaTianfa Technology Co., Ltd, Tianjin, China) according to the USP paddle method. The temperature was maintained at 37 ± 0.5°C, and the stirring rate was at 100 rpm. The commercial CAR tablet (Beijing Juneng Pharmaceutical Co. Ltd), accurately weighed bulk drug and nanosuspensions (all equivalent to 25 mg of CAR) were dispersed in 900 ml of dissolution medium. Five-milliliter samples were drawn, and the same volume of fresh dissolution medium was added at 5, 15, 30, and 60 min, respectively. Then, the samples were filtered through a 0.1-μm syringe filter (Shanghai Huan’ao Trading Company, Shanghai, China) immediately before dilution, when necessary. Drug content was determined with a UV spectrophotometer at 242 nm.
Pharmacokinetic Study in Rats
The study was conducted in accordance with the Ethical Guidelines for Investigations in Laboratory Animals and was approved by the Ethics Review Committee for Animal Experimentation of Shenyang Pharmaceutical University (Shenyang, China). Male Wistar rats weighing 200 ± 20 g were obtained from Experimental Animal Center of Shenyang Pharmaceutical University (Shenyang, China). All the rats were divided randomly into two groups comprising five animals in each and fasted overnight but allowed to free access to water before experiment. Two types of CAR formulations at a dose of 10 mg/body weight were orally administrated to two groups of rats, i.e., CAR nanosuspension of optimized formulation and suspension prepared by dispersing commercial CAR tablets in 0.5% CMC–Na solution and sonicated for 10 min.
Blood samples (0.5 ml) of each animal were sampled via the suborbital vein at 0, 0.083, 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 12, 16, 24, 36 h after administration. All the blood samples were immediately centrifuged at 10,000×g for 5 min to separate the plasma. The plasma obtained was stored at −20°C until analysis.
The concentration of CAR in rat plasma was determined by HPLC(22). The HPLC system was composed of a model LC-20A pump (Shimadzu, Kyoto, Japan) and a model SPD-M20A programmable photodiode array detector (Shimadzu, Kyoto, Japan). The analytical column was Diamonsil C18 (200 × 4.6 mm, 5 μm) (Dikma, USA). The HPLC mobile phase was a mixture of methanol, NaH2PO4/Na2HPO4 (12.3:1 M ratio) at 33 mM total concentration in water and glacial acetic acid (50:50:0.8, v/v/v). The flow rate was 1.0 ml/min. The UV detector wavelength was 242 nm. Plasma samples were processed as follows: a 200-μl plasma sample was mixed with 40 μl of an internal standard (propranolol) solution (30 μg/ml) and 80 μl 0.1 M NaOH and vortexed for 1 min. Then 320 μl of diethyl ether was added and the mixture was vortexed at room temperature for 5 min. After centrifugation at 10,000×g for 5 min, the organic layer was transferred into another clean tube and evaporated under nitrogen gas flow. Then, the residue was reconstituted using 80 μl of mobile phase and 20 μl of the sample was injected into the HPLC for analysis.
The main pharmacokinetic (PK) parameters were acquired with the help of a PK program DAS 2.0. The various PK parameters that were analyzed included maximum peak concentration of the drug in plasma (Cmax), the time to reach maximum concentration (Tmax), and the area under the plasma concentration–time curve (AUC0–36). All results were presented as mean ± S.D. values. Student’s t tests and ANOVA were performed to determine the significance of any differences.
RESULTS AND DISCUSSION
Effects of Process Parameters on Particle Size
In this study, CAR nanosuspensions were prepared successfully through the anti-solvent precipitation–ultrasonication process. When the drug, VES and SDS concentrations were fixed at 300 mg/ml, 180 mg/ml, and 0.5% w/v, respectively, the effects of process parameters on the particle size of the nanosuspensions were investigated. When the power inputs and the time length of ultrasonication were fixed at 400 W and 15 min, the influence of precipitation temperature on the average diameter of CAR nanoparticles is shown in Fig. 3a. The nanoparticles at the process temperature of lower than 10°C had smaller size than that at 25°C. This was caused by the rapid growth of cores. When the other process parameters were fixed, it was found that the particle size was hardly changed when the power inputs was over 400 W (Fig. 3b). When the time length of ultrasonication was changed in the range of 5–60 min, the smallest particle size was observed after 15 min; however, a longer time did not help to reduce the size (Fig. 3c). Therefore, the optimal values of the precipitation temperature, power inputs, and the time length of ultrasonication were selected as 10°C, 400 W, and 15 min, respectively.
Fig. 3.

Effects of a precipitation temperature, b ultrasonic power input, and c time length of ultrasonic treatment on particle size of CAR nanosuspensions (n = 3)
Formulation Optimization and Statistical Analysis
A total of 20 experiments were carried out to study the formulation factors that affect the particle size of nanosuspensions. Response data for all experimental runs of CCD are presented in Table II.
Table II.
Factor Levels and Observed Responses For CCD
| No. | Levels of independent factors | Response | ||
|---|---|---|---|---|
| CAR (mg/mL) | VES (mg/mL) | SDS (% w/v) | Particle size (nm) | |
| 1 | 200 | 180 | 0.50 | 116 |
| 2 | 241 | 216 | 0.68 | 177 |
| 3 | 241 | 216 | 0.32 | 529 |
| 4 | 241 | 144 | 0.68 | 164 |
| 5 | 241 | 144 | 0.32 | 629 |
| 6 | 300 | 180 | 0.50 | 233 |
| 7 | 300 | 180 | 0.50 | 222 |
| 8 | 300 | 240 | 0.50 | 489 |
| 9 | 300 | 180 | 0.50 | 240 |
| 10 | 300 | 120 | 0.50 | 514 |
| 11 | 300 | 180 | 0.20 | 832 |
| 12 | 300 | 180 | 0.50 | 218 |
| 13 | 300 | 180 | 0.80 | 171 |
| 14 | 300 | 180 | 0.50 | 203 |
| 15 | 300 | 180 | 0.50 | 222 |
| 16 | 359 | 216 | 0.68 | 567 |
| 17 | 359 | 144 | 0.32 | 1,558 |
| 18 | 359 | 216 | 0.32 | 684 |
| 19 | 359 | 144 | 0.68 | 478 |
| 20 | 400 | 180 | 0.50 | 1,425 |
The responses were fitted into a second-order polynomial model. The obtained model was validated using an ANOVA. The coefficient of determination (R2) closest to unity indicated a good model. P values lower than 0.05 indicated that the regression equation was statistically significant.
According to the results, a two second-order polynomial representing particle size was generated, which is shown below in terms of coded factors.
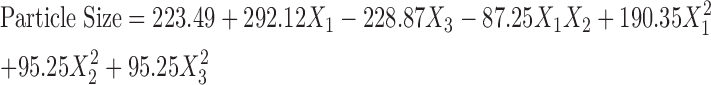 |
3 |
The R2 value is 0.9260, i.e., 92.60% of the sample variation in particle size was attributed to the experimental variables studied. Accordingly, the fitted equation can predict the best formulation for CAR nanosuspensions.
As shown in Fig. 4a, b, after a slight decrease, the particle size increased dramatically with increasing amounts of CAR. An increment in drug concentration had two opposite effects: on one hand, a higher supersaturation ratio from higher concentrations favor the formation of a large number of nuclei and tend to decrease the particle size(23); on the other hand, a higher drug level accelerates crystal growth by promoting condensation and/or coagulation(10). When the CAR concentration was low, the first effect prevailed; thus, a smaller drug particle size was obtained. However, at higher drug concentrations, the second effect becomes the dominant mechanism, resulting in larger particles. In Fig. 4a, c, the particle size shows an initial decrease and then a slight increase with increasing VES content. The ability of VES to stabilize the drug nanoparticles could be explained through the following: First, VES particles precipitate and remain preferentially in the interfacial area between the drug particles and the surrounding solution, effectively coating the particle with a negative charge and thereby preventing the nanoparticles from adhering to each other by repulsive electrostatic forces(12). Second, Ostwald ripening could be inhibited by incorporating a second water-insoluble component, which may lead to a difference in composition between the large and small particles during the said process. This difference may counter balance the driving force for Ostwald ripening, resulting in termination(2). VES possibly co-precipitated with the CAR nanoparticles during the preparation process, which led to a difference in composition between the particles, which in turn improved the stability of the nanoparticles by inhibiting Ostwald ripening. When the concentration of VES was low, it was not enough to fully cover the newly formed surfaces of the nanoparticles or effectively inhibit the Ostwald ripening process, resulting in the growth of particle size. However, an excessive amount of VES would increase the particle size by thickening the coat and inhibit the diffusion between the solvent and the anti-solvent during precipitation. As shown in Fig. 4b, c, an increase in SDS level decreases, and then slightly increases the particle size. SDS, a kind of anionic surfactant, was added in water as a stabilizer during the preparation process, which helps improve wetting and electrostatic stabilization of the precipitated drug particles via electrostatic repulsion(24). When the SDS level was low, flocculation can be minimized by raising the level. However, too much SDS would increase drug solubility in the stabilizer solution and increase particle size from Ostwald ripening(24).
Fig. 4.

Response surfaces for particle size. a The amount of SDS was fixed at 0.5%. b The amount of VES was fixed at 180 mg/mL. c The amount of CAR was fixed at 300 mg/mL
The optimum ranges for each factor were found to generate nanosuspensions with minimum particle size, maximum drug concentration, and minimum SDS concentration, using the equation and the responds in response surface. The optimum formulation conditions were as follows: CAR 296 mg/ml, VES 195 mg/ml, and SDS 0.47%. The nanosuspensions prepared with the optimized formulation yielded a mean particle size of 212 nm, which is in good agreement with the value predicted by the model (225 nm). The results confirm that the model was effective for predicting the impact of formulation composition on the particle size reduction of CAR nanosuspensions.
Particle Size and Zeta Potential
Optimized CAR nanosuspensions were successfully prepared and used to determine particle size distribution and zeta potential. The mean particle size, SPAN value, and zeta potential of three batches of nanosuspensions were 212 nm, 0.37, and −42 mV, respectively (Table III). Typically, surface charges can arise from the ionization of the particle surface or adsorption of anionic surfactants onto the surface(24). In the present study, the zeta potential of CAR nanosuspensions showed large negative values, mainly due to the adsorption of SDS and VES onto the particle surfaces, which would be beneficial for the storage stability of the nanosuspensions.
Table III.
Particle Size and Zeta Potential of the Optimum CAR Nanosuspensions (Mean ± S.D., n = 3)
| Mean particle size (nm) | Size distribution (nm) | SPAN | Zeta potential (mV) | ||
|---|---|---|---|---|---|
| D 10 | D 50 | D 90 | |||
| 212 ± 12 | 177 ± 7 | 212 ± 12 | 255 ± 15 | 0.37 ± 0.02 | −42 ± 3 |
Morphology
Figure 5 presents the morphologies of raw CAR, pure VES, SDS recrystallized from water, drug particles precipitated without the aid of VES, and nanoparticles of optimized formulation obtained by drying at room temperature. Figure 5a, b shows that bulk CAR and pure VES have irregular shapes and wide particle size distribution. SDS recrystallized from pure water exhibited a spine-like shape (Fig. 5c). Drug particles precipitated without the aid of VES in the organic phase during the preparation had a flaky shape, several micrometers in size. The SDS particles were spine-like and unevenly distributed over the surface of drug particles (Fig. 5d). Therefore, SDS alone was unable to form a robust interfacial film and effectively suppress the growth of the newly formed drug particles. As shown in Fig. 5e, the particles precipitated from the optimized formulation were flaky in shape with a narrow particle size distribution.
Fig. 5.

SEM micrographs of a raw CAR, b pure VES, c recrystallized SDS from pure water, d drug particles precipitated without VES, and e nanosuspensions of optimized formulation
PXRD and DSC Analysis
The changes in the crystalline state of CAR in the nanosuspensions were confirmed by powder X-ray diffraction (PXRD). Samples used for measurement were pure CAR, VES, SDS, their physical mixture, and lyophilized nanosuspensions powder of optimized formulation without cryoprotectant. As shown in Fig. 6, the typical crystalline peaks of CAR (5.84°, 14.84°, 18.44°, 24.32°, and 26.28°) were detected in the PXRD patterns of pure CAR and physical mixtures. This indicated the crystalline structure of CAR in the two samples above. However, no sharp peak for pure CAR was observed in the profile of the nanoparticles powder. The crystalline structure of CAR was lost because of the precipitation. The peaks of VES (15.80° and 18.32°) observed in pure VES and physical mixtures were absent in the PXRD spectrum of the dried nanosuspensions. This indicated that VES was converted into the amorphous form after precipitation. The characteristic peaks (4.48°, 6.72°, and 20.48°) of SDS were observed in the curves of SDS, physical mixtures, and nanosuspensions powder. This confirmed that the crystalline nature of SDS was not changed during preparation.
Fig. 6.

Powder X-ray diffraction patterns of CAR nanosuspensions and its ingredients. a Lyophilized nanosuspensions of optimized formulation without cryoprotectant, b physical mixture, c SDS, d VES, and e bulk CAR
DSC (Fig. 7) was also performed to confirm further the physical state of the samples. The data from DSC reasonably agreed with the results obtained by PXRD. The DSC profiles of CAR and VES exhibited sharp melting peaks at 116.47°C and 76.91°C, respectively. For SDS, the endothermic peak was at 193.38°C. In the physical mixture, the melting peaks all appeared but drifted slightly because of mixing. In contrast, the endothermic peaks of CAR and VES disappeared completely in the profiles of the nanosuspensions, suggesting a transition from the crystalline state to the amorphous state took place during preparation. Meanwhile, the crystalline state of SDS was not changed but the melting peak drifted slightly because of the precipitation.
Fig. 7.

DSC curves of CAR nanosuspensions and their ingredients. a Lyophilized nanosuspensions of optimized formulation without cryoprotectant, b physical mixture, c SDS, d VES, and e bulk CAR
FTIR Analysis
FTIR analysis was used to evaluate the possible intermolecular interactions between CAR and the excipients. The spectra of pure CAR, VES, SDS, their physical mixture, and lyophilized CAR nanosuspensions of optimized formulation without cryoprotectant are shown in Fig. 8. In the spectrum of pure CAR, the absorption band observed at 3,344.6 cm−1 was assigned to the N–H or O–H stretching vibration(18). For VES, the peaks at 1,753.7 and 1,714.0 cm−1 are due to the stretching vibration of the carbonyl group on ester and carboxylic acid, respectively(25). These absorption bands all appeared and almost have the same value as the curve of the physical mixture. However, the N–H or O–H stretching vibration peak (3,344.6 cm−1) of CAR, and the peak (1,714.0 cm−1) of carbonyl on the carboxylic acid group of VES disappeared for the CAR nanosuspensions. Meanwhile, the ester carbonyl-stretching band of VES was found to be a weak and broad peak (1,752.3 cm−1). Accordingly, the most possible reaction between CAR and VES could be the formation of intermolecular hydrogen bonding(18).
Fig. 8.

FTIR spectra of CAR nanosuspensions and its ingredients. a Lyophilized nanosuspensions of optimized formulation without cryoprotectant, b physical mixture, c SDS, d VES, and e bulk CAR
Short-Term Physical Stability
The short-term physical stability of CAR nanosuspension at 25°C was investigated to evaluate whether the nanosuspension was sufficiently stable for further processing such as drying. After 1 week, the nanosuspension remained homogeneous and no sediment was observed. At time zero, the mean particle size was 212 ± 12 nm with a SPAN value of 0.37 ± 0.02. And the CAR concentration was 5.82 ± 0.11 mg/ml. At the 1-week time point, the mean particle size was 225 ± 7 nm with a SPAN value of 0.35 ± 0.05. The drug concentration was 5.80 ± 0.08 mg/ml. The results demonstrated that VES combined with SDS was able to prevent aggregation of the nanoparticles effectively and prohibit the crystal growth caused by Ostwald ripening.
In Vitro Dissolution Studies
Figure 9 represents a comparison of the dissolution profiles of bulk CAR, the nanosuspension and the commercial tablet. Buffer medium with pH 1.0 was used to ensure sink condition during the dissolution rate testing. The experiment revealed higher dissolution rate for the nanosuspension compared to the bulk drug and the commercial tablet. The CAR nanosuspensions dissolved up to 91.94% within 5 min. In contrast, only 20.63% of the bulk CAR and 38.16% of the commercial tablet dissolved, respectively, during the same period. Another medium with pH 6.8 was used to compare the drug dissolution rate at more physiological pH. In that case, the difference between dissolution rates of nanosized particles, raw crystals, and commercial tablet was even higher (Fig. 9b) compared to results of Fig. 9a. After 60 min, 90.15% of the CAR in nanosuspensions dissolved in the phosphate buffer. However, only 8.13% of the bulk CAR and 23.89% of the commercial tablet dissolved, respectively, in the medium. These results demonstrate that the rate and extent of drug dissolution were markedly enhanced by the nanosuspensions. The increased dissolution rate of CAR nanosuspensions could be attributed to the pronounced reduction in particle size, the corresponding increased surface area, the enhanced solubility, and the amorphous nature of the drug in the preparation(24).
Fig. 9.

Dissolution profiles of bulk CAR, the nanosuspension and the commercial tablet: a buffer of pH 1.0; b buffer of pH 6.8. Each value represents the mean ± S.D. (n = 3)
In Vivo Studies
The plasma concentration–time profiles and the main PK parameters of CAR resulted from the oral administration of the nanosuspension and the reference formulation in Wistar rats are presented in Fig. 10 and Table IV, respectively. As expected, the oral absorption of the commercial tablet was found to be very low due to its poor dissolution properties. Significant increase in absorption was observed with the nanosuspension. As shown in Table IV, the Cmax and AUC0–36 values of nanosuspension were approximately 3.3- and 2.9-fold greater than that of reference preparation (p < 0.05), respectively. The Tmax was shorter for the nanoparticle than that of the tablet (p < 0.05). These findings were consistent with the results from the dissolution tests, indicating that the differences in CAR absorption are primarily attributed to the dissolution behavior of CAR. Meanwhile, the direct uptake of the intact nanoparticles by mechanisms involving M-cells in Peyer’s patches of the gastrointestinal (GI) lymphoid tissue might be another possible reason for the improved absorption of the nanosized drug particles. This approach may provide a route for avoiding first-pass metabolism of CAR, thus, the low drug uptake by this pathway might be enhanced(26).
Fig. 10.

Plasma concentration–time profiles of CAR after oral administration of nanosuspensions and the reference formulation in rats. Each value represents the mean ± S.D. (n = 5)
Table IV.
Pharmacokinetics Parameters of CAR in Rat Plasma After Oral Administration, Each Value Represents the Mean ± S.D. (n = 5)
| Parameters | Nanosuspension | Commercial tablet |
|---|---|---|
| C max (μg/mL) | 6.30 ± 1.34a | 1.90 ± 0.27 |
| T max (h) | 0.85 ± 0.67a | 2.60 ± 1.14 |
| AUC0–36 (μg h/mL) | 67.47 ± 14.96a | 23.64 ± 6.50 |
aStatistically significant compared with the commercial tablet (p < 0.05)
CONCLUSION
The present study proposed a novel formulation by applying VES as the co-stabilizer in the organic phase to fabricate CAR nanosuspensions via the anti-solvent precipitation–ultrasonication technique. The particle size of nanosuspensions was highly dependent on process parameters. With the optimized formulation, nanoparticles with a mean particle size of 212 nm were obtained. The nanosuspension was physically stable in terms of the particle size at 25°C over 1 week. CAR nanosuspensions exhibited markedly enhanced dissolution rates compared with the raw drug and the commercial tablet. The in vivo test demonstrated that the Cmax and AUC0–36 values of nanosuspension were approximately 3.3- and 2.9-fold greater than that of that of the commercial tablets (p < 0.05), respectively. The results demonstrate that VES is an efficient co-stabilizer for fabrication of stable aqueous nanosuspensions via the anti-solvent precipitation–ultrasonication technique. The method is rapid, easy to control, and inexpensive. Further studies are needed to determine whether VES can also be applied to make nanosuspensions of other drugs.
ACKNOWLEDGMENTS
This work was supported by National Ministry of Science and Technology for 973 major research projects of China (no. 2009CB930300).
Contributor Information
Dandan Liu, Email: liudandan1124@126.com.
Weisan Pan, Phone: +86-24-23986313, FAX: +86-24-23953241, Email: pwstfzy@163.com.
REFERENCES
- 1.Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 2.Lindfors L, Skantze P, Skantze U, Rasmusson M, Zackrisson A, Olsson U. Amorphous drug nanosuspensions. 1. Inhibition of Ostwald ripening. Langmuir. 2006;22(3):906–910. doi: 10.1021/la0523661. [DOI] [PubMed] [Google Scholar]
- 3.Keck C, Muller R. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur J Pharm Biopharm. 2006;62(1):3–16. doi: 10.1016/j.ejpb.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Vaneerdenbrugh B, Vandenmooter G, Augustijns P. Top-down production of drug nanocrystals: nanosuspension stabilization, miniaturization and transformation into solid products. Int J Pharm. 2008;364(1):64–75. doi: 10.1016/j.ijpharm.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 5.Dewaard H, Hinrichs W, Frijlink H. A novel bottom–up process to produce drug nanocrystals: controlled crystallization during freeze-drying. J Control Release. 2008;128(2):179–183. doi: 10.1016/j.jconrel.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Michal EM, Margaret AH, Keith PJ, Robert OWI. Drug nanoparticles by antisolvent precipitation mixing energy. Langmuir. 2006;22:8951–8959. doi: 10.1021/la061122t. [DOI] [PubMed] [Google Scholar]
- 7.Zhang H-X, Wang J-X, Zhang Z-B, Le Y, Shen Z-G, Chen J-F. Micronization of atorvastatin calcium by antisolvent precipitation process. Int J Pharm. 2009;374(1–2):106–113. doi: 10.1016/j.ijpharm.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Dong Y, Ng WK, Shen S, Kim S, Tan RBH. Preparation and characterization of spironolactone nanoparticles by antisolvent precipitation. Int J Pharm. 2009;375(1–2):84–88. doi: 10.1016/j.ijpharm.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 9.Cho E, Cho W, Cha K-H, Park J, Kim M-S, Kim J-S, et al. Enhanced dissolution of megestrol acetate microcrystals prepared by antisolvent precipitation process using hydrophilic additives. Int J Pharm. 2010;396(1–2):91–98. doi: 10.1016/j.ijpharm.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 10.Xia D, Quan P, Cui F. Preparation of stable nitrendipine nanosuspensions using the precipitation–ultrasonication method for enhancement of dissolution and oral bioavailability. Eur J Pharm Sci. 2010;40(4):325–334. doi: 10.1016/j.ejps.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Castro MDLd, Priego-Capote F. Ultrasound-assisted crystallization (sonocrystallization) Ultrason Sonochem. 2007;14:717–724. doi: 10.1016/j.ultsonch.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Chen AX. Vitamin E succinate stabilized pharmaceutical compositions, methods for the preparation and the use thereof. US Patent 20070207173.
- 13.Frishman WH. Carvedilol. N Engl J Med. 1998;339(24):1759–1765. doi: 10.1056/NEJM199812103392407. [DOI] [PubMed] [Google Scholar]
- 14.GlaxoSmithKline. Coreg® (carvedilol) tablets. January 2011. Available from: http://us.gsk.com/products/assets/us_coreg.pdf
- 15.Decker S, Gabel R-d, Lapotnikoff J, Wirl A, Zimmermann I. Carvedilol-lipophilic solutions. US Patent 20040204474. October 14.
- 16.Ini S, Levi S, Abramov M, Turgeman E. Carvedilol phosphate. US Patent 20090286844. November 19.
- 17.Wen X. Preparation and study the 1:2 inclusion complex of carvedilol with β-cyclodextrin. J Pharm Biomed Anal. 2004;34(3):517–523. doi: 10.1016/S0731-7085(03)00576-4. [DOI] [PubMed] [Google Scholar]
- 18.Wei L, Li J, Guo L, Nie S, Pan W, Sun P, et al. Investigations of a novel self-emulsifying osmotic pump tablet containing carvedilol. Drug Dev Ind Pharm. 2007;33(9):990–998. doi: 10.1080/03639040601150328. [DOI] [PubMed] [Google Scholar]
- 19.Planinšek O, Kovačič B, Vrečer F. Carvedilol dissolution improvement by preparation of solid dispersions with porous silica. Int J Pharm. 2011;406(1–2):41–48. doi: 10.1016/j.ijpharm.2010.12.035. [DOI] [PubMed] [Google Scholar]
- 20.Sun W, Mao S, Shi Y, Li LC, Fang L. Nanonization of itraconazole by high pressure homogenization: stabilizer optimization and effect of particle size on oral absorption. J Pharm Sci. 2011;100:3365–3373. doi: 10.1002/jps.22587. [DOI] [PubMed] [Google Scholar]
- 21.Torrado JJ, Illurn L, Davis SS. Particle size and size distribution of albumin microspheres produced by heat and chemical stabilization. Int J Pharm. 1989;51:85–93. doi: 10.1016/0378-5173(89)90079-3. [DOI] [Google Scholar]
- 22.Wei L, Sun P, Nie S, Pan W. Preparation and evaluation of SEDDS and SMEDDS containing carvedilol. Drug Dev Ind Pharm. 2005;31(8):785–794. doi: 10.1080/03639040500216428. [DOI] [PubMed] [Google Scholar]
- 23.Kakran M, Sahoo NG, Li L, Judeh Z, Wang Y, Chong K, et al. Fabrication of drug nanoparticles by evaporative precipitation of nanosuspension. Int J Pharm. 2010;383(1–2):285–292. doi: 10.1016/j.ijpharm.2009.09.030. [DOI] [PubMed] [Google Scholar]
- 24.Kesisoglou F, Panmai S, Wu Y. Nanosizing—oral formulation development and biopharmaceutical evaluation. Adv Drug Deliv Rev. 2007;59(7):631–644. doi: 10.1016/j.addr.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Jingxi X. Application of Fourier transform spectroscopy in organic and pharmaceutical chemistry. Science Press; 1987.
- 26.Rabinow BE. Nanosuspensions in drug delivery. Nat Rev Drug Discov. 2004;3(9):785–796. doi: 10.1038/nrd1494. [DOI] [PubMed] [Google Scholar]