

Abstract
The purpose of this study was to develop and validate a rapid, sensitive, and specific reversed-phase high-performance liquid chromatography method for the quantitative determination of native tenofovir (TNF) for various applications. Different analytical performance parameters such as linearity, precision, accuracy, limit of quantification (LOQ), limit of detection (LOD), and robustness were determined according to International Conference on Harmonization (ICH) guidelines. A Bridge™ C18 column (150 × 4.6 mm, 5 μm) was used as stationary phase. The retention time of TNF was 1.54 ± 0.03 min (n = 6). The assay was linear over the concentration range of 0.1–10 μg/mL. The proposed method was sensitive with LOD and LOQ values equal to 50 and 100 ng/mL, respectively. The method was accurate with percent mean recovery from 95.41% to 102.90% and precise as percent RSD (relative standard deviation) values for intra-day, and inter-day precision were less than 2%. This method was utilized for the estimation of molar absorptivity of TNF at 259 nm (ε259 = 12,518 L/mol/cm), calculated from linear regression analysis. The method was applied for determination of percentage of encapsulation efficiency ( 22.93 ± 0.04%), drug loading (12.25 ± 1.03%), in vitro drug release profile in the presence of enzyme (43% release in the first 3 h) and purification analysis of hyaluronic acid-based nanomedicine.
KEY WORDS: high-performance liquid chromatography, hyaluronic acid, molar absorptivity, nanomedicine, tenofovir
INTRODUCTION
Antiretroviral drugs are one of the most important therapeutic families used to treat human immunodeficiency viral (HIV) infections (1). Tenofovir (TNF) belongs to the antiretroviral drugs under the category of nucleotide analog reverse transcriptase inhibitors (Fig. 1a) (2). In its prodrug form it has already been approved by the US Food and Drug Administration (FDA) to treat HIV infections (1,3). It is a weak acidic, water-soluble drug with molecular weight of 287.21 g/mol, log P value of −1.6 and two pKa values of 3.8 and 6.7 (3).
Fig. 1.

Chemical structures of: a tenofovir and b adefovir
Microbicides are currently the principal focus for HIV prevention strategies (4,5). These are the agents used topically within the vagina or rectum in order to prevent infections caused by HIV and other enveloped viruses and sexually transmitted pathogens. Several agents have been tested in clinical trials for their safety and microbicidal effects (6). The effectiveness and safety of TNF as a microbicide for the prevention of HIV infections have already been approved in some of the recent studies (7,8). TNF formulations as solid lipid nanoparticles (9), vaginal gel (10,11), and vaginal ring (12) have already been developed.
Several assays that measured the native TNF in human plasma, such as high-performance liquid chromatography (HPLC) with UV detection (13–15), liquid chromatography–mass spectrometry (LC–MS) detection (16–19), and for the estimation of the prodrug form of TNF, named tenofovir disoproxil fumarate (TDF) in human plasma in combination with other drugs by using HPLC methods have been described in the literature (20,21). But most of these methods are tedious, time consuming, and involve complex sample preparations.
The development of LC–MS/MS method for the detection of adefovir (structurally very close to TNF, Fig. 1b) in human serum and urine (22), and the HPLC method for the determination of a nucleoside analog acyclovir and its related impurities have also been reported (23). However, to our knowledge there is no HPLC method that has been reported so far for simple and rapid quantitative estimation of TNF in nanomedicines (NMs) formulation. Moreover, no information is available for the molar absorptivity (ε) value of TNF, which could be very useful for purity evaluation when sufficiently pure standards of TNF are not available. Therefore, in the present study, the authors intend to develop a reversed phase-HPLC method for the determination of TNF in its native form. The authors further used the developed HPLC method to estimate the ε value of TNF, percentage of encapsulation efficiency (% EE), in vitro release profile, and purity analysis of hyaluronic acid (HA)-NMs.
HA is an anionic polysaccharide composed of alternating units of D-glucuronic acid and N-acetyl-d-glucosamine (24,25). We selected the HA in this work because it has received much attention these days for drug delivery applications due of its biodegradable, biocompatible, non-immunogenic, and mucoadhesive properties (24–26). The hyaluronidase (HAase) enzyme, present abundantly in human seminal fluid (27) was utilized as the triggering component for degradation of HA-NMs and thus resulting in burst release of encapsulated TNF. It has already been proven that HAase hydrolyzes the β(1-4)glycosidic bonds of HA (26).
A short run time in the HPLC method always facilitate analysis of several samples per day as already published in some of the studies with HPLC methods, (13,28–30). The run time of 2.5 min in this study also facilitates analysis of several hundred samples of TNF per day. This newly described procedure offers significant advantages over previously published methodologies by its simplicity and rapidness for the detection of TNF. The ε value of TNF determined by using this HPLC method is an intrinsic property of a drug and measures how strongly a chemical species absorbs light at a given wavelength. It is based on the concept of Lambert–Beer's law according to which the absorbance (Ab) of a substance depends on the path length (l), the length of solution through which the light passes (for cells used in this study, l = 1 cm), and the concentration (c, usually expressed in moles per liter) of the substance (Eq. 1) (31).
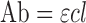 |
1 |
EXPERIMENTAL
Reagents and Chemicals
TNF (99% purity) was from Beijing Zhongshuo Pharmaceutical Technology Development Co., Ltd. (Beijing, China). HA of molecular weight 11,624 Da was kindly provided by Zhenjiang Dong Yuan Biotech Co., Ltd. (Jiangsu, China). HAase from bovine testes with specific activity of 810 units/mg, acetone, N-hydroxysuccinimide (NHS), and adipic acid dihydrazide (ADH) were from Sigma–Aldrich (St. Louis, MO). Acetonitrile was from Fischer Scientific (Pittsburgh, PA). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) was from Thermo Fisher Scientific Inc. (Rockford, IL). Milli Q water used throughout the study was the purified water with Millipore water purification system (Millipore Corporation, Danvers, MA). All other chemicals were of analytical grade and used without further purification.
Instrumentation
The HPLC system (Waters, Milford, MA) consisted of a 1575 binary pump system, 717 plus auto sampler, 2487 dual wavelength absorbance detector, and a BridgeTM C18 column (150 × 4.6 mm, 5 μm). The results were acquired and processed with Breeze™ software (version 3.3). A Genesys 10 Bio UV-vis Spectrophotometer (Thermo Electron Scientific Instruments LLC, Madison, WI) was used to determine the ε value of TNF. Spectra/Por cellulose ester membrane dialysis bags (Spectra/Por Float-A-Lyzer G2, MWCO 3.5–5 kDa) were purchased from Spectrum Laboratories Inc. (Rancho Dominguze, CA). The freeze dryer system was of Labconco Corporation (Kansas City, MO). The in vitro drug release study was performed using a thermostatically controlled shaking water bath (BS-06, Lab Companion, Seoul, Korea). The instrument to determine pH was from Mettler-Toledo Inc. (Columbus, OH).
Analytical Conditions
The HPLC assay was performed isocratically at ambient temperature (23°C). The optimization of the method was done by using various v/v ratios of mobile phases consisting of water–methanol or water–acetonitrile at different pH and flow rates, at the detection wavelength of 259 nm. The sample volume of 10 μL was injected for each run. Mobile phase solvents were degassed in an ultrasonic bath (Sonicator VWR model 150 D; VWR International., West Chester, PA) for 10 min before their use.
Preparation of Standard and Sample Solutions of TNF
The standard stock solution of TNF (100 μg/mL) was prepared by dissolving 1 mg of drug in 10 mL of milli Q water. Serial dilutions of stock solution with the same diluent led to the solutions in the concentration range of 0.1–10 μg/mL for HPLC analysis.
Method Validation
Nine samples with different levels of drug concentrations in the range of 0.1–10 μg/mL were selected to perform the linearity experiment and to construct the calibration curve of the method. Moreover four different levels of drug concentrations (0.5, 1, 5, 10 μg/mL) were utilized to perform various other validation parameters according to International Conference of Harmonization (ICH, Q2:R1) guidelines (32). The variations in the HPLC peak area were reported as percentage of relative standard deviation (% RSD) for each validation parameters except the accuracy, where it was reported in terms of percent mean recovery. The selected validation parameters were described below.
Linearity
Linearity is the ability of the method to elicit test results that are directly proportional to analyte concentration within a given range. For the establishment of linearity, a minimum of five concentrations is recommended as per the ICH guidelines. In this analysis, a calibration plot was prepared by analyzing nine solutions in the concentration range of 0.1–10 μg/mL. The intercept, slope, and correlation coefficient (R2) were determined by linear regression data analysis.
Accuracy
Accuracy is the percent of analyte recovered by assay. It was assessed in triplicate by using four concentration levels (0.5, 1, 5, 10 μg/mL). Accuracy was reported as percent mean recovery calculated for each concentration levels selected to perform validation of the method.
Precision
Precision was assessed in triplicate by using four concentration levels (0.5, 1, 5, 10 μg/mL) and was reported as percentage of RSD. Similarly, the intra-day precision was determined and the inter-day precision was based on analysis of these concentrations over a period of 1 week.
Limit of Quantification and Limit of Detection
The limit of quantification (LOQ) is defined as the lowest concentration of an analyte that can be determined with acceptable precision and accuracy under the stated operational conditions of the method. Limit of detection (LOD) is defined as the concentration of an analyte that can be detected but not be quantified. LOQ and LOD values can also be defined as the concentrations that could be detected and yield a signal-to-noise ratio of 10:1 and 3:1, respectively (32).
Robustness
The robustness of a method is its ability to remain unaffected by small deliberate variations in method parameters. The following changes in the optimum parameter values were examined: the flow rate of the mobile phase (adjusted by ± 10%), pH (adjusted by ± 0.1 unit), composition of mobile phase (adjusted by ± 1 unit), and temperature (±3°C). The variation in the HPLC chromatogram area was calculated for each robustness parameter.
Specificity
Specificity of the method was determined by analyzing potential interfering peaks of the formulation components at retention time of TNF. In addition, empty NMs (without the drug) were prepared, purified, and ultra-centrifuged using Beckman L8-70M (Beckman Instruments Inc., Palo Alto, CA) at 20,000 rpm for 45 min at 10°C to isolate the NMs. The analysis of the supernatant was performed in triplicate (n = 3) with the same conditions as used for TNF detection. The purpose of this analysis was to detect any significant peak of formulation components EDC, NHS, and ADH at the retention time of TNF.
Applicability of the HPLC Method
Determination of Molar Absorptivity of TNF
The proposed HPLC method was applied for the estimation of ε value of TNF. The calculation of the ε by HPLC analysis was done by using Eq. 1 that has been rearranged to give Eq. 2, as previously explained by Pelillo et al. (33).
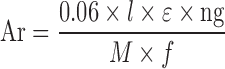 |
2 |
Where, Ar is the area of the drug chromatogram (milli-absorbance unit per second or mAU.sec) recorded by the HPLC detector, l is the path length in cm, M is the molecular weight of the drug in Da, f is the mobile phase flow rate in milliliter per minute, and ng is the injected drug amount for each injection volume of 10 μL.
In Eq. 2, all other parameters except Ar and ε are constant for a given drug and analytical conditions, thus they can be referred to as a constant k leading to Eq. 3.
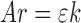 |
3 |
The elaborated Eqs. 2 and 3, could be a useful and alternative tool to better estimate the ε value of any molecule by using HPLC data. The ε value is estimated by using the slope of the regression curve generated using the injected amounts, leading to k (mole centimeter second per liter) and the corresponding areas (Ar).
The ε value of TNF was also calculated using a UV-vis spectrophotometer by preparing a calibration curve in the concentration range of 1–10 μg/mL. For each concentration (molar concentration), Ab was calculated and a plot was constructed by taking the absorbance on y-axis and the concentration on x-axis. The linear regression data analysis, performed through the experimental points by setting the intercept of the calibration curve to zero, allowed obtaining the ε value from the slope of this curve by using Eq. 1.
Statistical Analysis
A student t test was performed for comparison of the results obtained for the ε value of TNF by HPLC and UV-vis spectrophotometer. The p value either less than or equal to 0.05 was considered statistically significant.
Preparation and Purification of HA-NMs
HA-NMs were prepared by surfactant free cross-linking method, adapted from a previous report (34). HA was cross-linked with adipic acid dihydrazide (ADH) using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), and carbodiimide chemistry. Briefly, 1 mg/mL of aqueous solution of HA was prepared in milli Q water and mixed well by continuous stirring for 1 h. A small amount of acetone was added in this solution and stirred for 1 h to make sure that all the components were well dispersed. Then the aqueous solutions of EDC, NHS, and ADH were added in the above solution and stirred further for 15 h, leading to the formation of an amide bond. Finally, more acetone was added and the solution was stirred continuously for 6 h. The organic solvent (acetone) was evaporated by continuous stirring for several hours. The HA-NMs dispersion in water was subjected to dialysis to remove the non-reacted formulation components (EDC, NHS, ADH, and intermediate products formed). One milliliter of sample from dialysis media was taken at predetermined intervals (3, 6, 9, 12, 24 h) and analyzed by this HPLC method.
Physicochemical Characterization of HA-NMs
The particle mean diameter (PMD: nm), size distribution, polydispersity index (PDI), and zeta potential (millivolts) of the NMs were measured at 25°C by dynamic light scattering method using Zetasizer Nano ZS, Malvern Instruments Ltd., Worcestershire, UK. The PMD of the NMs was represented as Z-average diameter. According to the National Institute Standard, a sample with PDI value less than 0.05 was considered monodispersed (35). The zeta potential of the NMs dispersion was measured using the zeta potential analysis mode of the instrument. Nanosphere™ size standard (59.0 ± 2.5 nm) and zeta potential standard (−68.0 ± 6.8 mV) were used to calibrate the instrument prior to the NMs sample analysis. The magnitude of the zeta potential (positive or negative) gives an indication of the stability of the colloidal system. Colloidal systems with greater than ± 30 mV are considered stable (36).
Transmission electron microscopy (TEM) analysis was utilized to determine the morphology of NMs. A Philips STEM CM12 instrument (Philips Electronic Instruments, Inc., Mahwah, NJ) was used for TEM analysis. Digital images were acquired with a large format (II Megapixel) retractable and fiber-optical coupled SC100 ORIUS© CCD camera. To obtain the specimens, a drop of NMs dispersion was placed on a copper grid coated with carbon film and air dried. The observation was performed at an accelerating voltage of 80 kV.
Drug Encapsulation Method and Evaluation of Drug Content of HA-NMs
The encapsulation of TNF inside HA-NMs was done by using soaking method of drug encapsulation which occur by passive diffusion and non covalent binding. Briefly, HA-NMs dispersion after purification by dialysis method was freeze dried, immersed (10 mg) in a concentrated aqueous solution of TNF (5 mg/mL) and left to soak for 3 days at room temperature. After soaking process, the TNF content of the NMs was determined after ultra-centrifugation, indirectly from the drug content of the supernatant by HPLC analysis. The percentage of encapsulation efficiency (% EE) and drug loading (DL) in percentage of w/w of NMs were calculated using following Eqs. 4 and 5:
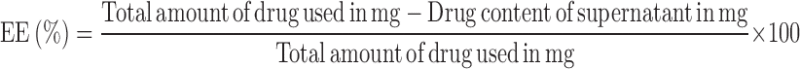 |
4 |
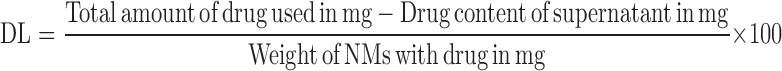 |
5 |
In Vitro Drug Release Analysis
The release study was performed with equal amount of HAase as found in the seminal fluid (1.08 units approximately in 3 mL of ejaculate) (26). The analysis was performed with mixture of vaginal fluid simulant (VFS, pH 4.2) and seminal fluid simulant (SFS, pH 7.8) prepared in 1:3 ratios, respectively, and in VFS alone. These simulants were prepared according to previous reports (37,38). Three milliliters of the NMs dispersion containing 10 mg of NMs in the simulant mixture (pH 7.1) was put into a dialysis bag and immersed into 20 mL of the simulant mixture. The dialysis bag was incubated in a thermostatically controlled shaking water bath with 60 rpm at 37°C. Aliquots of samples were taken at different intervals of time (1, 3, 6, 9, 24, and 48 h) from the release media. Fresh simulant mixture was added at the same rate, in order to maintain the sink condition. The concentration of drug in the solution was determined by this HPLC method with each experiment run in triplicate (n = 3).
RESULTS AND DISCUSSION
HPLC Method Development and Validation
The best composition of the mobile phase was found to be water (pH 5.1)/acetonitrile (35:65 v/v) delivered at a flow rate of 1 mL/min. For the preparation of the aqueous phase (pH 5.1), 1 mL of triethylamine was added to 1,000 mL of water and the final pH was adjusted with orthophosphoric acid (85%) to 5.1. The retention time of TNF was 1.54 ± 0.03 min (n = 6) as shown by the representative chromatogram of TNF (Fig. 2) at the detection wavelength of 259 nm. The quantification of TNF was based on the calibration curve constructed between the areas of the drug chromatogram (y-axis) versus concentration (x-axis). The calibration curve was linear with regression Equation; y = 25.847x + 2145.1, and correlation coefficient (R2) = 0.9989. The LOQ and LOD values of TNF were found to be 100 ng/mL and 50 ng/mL, respectively. The method was found to be accurate (as the percent mean recovery was within the acceptable limits of 90-110%) and precise (% RSD less than 2%), as showed in Tables I and II, respectively. The percentage RSD values for each robustness parameter was less than 2%, which indicated that the impact of minor variations in the method parameters was within the acceptable limits and results were unaffected (Table III).
Fig. 2.

Typical chromatogram of TNF (concentration, 5 μg/mL) at detection wavelength of 259 nm
Table I.
Accuracy (Percent Mean Recovery) Results for Different Levels of TNF (n = 3)
| Level of standard solutions (μg/mL) | Amount recovered (μg/mL) | Mean recovery ± SD (%) | RSD (%) |
|---|---|---|---|
| 0.5 | 0.481 | 95.41 ± 0.70 | 0.73 |
| 0.476 | |||
| 0.474 | |||
| 1 | 0.989 | 98.53 ± 0.38 | 0.39 |
| 0.985 | |||
| 0.982 | |||
| 5 | 5.224 | 102.9 ± 1.38 | 1.34 |
| 5.134 | |||
| 5.089 | |||
| 10 | 9.710 | 97.85 ± 0.71 | 0.73 |
| 9.853 | |||
| 9.791 |
Table II.
Precision of the HPLC Method
| Concentration (μg/mL) | Precision | |||||||
|---|---|---|---|---|---|---|---|---|
| Intra-day | Inter-day | |||||||
| Within-day | Day 1 | Day 2 | Day 2 | |||||
| Peak area (mAU) | % RSD | Peak area (mAU) | % RSD | Peak area (mAU) | % RSD | Peak area (mAU) | % RSD | |
| 0.5 | 13,458 | 0.574 | 14,077 | 1.090 | 14,258 | 1.531 | 12,758 | 0.555 |
| 13,407 | 14,357 | 14,569 | 12,900 | |||||
| 13,559 | 14,334 | 14,688 | 12,819 | |||||
| 1 | 26,626 | 0.619 | 27,408 | 0.595 | 30,122 | 0.125 | 26,771 | 0.444 |
| 26,326 | 27,548 | 30,069 | 26,519 | |||||
| 26,363 | 27,223 | 30,142 | 26,686 | |||||
| 5 | 131,704 | 0.608 | 135,072 | 0.104 | 129,824 | 0.580 | 126,704 | 1.044 |
| 130,350 | 134,838 | 128,825 | 127,985 | |||||
| 131,758 | 134,822 | 130,299 | 129,377 | |||||
| 10 | 229,528 | 0.925 | 252,158 | 0.954 | 232,573 | 0.865 | 228,613 | 1.158 |
| 225,377 | 256,904 | 229,744 | 223,880 | |||||
| 226,863 | 255,445 | 228,729 | 224,328 | |||||
Table III.
Robustness Analysis (n = 3) by HPLC Method
| Robustness parameters | Flow rate (mL/min) | pH (water) | ACN/water (pH 5.1) | Temperature (°C) | ||||
|---|---|---|---|---|---|---|---|---|
| 1.1 | 0.9 | 5.0 | 5.2 | (66:34) | (64:36) | 20 | 26 | |
| % RSD | 0.147 | 0.574 | 1.598 | 1.552 | 1.428 | 1.438 | 1.332 | 1.403 |
Applicability of the Method for Determination of Molar Absorptivity (ε) of TNF
The calibration curves were constructed in the concentration range of 1–10 μg/mL for calculation of the ε value using UV-vis Spectrophotometer and HPLC method. The curves were found to be linear with the mean value (n = 3) of R2 = 0.9993 (Fig. 3a) and 0.9972 (Fig. 3b) for UV and HPLC methods, respectively. The ε value was estimated by calculating the slope of the calibration curves as shown in Table IV. The results for the average value of the ε calculated by HPLC (12,518 L/mol/cm) and UV-vis spectrophotometer (11,571 L/mol/cm) were compared using the Student t test. A significant difference between these two results was observed (p value = 0.001595, less than 0.05), which led to the rejection of the null hypothesis. The HPLC allowed producing more accurate and reliable results (low relative standard error of the mean; SEM); also, the peak of the drug could be well separated from other potential impurities in the sample and selectively estimated by using the HPLC method rather than UV method. The foregoing considerations finally governed our decision to use the value of 12,518 L/mol/cm obtained by HPLC analysis as the ε final value of TNF. It was noteworthy that this value was somewhat close to the reported ε value of TDF (15,860 L/mol/cm, at the wavelength of 259 nm), a prodrug of TNF (39) and of adefovir (14,100 L/mol/cm, at the wavelength of 260 nm): a molecule that is structurally close to TNF as shown in Fig. 1b (40).
Fig. 3.

The calibration curves for calculation of molar absorptivity of TNF by: a UV spectrophotometer and b HPLC method. Each data point represents the mean of triplicate readings (n = 3)
Table IV.
Molar Absorptivity (ε) of TNF by UV Spectrophotometer and HPLC Method (n = 3)
| Method | Calibration range (μg/mL) | Equation | Correlation (R 2) | Slope (ε) (L/mol/cm) | Mean value |
|---|---|---|---|---|---|
| UV | 1–10 | y = 11,433c | 0.9957 | 11,433 | 11,571 |
| y = 11,731c | 0.9997 | 11,731 | |||
| y = 11,551c | 0.9997 | 11,551 | |||
| HPLC | 1–10 | Ar = 12,407 k | 0.9967 | 12,407 | 12,518 |
| Ar = 12,582 k | 0.9967 | 12,582 | |||
| Ar = 12,565 k | 0.9981 | 12,565 |
Application of the Method for HA-NMs
Characterization of HA-NMs
The PMD, PDI, and zeta potential value for HA-NMs analyzed in triplicate (n = 3) were 96 ± 0.76 nm, 0.048 ± 0.005, and −52.47 ± 0.89 mV, respectively. The low PDI value (less than 0.05) indicated that the NMs were monodispersed. The high zeta potential value (with absolute values greater than 30 mV) explained the stability of NM dispersion in water. Morphology (assessed by TEM analysis) and size distribution of HA-NMs were shown in Fig. 4a and b, respectively. The results confirmed that NMs have a spherical shape and uniform size distribution.
Fig. 4.

a Transmission electron micrograph; Scale bar represents 500 nm and b size distribution of HA-NMs; PMD = 96 ± 0.76 nm; and PDI = 0.048 ± 0.005
Determination of the Percentage of EE, DL, In Vitro Drug Release Profile and Purity Analysis of HA-NMs
Figure 5 showed the HPLC chromatogram of dialysis media. The dialysis was performed against 1 L of milli Q water for 24 h with three water changes. The data clearly indicated that after this time, there were no detectable levels of non-reacted formulation components present which could adversely affect the TNF analysis using this HPLC method.
Fig. 5.

Typical overlay chromatogram for dialysis media, before (at time; t = 0 h and after 24 h of dialysis)
The percentage of EE and DL of HA-NMs (10 mg) was found to be 22.93 ± 0.04% and 12.25 ± 1.03% after incubation with 5 mg/mL aqueous solution of TNF for 3 days. The in vitro release profile was depicted in Fig. 6. In the presence of HAase an initial burst effect was observed in the first 3 h, with approximately 43% of the drug content being released. Almost 53% of the drug was released in 6 h. After that, a stable plateau phase was observed with nearly 82% of drug being released by 48 h. While in the absence of HAase, nearly 31% of drug was released at the end of the first 3 h followed by a slow drug release kinetics reaching a cumulative value of 49% in 48 h. The TNF release in VFS was somewhat lower than release in the absence of HAase. Almost 20% of TNF was released in first 3 h followed by a slow drug release kinetics reaching a cumulative value of 49% in 48h. No interfering peaks of formulation components were observed at the retention time of TNF either during NMs drug content quantification or in vitro release study by this HPLC method.
Fig. 6.

In vitro drug release profile of HA-NMs in the presence and absence of HAase as function of the time in hours. Results are given as means ± SEM of at least three experiments (n = 3)
CONCLUSIONS
A simple, isocratic, reversed-phase HPLC assay was developed and validated for the quantitative analysis of TNF in its native form. The method was demonstrated to be sensitive, rapid, accurate, and precise. The assay provides a linear response across a wide range of concentrations. The robustness values for variation in the flow rate, temperature, mobile phase pH, and mobile phase composition were well within the acceptance limit, which showed that the method was robust with a good system suitability and precision under a given set of conditions. This HPLC method was found to have a suitable application for the determination of the ε value of TNF estimated for the first time. The assay was successfully applied for the determination of the DL, percentage of EE, in vitro drug release profile and purification analysis of HA-NMs.
ACKNOWLEDGMENTS
The project described was supported by Grant Number R01AI087304 from the National Institute of Allergy And Infectious Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy And Infectious Diseases or the National Institutes of Health. We gratefully acknowledge Mr. Jack Liu (Zhenjiang Dong Yuan Biotech Co., Ltd., Jiangsu, China) for providing samples of hyaluronic acid.
REFERENCES
- 1.Checa A, Oliver R, Hernández-Cassou S, Saurina J. Determination of HIV drugs in biological matrices: a review. Anal Chim Acta. 2009;647(1):1–13. doi: 10.1016/j.aca.2009.05.028. [DOI] [PubMed] [Google Scholar]
- 2.Zidan AS, Spinks C, Fortunak J, Habib M, Khan MA. Near-infrared investigations of novel anti-HIV tenofovir liposomes. AAPS J. 2010;12(2):202–214. doi: 10.1208/s12248-010-9177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi SU, Bui T, Ho RJ. pH-Dependent interactions of indinavir and iipids in nanoparticles and their ibility to entrap a solute. J Pharm Sci. 2008;97(2):931–943. doi: 10.1002/jps.21020. [DOI] [PubMed] [Google Scholar]
- 4.Rohan LC, Sassi AB. Vaginal drug delivery systems for HIV prevention. AAPS J. 2009;11(1):78–87. doi: 10.1208/s12248-009-9082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ndesendo VM, Pillay V, Choonara YE, Buchmann E, Bayever DN, Meyer LC. A review of current intravaginal drug delivery approaches employed for the prophylaxis of HIV/AIDS and prevention of sexually transmitted infections. AAPS PharmSciTech. 2008;9(2):505–520. doi: 10.1208/s12249-008-9073-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D'Cruz OJ, Uckun FM. Clinical development of microbicides for the prevention of HIV infection. Curr Pharm Des. 2004;10(3):315–336. doi: 10.2174/1381612043386374. [DOI] [PubMed] [Google Scholar]
- 7.Abdool Karim Q, Abdool Karim SS, Frohlich JA, Grobler AC, Baxter C, Mansoor LE, et al. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science. 2010;329(5996):1168–1174. doi: 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rohan LC, Moncla BJ, Kunjara Na Ayudhya RP, Cost M, Huang Y, et al. In vitro and ex vivo testing of tenofovir shows it is effective as an HIV-1 microbicide. PLoS One. 2010;5(2):1–13. doi: 10.1371/journal.pone.0009310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alukda D, Sturgis T, Youan BB. Formulation of tenofovir-loaded functionalized solid lipid nanoparticles intended for HIV prevention. J Pharm Sci. 2011;100(8):3345–3356. doi: 10.1002/jps.22529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayer KH, Maslankowski LA, Gai F, El-Sadr WM, Justman J, Kwiecien A, et al. Safety and tolerability of tenofovir vaginal gel in abstinent and sexually active HIV-infected and uninfected women. AIDS. 2006;20(4):543–551. doi: 10.1097/01.aids.0000210608.70762.c3. [DOI] [PubMed] [Google Scholar]
- 11.Rosen RK, Morrow KM, Carballo-Dieguez A, Mantell JE, Hoffman S, Gai F, et al. Acceptability of tenofovir gel as a vaginal microbicide among women in a phase I trial: a mixed-methods study. J Womens Health (Larchmt) 2008;17(3):383–392. doi: 10.1089/jwh.2006.0325. [DOI] [PubMed] [Google Scholar]
- 12.Saxena BB, Han YA, Fu D, Rathnam P, Singh M, Laurence J, Lerner S. Sustained release of microbicides by newly engineered vaginal rings. AIDS. 2009;23(8):917–922. doi: 10.1097/QAD.0b013e32832af57c. [DOI] [PubMed] [Google Scholar]
- 13.Kandagal PB, Manjunatha DH, Seetharamappa J, Kalanur SS. RP-HPLC method for the determination of tenofovir in pharmaceutical formulations and spiked human plasma. Anal Lett. 2008;41(4):561–570. doi: 10.1080/00032710801910742. [DOI] [Google Scholar]
- 14.Sentenac S, Fernandez C, Thuillier A, Lechat P, Aymard G. Sensitive determination of tenofovir in human plasma samples using reversed-phase liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;793(2):317–324. doi: 10.1016/S1570-0232(03)00333-7. [DOI] [PubMed] [Google Scholar]
- 15.Jullien V, Tréluyer JM, Pons G, Rey E. Determination of tenofovir in human plasma by high-performance liquid chromatography with spectrofluorimetric detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;785(2):377–381. doi: 10.1016/S1570-0232(02)00933-9. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi M, Kudaka Y, Okumura N, Hirano A, Banno K, Kaneda T. Determination of plasma tenofovir concentrations using a conventional LC-MS method. Biol Pharm Bull. 2007;30(9):1784–1786. doi: 10.1248/bpb.30.1784. [DOI] [PubMed] [Google Scholar]
- 17.Yadav M, Mishra T, Singhal P, Goswami S, Shrivastav PS. Rapid and specific liquid chromatographic tandem mass spectrometric determination of tenofovir in human plasma and its fragmentation study. J Chromatogr Sci. 2009;47(2):140–148. doi: 10.1093/chromsci/47.2.140. [DOI] [PubMed] [Google Scholar]
- 18.Delahunty T, Bushman L, Robbins B, Fletcher CV. The simultaneous assay of tenofovir and emtricitabine in plasma using LC/MS/MS and isotopically labeled internal standards. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877(20–21):1907–1914. doi: 10.1016/j.jchromb.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennetto-Hood C, Long MC, Acosta EP. Development of a sensitive and specific liquid chromatography/mass spectrometry method for the determination of tenofovir in human plasma. Rapid Commun Mass Spectrom. 2007;21(13):2087–2094. doi: 10.1002/rcm.3056. [DOI] [PubMed] [Google Scholar]
- 20.Raju NA. Simultaneous estimation of tenofovir disproxil fumerate, emtricitabine & efavirenz in tablet dosage forms by isocratic-RP-HPLC. J Pharm Res. 2009;2(6):1103–1106. [Google Scholar]
- 21.Rezk NL, Crutchley RD, Kashuba AD. Simultaneous quantification of emtricitabine and tenofovir in human plasma using high-performance liquid chromatography after solid phase extraction. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;822(1–2):201–208. doi: 10.1016/j.jchromb.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 22.Sun D, Wang H, Wang B, Guo R. Development and validation of a sensitive LC-MS/MS method for the determination of adefovir in human serum and urine. J Pharm Biomed Anal. 2006;42(3):372–378. doi: 10.1016/j.jpba.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 23.Huidobro AL, Rupérez FJ, Barbas C. LC methods for acyclovir and related impurities determination. J Pharm Biomed Anal. 2005;37(4):687–694. doi: 10.1016/j.jpba.2004.11.039. [DOI] [PubMed] [Google Scholar]
- 24.Choi KY, Chung H, Min KH, Yoon HY, Kim K, Park JH, Kwon IC, Jeong SY. Self-assembled hyaluronic acid nanoparticles for active tumor targeting. Biomaterials. 2010;31(1):106–114. doi: 10.1016/j.biomaterials.2009.09.030. [DOI] [PubMed] [Google Scholar]
- 25.Al-Ghananeem AM, Malkawi AH, Muammer YM, Balko JM, Black EP, Mourad W, et al. Intratumoral delivery of Paclitaxel in solid tumor from biodegradable hyaluronan nanoparticles formulations. AAPS PharmSciTech. 2009;10(2):410–417. doi: 10.1208/s12249-009-9222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carole E, Schanté GZ, Herlin C, Vandamme TF. Chemical modifications of hyaluronic acid for the synthesis of derivatives for a broad range of biomedical applications. Carbohyd Polym. 2011;85(3):469–489. doi: 10.1016/j.carbpol.2011.03.019. [DOI] [Google Scholar]
- 27.Swyer GI. The hyaluronidase content of semen. Biochem J. 1947;41(3):409–413. doi: 10.1042/bj0410409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nirogi RVS, Kandikere VN, Shukla M, Mudigonda K, Maurya S, Boosi R, et al. Sensitive and selective liquid chromatography–tandem mass spectrometry method for the quantification of azithromycin in human plasma. Anal Chim Acta. 2005;553(1–2):1–8. doi: 10.1016/j.aca.2005.08.007. [DOI] [Google Scholar]
- 29.Khan A, Khan MI, Iqbal Z, Shah Y, Ahmad L, Watson DG. An optimized and validated RP-HPLC/UV detection method for simultaneous determination of all-trans-retinol (vitamin A) and alpha-tocopherol (vitamin E) in human serum: comparison of different particulate reversed-phase HPLC columns. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878(25):2339–2347. doi: 10.1016/j.jchromb.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 30.Xu M, Ju W, Xia X, Tan H, Chen M, Zhang J, et al. Determination of rimantadine in rat plasma by liquid chromatography/electrospray mass spectrometry and its application in a pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;864(1–2):123–128. doi: 10.1016/j.jchromb.2008.01.053. [DOI] [PubMed] [Google Scholar]
- 31.Liebhafsky HA, Pfeiffer HG. Beer's law in analytical chemistry. J Chem Educ. 1953;30(9):450–452. doi: 10.1021/ed030p450. [DOI] [Google Scholar]
- 32.ICH Guideline . Q2 (R1). Validation of analytical procedures: text and methodology. Geneva Switzerland: ICH Secretariat; 2005. [Google Scholar]
- 33.Pelillo M, Cuvelier ME, Biguzzi B, Gallina Toschi T, Berset C, Lercker G. Calculation of the molar absorptivity of polyphenols by using liquid chromatography with diode array detection: the case of carnosic acid. J Chromatogr A. 2004;1023(2):225–229. doi: 10.1016/S0021-9673(03)01206-8. [DOI] [PubMed] [Google Scholar]
- 34.Hu Z, Xia X, Tang L (inventors). Process for synthesizing oil and surfactant-free hyaluronic acid nanoparticles and microparticles. United States patent 7,601,704 B2. 2009 October 13.
- 35.Hackley V, Ferraris C. The use of nomenclature in dispersion science and technology, NIST Recommended Practice Guide. NIST SP. 2001; 960-3; 72; SP
- 36.Romero-Pérez A, García-García E, Zavaleta-Mancera A, Ramírez-Bribiesca JE, Revilla-Vázquez A, Hernández-Calva LM, et al. Designing and evaluation of sodium selenite nanoparticles in vitro to improve selenium absorption in ruminants. Vet Res Commun. 2010;34(1):71–79. doi: 10.1007/s11259-009-9335-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Owen DH, Katz DF. A review of the physical and chemical properties of human semen and the formulation of a semen simulant. J Androl. 2005;26(4):459–469. doi: 10.2164/jandrol.04104. [DOI] [PubMed] [Google Scholar]
- 38.Owen DH, Katz DF. A vaginal fluid simulant. Contraception. 1999;59(2):91–95. doi: 10.1016/S0010-7824(99)00010-4. [DOI] [PubMed] [Google Scholar]
- 39.Patel S, Baghel US, Rajesh P, Prabhakar D, Engla G, Nagar PN. Spectrophotometric method development and validation for simultaneous estimation of tenofovir disoproxil fumarate and emtricitabine in bulk drug and tablet dosage form. Int J Pharm Clin Res. 2009;1:28–30. [Google Scholar]
- 40.O'Neil MJ, Smith A, Heckelman PE. The Merck index. 13. New Jersey: Merck research laboratories; 2001. [Google Scholar]