

Abstract
Loratadine is a class II water-insoluble drug and its dissolution rate and, consequently, absorption are dependent on the gastrointestinal pH. The resulting very high variability in bioavailability and related inter- and intra-subject absorption variations present a major challenge that hinders the realization of an effective and uniform therapy. Among the several techniques that have been used to minimize pH dependency of dissolution rate, liquisolid compacts technique can be suggested as a promising solution. In this study, it was hypothesized that the formulation of loratadine using liquisolid compacts technique may reduce the effect of pH variation on the drug dissolution rate. Solubilities of loratadine in propylene glycol, Tween 80, and polyethylene glycol 400 were first measured and propylene glycol was selected as for producing the highest solubility among the tested solvents. Several liquisolid tablet formulations containing various ratios of drug: propylene glycol (5%, 10%, and 20% w/w) were prepared. The ratio of microcrystalline cellulose (carrier) to silica (coating powder material) was kept constant in all formulations. The dissolution behavior of loratadine from liquisolid compacts was investigated in several buffered media with different pH values (pH 1.2, 2.5, and 5). The results showed that the drug release rates produced by liquisolid compacts were significantly higher and less affected by pH variation compared with conventionally made (direct compression) and commercial (Clarityn) tablets. In conclusion, liquisolid compacts technique may be used as a tool to minimize the effects of pH variation on the dissolution rate of drugs with poor water solubility.
Key words: dissolution rate, liquisolid compacts, loratadine, pH variations, solubility
INTRODUCTION
Loratadine is a tricyclic piperidine derivative, orally effective, long-acting, non-sedating, second-generation antihistamine with no significant antimuscarinic activity. It is used for the symptomatic relief of allergic conditions including rhinitis and chronic urticaria (1, 2). According to the Biopharmaceutics Classification System and based on its poor water solubility and high permeability, loratadine is classified as a class II drug (3). As a member of this class of drugs, the rate of oral absorption of loratadine is controlled by its dissolution rate in the gastrointestinal (GI) tract and, as a result, no more than 80% of the total dose administered is absorbed (3). Additionally, loratadine is an ionizable weak base and its solubility is pH dependent; decreasing exponentially with the increase in pH (3). The oral bioavailability of the drug was found to be highly variable due to pH variations in the fluids of GI tract (3). Similarly, a very high degree of inter-subject variability in the pharmacokinetic parameters of loratadine was reported under fed and fasting conditions (4). The concomitant administration of this drug with food was accompanied with increased bioavailability and delayed time to peak plasma concentrations. Recent reports (5, 6) investigated the inclusion complexation with cyclodextrin derivatives to improve the aqueous solubility and bioavailability of loratadine.
One suggested technique to improve the dissolution properties of poorly water-soluble drugs is “liquisolid compacts”, which was first described by Spireas (7). This new formulation technique involves the conversion of a liquid medication into a dry-looking, freely flowable, and readily compressible powder. This is achieved by dissolving/dispersing the insoluble drug in an orally safe water-miscible solvent which is then loaded by simple blending to a mixture of two powder compounds, referred to as carrier and coating materials. Cellulose derivatives are the most studied as the carrier, whereas very fine particle size silica powder is often employed as the coating material. Liquisolid compacts showed a promise for improving the dissolution rate and bioavailability of many water-insoluble drugs such as prednisolone (8), methchlorothiazide (9), hydrochlorothiazide (10), piroxicam (11, 12), indomethacine (13), carbamazepine (14), famotidine (15), naproxen (16), and repaglinide (17). More recently, the potential of liquisolid compacts technique in producing sustained release systems using propranolol hydrochloride (18) and theophylline (19) was explored.
Although the enhanced dissolution rate achieved by means of liquisolid compacts technique has been extensively studied, no reports, thus far, explored whether this technique can be beneficial for producing tablets with pH-independent drug release. Thus, the aim of this work was to examine the hypothesis that liquisolid compacts technique could be exploited to minimize the influence of pH variations on the dissolution rate of loratadine, as a model of class II drugs. In the current study, 10 mg loratadine was formulated into liquisolid tablets, by first dissolving/dispersing in propylene glycol with different drug concentrations, followed by mixing with similar powder excipients (Avicel PH 102/ Aerosil 200; 20:1) to form liquisolid compacts prior to compression. The in vitro drug dissolution rates of liquisolid formulations, direct compression, and commercial tablets were compared using a USP dissolution apparatus II at different dissolution media (pH 1.2, 2.5, and 5).
MATERIALS AND METHODS
Materials
The following materials were used as received: loratadine (Cadila Pharmacueticals Limited, India); Vivapur microcrystalline cellulose 102 (Avicel PH 102, FMC Co., Philadelphia, PA, USA); colloidal silicone dioxide (Aerosil 200, Degussa, Belgium); sodium starch glycolate (Explotab, FMC Co., Philadelphia, PA, USA); polysorbate 80 (Tween 80, Sigma, USA); polyethylene glycol 400 (PEG 400 Interpharm Ltd, UK); propylene glycol (PG, Interpharm Ltd, UK). All other reagents and chemicals were of analytical grade.
Solubility Studies
The solubility of loratadine in three non-volatile solvents, namely, propylene glycol, polyethylene glycol 400, and polysorbate 80, was studied. In 10-ml test tubes, saturated solutions of the drug in the above solvents were prepared by mixing loratadine with such amounts of each of the solvents in order to produce systems containing an excess of the drug. The mixtures were stirred for 30 min, followed by sonication for another 30 min, and then maintained for 48 h at 25°C under constant agitation to ensure that equilibrium state was reached. The mixtures were then passed through 0.45 μm filters (Millipor HA, USA) to remove the undissolved excess of the drug. Accurately measured volumes of the filtrates were further diluted with methanol and analyzed spectrophotometrically at 247 nm (7800 Jasco, Japan) to determine drug content in each solvent. Three measurements were performed for each sample to compute the solubility of loratadine. The whole work was conducted under ambient room temperature at 25°C.
Preparation of Direct Compression Tablets
Loratadine conventional tablets (formulation F0) were produced using a direct compression method. Ten-milligram drug was mixed with 100 mg Avicel PH 102 and 5 mg Aerosil 200 (ratio of microcrystalline cellulose:Aerosil 200 was 20, R) for a period of 10 min in a cubic mixer (ERWEKA GmbH, Type UG, Germany). The mixture was further mixed with 5 mg sodium starch glycolate (as the disintegrating agent) for another 10 min. A single punch machine (ERWEKA GmbH, EK0, Germany) was used to compress the mixture. Compression load was adjusted in such a way to produce tablets with a hardness of 5–7 kg cm−1.
Preparation of Liquisolid Tablets
Calculation of Liquid Load Factor
In the current study, propylene glycol liquid vehicle (PG) was used as the liquid vehicle, microcrystalline cellulose Avicel PH 102 as the carrier material, and Aerosil 200 as the coating powder. Liquid load factor, Lf, defined as the ratio of weight of the liquid medication and carrier powder in the liquisolid compacts, refers to the maximum amount of the liquid medication that can be loaded to the carrier material to produce an acceptably flowable and compressible liquid/powder admixture.
Lf is given by the following equation (7):
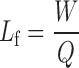 |
1 |
where:
- W
is the weight of liquid medication and
- Q
is the weight of carrier material.
It can also be calculated using the following equation:
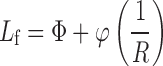 |
2 |
where:
- Φ
is the flowable liquid retention potential for Avicel PH 102; it equals to 0.15,
- φ
is the flowable liquid retention potential for Aerosil 200; it equals to 1.5, and
- R
is the ratio of carrier (Q) to coating (q) materials; it is given by the following equation:
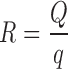 |
3 |
The ratio of Avicel PH 102 to Aerosil 200 was selected at R = 20. Thus,
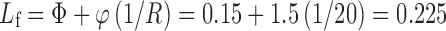 |
This calculated Lf is general and does not take in account drug factors. Therefore, it should be adjusted experimentally according to each drug substance and its concentration in the liquid.
The experimental liquid load factor was calculated starting from the Lf value calculated above. Following the calculations of Q and q amounts, a mixture of half of these quantities was added to the liquid medication in a mortar. Two grams of the carrier-coating materials admixture (R = 20) was added the above mixture and angle of repose, Carr’s index, and Hausner’s ratio were measured. This procedure was repeated until acceptable values of angle of repose, Carr’s index, and Hausner’s ratio were achieved.
Procedure of Preparation
Three liquisolid formulations (see Table I), namely, F1, F2, and F3, were prepared first by mixing quantities of 10 mg of the solid drug with 190, 90, or 40 mg of the liquid vehicle (PG) in such a way to produce liquid medication mixtures with varying concentrations of loratadine, 5%, 10%, and 20%, respectively. Each liquid mixture was heated to 50°C with continuous stirring, followed by sonication for 15 min, until a homogenous mixture was achieved. Afterwards, a binary mixture of the carrier material Avicel PH 102 and the coating powder Aerosil 200 (with a ratio of 20:1) was added to each of the above mixtures containing the drug and PG under constant mixing in a mortar. The quantities of the carrier and coating materials, calculated based on Lf value, are enough to maintain acceptable flow and compression properties. Depending on the ratio of drug to PG in each formulation, different liquid load factors (Lf) ranging from 0.287 to 0.302 were employed in the preparation of the liquisolid formulations, computed as previously described (see “Calculation of Liquid Load Factor” section). Finally, 5% (w/w) of the disintegrating material sodium starch glycolate was mixed with each combination for a period of 10 min. The final mixtures were compressed using a single punch tablet press machine (ERWEKA GmbH, EK0, Germany) to achieve a tablet hardness of 5–7 kg cm−1. Key formulation characteristics of the prepared loratadine liquisolid compacts are shown in Table I.
Table I.
Formulation Characteristics of Loratadine Direct Compression and Liquisolid Tablets
| Formulationa | Drug concentration in liquid medication (% w/w) | Loratadine strength in unit dose (mg) | Propylene glycol (mg) | expL f | Avicel PH 102 (mg) | Aerosil 200 (mg) | Sodium starch glycolate (mg) | Tablet weight (mg) |
|---|---|---|---|---|---|---|---|---|
| F0 | N/A | 10 | N/A | N/A | 100 | 5 | 5 | 120 |
| F1 | 5 | 10 | 190 | 0.287 | 697 | 35 | 49 | 980 |
| F2 | 10 | 10 | 90 | 0.292 | 342.5 | 17.12 | 23.7 | 484 |
| F3 | 20 | 10 | 40 | 0.302 | 165.6 | 8.28 | 11.25 | 235 |
expL f experimental L f
aIn all formulations, the ratio of carrier to coating material is R = 20
Quality Control of Prepared Tablets
Tablets were assessed by performing tests for weight variation (homogeneity of weight), friability, disintegration, and content uniformity. All these tests were performed in triplicate and in accordance with the compendial requirements (20).
Friability test was carried out using ERWEKA GmbH TA 20 (Germany) Friability tester. For each formulation, different numbers of tablets were used based on the tablets’ mean weight and the friability percentage was calculated.
The USP disintegration apparatus (ERWEKA GmbH ZT 502, Germany) was employed to perform the disintegration test. Each time, six tablets of each formulation were tested in distilled water at 37 ± 2 C and the average disintegration time was calculated (20). For content uniformity test, tablets should contain not less than 95% and not more than 105% of the labeled strength (20).
Dissolution Studies
The in vitro dissolution studies of loratadine from the different formulations were performed using the USP dissolution apparatus II—paddle method (Pharma Test, PT-DT7, Germany). In these studies, 0.1 N HCl aqueous solution (pH 1.2), HCl/sodium acetate buffer solution (pH 2.5), and acetate buffer solution (pH 5) were employed as the dissolution media. Each time, six tablets from each formulation were placed in 900 ml dissolution medium per vessel. The medium was maintained at 37 ± 0.1°C and the rate of stirring was 50 ± 2 rpm. The formulations were tested in the first two media, i.e., HCl aqueous solution and HCl/sodium acetate buffer solution, for a whole hour, and at appropriate time intervals (5, 10, 20, 40, and 60), 5 ml of the samples were withdrawn. Whereas, the dissolution test was continued for an extra hour (a total of 2 h) in the acetate buffer solution at pH 5, and 5-ml samples were collected at time intervals of 10, 20, 40, 60, 90, and 120. Additional comparisons were carried out by studying the drug release from the brand product Clarityn® (Schering-Plough), containing 10 mg loratadine, in all of the three dissolution media. To maintain a constant volume of dissolution media, 5 ml of fresh dissolution fluid was added. The collected samples were filtered through a 0.45-mm Millipore filter. The samples were then analyzed spectrophotometrically at 247 nm (7800 Jasco, Japan). For assessment and comparison, the dissolution results of liquisolid tablets and Clarityn were compared using a two-tailed unpaired Student’s t test at P < 0.05.
RESULTS
Solubility Studies
The solubility of loratadine in PEG 400, Tween 80, and propylene glycol is shown in Table II. The table demonstrates that, among all the three solvents, propylene glycol was the best solvent for loratadine. Thus, propylene glycol was selected as the non-volatile solvent in the preparation of liquisolid compacts.
Table II.
Solubility of Loratadine in Various Solvents
| Solvent | Solubility (% w/w) |
|---|---|
| PEG 400 | 5.53 (±1.12) |
| Tween 80 | 7.49 (±0.95) |
| PG | 16 (±1.39) |
PEG 400 polyethylene glycol 400, PG propylene glycol
Determination of Liquid Load Factor
Following flowability studies, experimental liquid load factor, for each formulation, that produces liquisolid compacts with good flowability was determined. Table III shows experimental liquid load factor values with the corresponding angle of repose, Carr’s index, and Hausner’s ratio values.
Table III.
Determination of the Liquid Load Factor, Angle of Repose, Carr’s Index, and Hausner’s Ratio
| Formulation | expL f | Angle of repose | Carr’s index | Hausner’s ratio |
|---|---|---|---|---|
| F1 | 0.287 | 27 | 14 | 1.16 |
| F2 | 0.292 | 30 | 17 | 1.2 |
| F3 | 0.302 | 26 | 10 | 1.11 |
expL f experimental L f
The experimental Lf values are close to the calculated value, which is 0.225, and to those values found in the literature (8, 13–15). The use of these optimized experimental values of Lf helps reducing the required amounts of carrier and coating materials and consequently reducing tablet size. To prepare the liquisolid tablets, the R value of 20 was selected because this value is proved to provide liquisolid compacts with ideal flowability characteristics (8, 14).
Quality Control of Prepared Tablets
The results of the quality control tests carried out to evaluate liquisolid tablets are shown in Table IV. It was apparent that the liquisolid tablets complied with the requirements that should be present in tablets including uniformity of weight, friability, disintegration time, and uniformity of drug content.
Table IV.
Results of the Quality Control Tests Performed on Liquisolid Tablets
| Formulation | Weight uniformity (mg) | Friability (%) | Disintegration (min) | Content uniformity (%) |
|---|---|---|---|---|
| F0 | 119.8 (±0.87) | 0.56 | 1.7 | 99.7 (±1.2) |
| F1 | 978.8 (±9.06) | 0.61 | 1.9 | 101.3 (±1.7) |
| F2 | 485.2 (±5.44) | 0.94 | 1.5 | 100.9 (±1.05) |
| F3 | 235.4 (±3.49) | 0.45 | 2 | 101.7 (±2.3) |
Dissolution Studies
Figures 1, 2, and 3 display the dissolution profiles of loratadine from the liquisolid, direct compression, and commercial tablets in different dissolution media. At pH 1.2 (Fig. 1), all formulations released more than 75% of the labeled strength within 5 min. In addition, no significant difference could be detected among different formulations at time intervals 10, 20, 40, and 60 min (P < 0.05). However, at time interval of 5 min, direct compression tablets (F0) produced significantly lower dissolution rates in comparison with other formulations.
Fig. 1.

Percentage of loratadine released from liquisolid formulations (F1, F2, and F3), direct compression, and Clarityn at pH 1.2 tablets over a period of 1 h
Fig. 2.

Percentage of loratadine released from liquisolid formulations (F1, F2, and F3), direct compression, and Clarityn at pH 2.5 tablets over a period of 1 h
Fig. 3.

Percentage of loratadine released from liquisolid formulations (F1, F2, and F3), direct compression, and Clarityn at pH 5 tablets over a period of 2 h
At pH 2.5 (Fig. 2), liquisolid formulations (F1, F2, and F3) showed a significantly higher dissolution rate comparable to direct compression (F0) and commercial tablets (Clarityn®) at P < 0.05. For example, the mean percentages of drug released from F1, F2, F3, F0, and commercial tablets after 10 min were 84.6%, 82.8%, 71.5%, 62.1%, and 53.7%, respectively.
Similar results were obtained at pH 5 with formulations F1 and F2 showing significantly higher release than other formulations (Fig. 3). For example, after 30 min, the mean percentages of drug release of 25.6%, 23.9%, 17.7%, 11.2%, and 6.2% were measured from F1, F2, F3, F0, and commercial tablets, respectively. However, over the 2-h long experiment, only approximately 50% of total drug was released from the best two formulations, i.e., F1 and F2.
DISCUSSION
According to the findings of this study and in comparison with other formulations, F2 was the best formulation producing the best drug release profile with acceptable values of amounts of carrier and coating materials, tablet weight, and cost. The dissolution behavior can be studied using the classic dissolution rate equation of Noyes–Whitney (21):
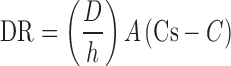 |
4 |
In the conditions of our experiment, diffusion coefficient (D) of the drug molecules and the thickness (h) of the static diffusion layer are expected to remain constant. Cs, the saturation concentration or solubility, of loratadine varies based on the pH, decreasing significantly as the pH rises from 1.2 to 5, and C, the actual concentration of the drug in the bulk solution, increases continuously as the drug diffuses to the dissolution medium. Liquisolid compacts contain either solutions or dispersions, with very fine particle size (high surface area A), of the drug. Ideally, if the drug is completely dissolved in the liquisolid compacts, it should diffuse readily to the surrounding environment skipping the dissolution step and resulting in rapid release. Nonetheless, the release profiles of loratadine (Figs. 1, 2, and 3) suggest that the release process does not take place instantly and becomes slower as the pH value increases from 1 to 5. This may be explained by a possible sudden release of the dissolved drug in the liquisolid compacts to the surrounding layers of the dissolution medium, causing these layers to become quickly saturated with the drug as the solvent, PG, becomes diluted with the medium; hence, precipitation may take place. If this occurred, the drug release would be delayed depending on the dissolution rate which, according to Eq. 4, is influenced by the concentration gradient (Cs – C). This effect appears to be more obvious with higher pH values. At pH 5, the pre-dissolved drug in the liquisolid formulations F1 and F2 produced a relatively quick drug release of about 25% at the first 10 min (Fig. 3). However, it needed an extra 110 min before the next 25% of the drug was released. This sharp decrease in release may be explained by drug precipitation and consequent slow dissolution rate of the precipitated drug. If 10 mg of loratadine, the labeled strength, is completely released in the medium with a volume of 900 ml, a concentration of 11.1 μg/ml will be reached, which is higher than the solubility of loratadine at a close pH value (7.6 μg/ml at pH 5.19 and 25°C) (22). This low saturation solubility (Cs) at pH 5 produces a small concentration difference (Cs – C) and would promote precipitation during drug release and subsequently a slow dissolution rate. Comparably, this effect was less apparent at pH 2.5 and practically undetectable at pH 1.2 as the solubilities of loratadine increased to far higher values of 0.60 and 4.59 mg/ml (3), respectively. Furthermore, the solubility of loratadine in propylene glycol, which is 16% w/w (as measured in this study), justifies the improved dissolution characteristics observed with formulations F1 and F2 where the drug in the liquisolid compacts was in the form of a solution; whereas, in formulation F3, the amount of the drug was above the saturation concentration, i.e., the drug was in a form of a suspension, thus this formulation showed slower release compared with the former two formulations.
When comparing the release profiles of liquisolid tablets with non-liquisolid (DC and commercial) ones, although no significant differences were detected at strongly acidic pH, differences started to become more apparent as the pH was increased to pH 2.5 to reach a difference of up to 200% at pH 5. This suggests that the liquisolid technique may promote drug dissolution in stomach regardless of the pH value which varies in approximate range of 1–5 between fasted and fed states.
Loratadine has a pKa of 4.9 (23), signifying that most loratadine, 100% and 99.6%, would be ionized at pH 1.2 and 2.5, respectively. This indicates that if the stomach pH is in this range, even though the drug is completely dissolved it will not be absorbed in the stomach, because it is ionized, and absorption will be delayed until the drug is transported to the small intestines. However, if the pH of stomach reached 5 (as in the fed state), 55.7% of the dissolved drug would be in the unionized form and ready for absorption. In both situations, liquisolid tablets are showing better release rates over conventional tablets and, therefore, they would be expected to reduce variabilities in bioavailability resulting from stomach pH variations.
One main advantage of minimizing pH dependency of drug release would be the potential reduction in inter- and intra-subject absorption variations between fasting and fed states. In a study conducted by Noumair’s group (4), loratadine administrated after a fat-rich meal showed a significant increase in bioavailability represented by an increase in Cmax and AUC. Although, the fat-rich meal is expected to increase the gastric pH (up to approximately pH 5), it provides an oily medium for dissolving the water-insoluble drug which is then absorbed mainly through the fat absorption pathway facilitated by bile salts. This explains the delay in Tmax as a result of the delayed gastric emptying time, resulted from fat-rich meal, and the time that the drug takes to pass to blood circulation, as fat is first absorbed into the lymphatic circulation. Liquisolid compacts technique can be suggested to improve the bioavailability of loratadine, and similar poorly soluble drugs, without a compromise in Tmax if the drug can still be released and absorbed by normal absorption pathway at a stomach pH value of as much high as 5. Further in vivo investigation is required to examine this hypothesis.
CONCLUSIONS
The major finding of this work is the improved dissolution of loratadine at different pH values, particularly at pH 2.5 and 5, in comparison with the brand product. In agreement with other studies (8, 13), propylene glycol was the best candidate as the liquid vehicle to be used to prepare the drug liquid medication and better release characteristics were observed when the liquisolid compacts contained completely dissolved drug in the liquid medication. As a conclusion, the liquisolid compacts technique can be presented as a promising tool for the formulation of loratadine, and similar class II drugs, into immediate release tablets with minimized pH influence on their dissolution rates.
REFERENCES
- 1.Ahn HS, Barnett A. Selective displacement of [3H]mepyramine from peripheral vs. central nervous system receptors by loratadine, a non-sedating antihistamine. Eur J Pharmacol. 1986;127(1–2):153–5. doi: 10.1016/0014-2999(86)90219-0. [DOI] [PubMed] [Google Scholar]
- 2.Hilbert J, Radwanski E, Weglein R, Luc V, Perentesis G, Symchowicz S, et al. Pharmacokinetics and dose proportionality of loratadine. J Clin Pharmacol. 1987;27(9):694–8. doi: 10.1002/j.1552-4604.1987.tb03090.x. [DOI] [PubMed] [Google Scholar]
- 3.Khan MZ, Rausl D, Zanoski R, Zidar S, Mikulcic JH, Krizmanic L, et al. Classification of loratadine based on the biopharmaceutics drug classification concept and possible in vitro–in vivo correlation. Biol Pharm Bull. 2004;27(10):1630–5. doi: 10.1248/bpb.27.1630. [DOI] [PubMed] [Google Scholar]
- 4.Nomeir AA, Mojaverian P, Kosoglou T, Affrime MB, Nezamis J, Rodwanski E, et al. Influence of food on the oral bioavailability of loratadine and pseudoephedrine from extended-release tablets in healthy volunteers. J Clin Pharmacol. 1996;36(10):923–30. doi: 10.1002/j.1552-4604.1996.tb04759.x. [DOI] [PubMed] [Google Scholar]
- 5.Nacsa A, Ambrus R, Berkesi O, Szabo-Revesz P, Aigner Z. Water-soluble loratadine inclusion complex: analytical control of the preparation by microwave irradiation. J Pharm Biomed Anal. 2008;48(3):1020–3. doi: 10.1016/j.jpba.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Szabados-Nacsa Á, Sipos P, Martinek T, Mándity I, Blazsó G, Balogh Á, et al. Physico-chemical characterization and in vitro/in vivo evaluation of loratadine:dimethyl-[beta]-cyclodextrin inclusion complexes. J Pharm Biomed Anal. 2011;55(2):294–300. doi: 10.1016/j.jpba.2011.01.024. [DOI] [PubMed] [Google Scholar]
- 7.Spireas SS, Jarowski CI, Rohera BD. Powdered solution technology: principles and mechanism. Pharm Res. 1992;9(10):1351–8. doi: 10.1023/A:1015877905988. [DOI] [PubMed] [Google Scholar]
- 8.Spireas S, Sadu S. Enhancement of prednisolone dissolution properties using liquisolid compacts. Int J Pharm. 1998;166(2):177–88. doi: 10.1016/S0378-5173(98)00046-5. [DOI] [Google Scholar]
- 9.Spireas S, Wang T, Grover R. Effect of powder substrate on the dissolution properties of methyclothiazide liquisolid compacts. Drug Dev Ind Pharm. 1999;25(2):163–8. doi: 10.1081/DDC-100102156. [DOI] [PubMed] [Google Scholar]
- 10.Khaled KA, Asiri YA, El-Sayed YM. In vivo evaluation of hydrochlorothiazide liquisolid tablets in beagle dogs. Int J Pharm. 2001;222(1):1–6. doi: 10.1016/S0378-5173(01)00633-0. [DOI] [PubMed] [Google Scholar]
- 11.Javadzadeh Y, Siahi-Shadbad MR, Barzegar-Jalali M, Nokhodchi A. Enhancement of dissolution rate of piroxicam using liquisolid compacts. Il Farmaco. 2005;60(4):361–5. doi: 10.1016/j.farmac.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 12.Javadzadeh Y, Siahi MR, Asnaashari S, Nokhodchi A. An investigation of physicochemical properties of piroxicam liquisolid compacts. Pharm Dev Technol. 2007;12(3):337–43. doi: 10.1080/10837450701247574. [DOI] [PubMed] [Google Scholar]
- 13.Nokhodchi A, Javadzadeh Y, Siahi-Shadbad MR, Barzegar-Jalali M. The effect of type and concentration of vehicles on the dissolution rate of a poorly soluble drug (indomethacin) from liquisolid compacts. J Pharm Sci. 2005;8(1):18–25. [PubMed] [Google Scholar]
- 14.Tayel SA, Soliman II, Louis D. Improvement of dissolution properties of carbamazepine through application of the liquisolid tablet technique. Eur J Pharm Biopharm. 2008;69(1):342–7. doi: 10.1016/j.ejpb.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 15.Fahmy RH, Kassem MA. Enhancement of famotidine dissolution rate through liquisolid tablets formulation: In vitro and in vivo evaluation. Eur J Pharm Biopharm. 2008;69(3):993–1003. doi: 10.1016/j.ejpb.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 16.Tiong N, Elkordy AA. Effects of liquisolid formulations on dissolution of naproxen. Eur J Pharm Biopharm. 2009;73(3):373–84. doi: 10.1016/j.ejpb.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 17.El-Houssieny BM, Wahman LF, Arafa NM. Bioavailability and biological activity of liquisolid compact formula of repaglinide and its effect on glucose tolerance in rabbits. Biosci Trends. 2010;4(1):17–24. [PubMed] [Google Scholar]
- 18.Javadzadeh Y, Musaalrezaei L, Nokhodchi A. Liquisolid technique as a new approach to sustain propranolol hydrochloride release from tablet matrices. Int J Pharm. 2008;362(1–2):102–8. doi: 10.1016/j.ijpharm.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 19.Nokhodchi A, Aliakbar R, Desai S, Javadzadeh Y. Liquisolid compacts: The effect of cosolvent and HPMC on theophylline release. Colloids Surf B Biointerfaces. 2010;79(1):262–9. doi: 10.1016/j.colsurfb.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 20.The United States Pharmacopoeia National Formulary, USP32 NF27: The United States Pharmacopoeial Convention. Rockville; MD; 2009.
- 21.Noyes AA, Whitney WR. The rate of solution of solid substances in their own solutions. J Am Chem Soc. 1897;19:930–4. doi: 10.1021/ja02086a003. [DOI] [Google Scholar]
- 22.Popovic G, Cakar M, Agbaba D. Acid–base equilibria and solubility of loratadine and desloratadine in water and micellar media. J Pharm Biomed Anal. 2009;49(1):42–7. doi: 10.1016/j.jpba.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 23.Delpóna E, Valenzuela C, Gay P, Franqueza L, Snyders D, Tamargo J. Block of human cardiac Kv1.5 channels by loratadine: Voltage-, time- and use-dependent block at concentrations above therapeutic levels. Cardiovasc Res. 1997;35:341–50. doi: 10.1016/S0008-6363(97)00121-1. [DOI] [PubMed] [Google Scholar]