

Abstract
Sodium fluoride (NaF) is used as a source of fluoride ions in diverse applications. Fluoride salt is an effective prophylactic for dental caries and is an essential element required for bone health. However, fluoride is known to cause cytotoxicity in a concentration-dependent manner. Further, no information is available on the effects of NaF on mouse embryonic stem cells (mESCs). We investigated the mode of cell death induced by NaF and the mechanisms involved. NaF treatment greater than 1 mM reduced viability and DNA synthesis in mESCs and induced cell cycle arrest in the G2/M phase. The addition of NaF induced cell death mainly by apoptosis rather than necrosis. Catalase (CAT) treatment significantly inhibited the NaF-mediated cell death and also suppressed the NaF-mediated increase in phospho-c-Jun N-terminal kinase (p-JNK) levels. Pre-treatment with SP600125 or z-VAD-fmk significantly attenuated the NaF-mediated reduction in cell viability. In contrast, intracellular free calcium chelator, but not of sodium or calcium ion channel blockers, facilitated NaF-induced toxicity in the cells. A JNK specific inhibitor (SP600125) prevented the NaF-induced increase in growth arrest and the DNA damage-inducible protein 45α. Further, NaF-mediated loss of mitochondrial membrane potential was apparently inhibited by pifithrin-α or CAT inhibitor. These findings suggest that NaF affects viability of mESCs in a concentration-dependent manner, where more than 1 mM NaF causes apoptosis through hydroxyl radical-dependent and caspase- and JNK-mediated pathways.
Keywords: Mouse embryonic stem cells, Sodium fluoride, Cell death, ROS, Cellular signaling
Introduction
Fluoride is an effective prophylactic for dental caries and is an essential element required for bone health. However, fluoride can have double-edged sword effects on bones depending not only on the concentrations and to which bones are exposed, but also on the absorption capacity, age, and nutritional status of the individual (Caverzasio et al., 1998; Fordyce, 2011). The treatment of osteoporosis with sodium fluoride (NaF) at 20–30 mg/day exerts mostly positive effects on bone formation and water fluoridation at concentrations ranging from 1 to 2 mg/l apparently reduces dental caries prevalence (Fordyce, 2011). Otherwise, such fluoride treatments result in several disorders including enamel and skeletal fluorosis, renal toxicity, diarrhea, epithelial lung cell toxicity, and heart rate disorders (Ba et al., 2010; Everett, 2011; Fordyce, 2011; Gazzano et al., 2010). Fluoride is also able to induce detrimental effects on cells, although it depends on the doses and duration exposed and types of cells (Chien et al., 2006; Qu et al., 2008; Wurtz et al., 2008).
Growth arrest and apoptosis induction are among the most common toxic effects of fluoride on many types of cells (Ren et al., 2011; Thrane et al., 2001; Wang et al., 2011; Yang et al., 2011). Accumulated evidence has suggested that toxic heavy metals lead to apoptosis and growth inhibition depending on the exposure dose where reactive oxygen species (ROS) are closely involved (Son et al., 2010; 2011). ROS are generated at low concentrations in a constant manner in living organisms and is an essential event for the function of immune cells. However, over-expression or decreased removal of intracellular ROS induces oxidative damage to cells and tissues (Klaunig et al., 2011). A few investigators have demonstrated that fluoride induces apoptosis by elevating oxidative stress-mediated lipid peroxidation with subsequent mitochondrial stress and the activation of downstream pathways (Karube et al., 2009; Wang et al., 2010). Fluoride was also shown to suppress proliferation and induce apoptosis through decreased insulin growth factor-I expression and oxidative stress in primary cultured mouse osteoblasts (Wang et al., 2011). These findings suggest that fluoride exposure can mediate apoptotic cell death, in which the resultant ROS played an important role.
There are reports supporting the role of fluoride in inducing oral fluorosis. Fluorosis of the maxillary central incisors is believed to be associated with fluoride ingestion at high concentrations (> 1.5 mg/l with drinking water contents) at an early age between 15 and 30 months (Buzalaf and Levy, 2011). Considering that this age range is the time when unerupted permanent teeth form, it is suggested that the proliferation and differentiation of stem-like cells are sensitive to fluoride, as shown in osteoblasts and ameloblasts (Kubota et al., 2005; Ren et al., 2011; Wang et al., 2011; Yang et al., 2011). Children aged 8 to 12 year, who born and raised in the area containing 1.8 mg/l of fluoride in drinking water, also showed dental fluorosis rate by 53%, compared to those of the control area (0% prevalence rate, < 1.0 mg/l fluoride) (Ba et al., 2010). However, little information is available on the effects of fluoride on embryonic stem cells.
In this study, we examined how fluoride affects the viability and proliferation of mouse embryonic stem cells (mESCs). We also investigated the mode of cell death induced by fluoride and the mechanisms involved. The current findings suggest that fluoride induces mainly apoptotic cell death through ROS-dependent and caspase- and c-Jun N-terminal kinase (JNK)-mediated signaling pathways.
Materials and methods
Chemicals and supplies
Inhibitors for pan-caspase (z-VAD-fmk, FK009) and mitogen- activated protein kinases (MAPKs) (SP600125, PD98059, and SB203580) were purchased from ICN Biomedicals (Costa Mesa, CA) and TOCRIS (Minneapolis, MN), respectively. These inhibitors were dissolved in dimethylsulfoxide (DMSO) or ethanol immediately before use. The concentrations of these organic solvents did not exceed 0.5% of the medium. The sodium and calcium channel blockers tetrodotoxin (TTX) and nifedipine (NFD), were obtained from Abcam® (Cambridge, UK). The acetoxymethylester of the calcium chelator BAPTA (BAPTA-AM) and fetal bovine serum (FBS) were supplied by Molecular Probes (Eugene, OR) and Gibco-BRL (Gaithersburg, MD), respectively. Unless otherwise specified, other chemicals and culture plastics used in this study were purchased from Sigma Chemical Co. (St. Louis, MO) and Falcon Labware (Becton-Dickinson, Franklin Lakes, NJ), respectively.
Cell culture and treatment
The mouse embryonic stem cell (mESC) line D3 was obtained from the American Type Culture Collection (Rockville, MD). The mESCs were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco-BRL) supplemented with 200 mM L-glutamine, 0.2 mM β-mercaptoethanol, 5 ng/ml mouse leukemia inhibitory factor (LIF), 10% FBS, and 1% penicillin/streptomycin, without a feeder layer at 37 °C in an atmosphere containing 5% CO2. Cell suspensions (2×105 cells/ml) were seeded in 6-, 24- or 96-well flat-bottomed plates with 2 ml, 500 μl, or 200 μl per well, respectively. When the cells reached 70–80% confluence, they were exposed to increasing concentrations (0–5 mM) of NaF in the presence and absence of each pharmacological inhibitor, ion channel blocker, or antioxidant. At various treatment times (0–72 h), cells were collected and processed for further experiments.
Measurement of cell viability
mESCs were processed for the determination of cell viability after various times of NaF exposure using the Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan). In this assay, water-soluble tetrazolium (WST)-8 is produced by living cells and thus the level of WST produced is proportional to the viability of cells. All experimental procedures were followed according to the manufacturer’s instructions and WST absorbance was measured at 450 nm using a microplate reader (Packard Instrument Co., Downers Grove, IL).
Measurement of DNA synthesis
The level of DNA synthesis in mESCs was measured by adding 1 μCi of 3H-thymidine deoxyribose (TdR, Amersham Pharmacia Biotech Inc., Piscataway, NJ) to the cells cultured in 96-well plates during the last 4 h prior to cell harvesting. Cells were collected using a harvester (Inotech Biosystems International, Inc., Switzerland) 24 h after NaF exposure. Beta emission from the 3H-TdR-incorporated cells was measured for 1 min using a liquid scintillation counter (Packard Instrument Co.).
Enzyme immunometric assay
JNK activity was determined using an immunometric assay kit according to the manufacturer’s instructions. In brief, mESCs were suspended in a cell lysis buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate). Protein concentrations were determined using a BCA protein assay kit (Thermo Fisher Scientific Inc., Portsmouth, NH) and samples containing equal amounts of protein were placed into p-SAPK/JNK sandwich ELISA kit microtiter plates (Cell Signaling Technology, MA). Finally, the absorbance was measured using a microplate reader (Packard Instrument Co.).
Cell cycle analysis
Cell cycle was determined by flow cytometric analysis after propidium iodide (PI) staining. In brief, NaF-treated cells (2×106 cells) were fixed with 70% ethanol for 24 h, and then incubated overnight at 4 °C with 500 μl of the PI staining mixture (50 μg/ml PI in a sample buffer containing 100 μg/ml RNase A). After staining, 10,000 cells per experiment were analyzed using the FACS Calibur® system (Becton Dickinson, San Jose, CA). Cell cycle progression was determined using the ModFit LT program (Verity Software House, Topsham, ME).
FITC-annexin V/propidium iodide (PI) staining
The mESCs were washed twice with phosphate-buffered saline (PBS) before suspension in 1× binding buffer (10 mM HEPES, pH 7.4, 140 mM NaOH, and 2.5 mM CaCl2). FITC-labeled annexin V (10 μl) was mixed with 100 μl of a cell suspension containing 2×105 cells, and the cells were incubated at room temperature for 5 min. Thereafter, 4 μl of PI solution (10 g/ml) was added into the cells followed by an additional 5 min incubation. The scatter parameters of the cells (5,000 cells per experiment) were analyzed using a FACS Calibur® system. Four cell populations were identified according to the following characteristics: the viable population in the lower-left quadrant (low PI and FITC signals); the early apoptotic population in the lower-right quadrant (low PI and high FITC signals); the necrotic population in the upper-left quadrant (high PI and low FITC signals); and the late apoptotic or necrotic population in the upper-right quadrant (high PI and high FITC signals).
ELISA assay of DNA fragmentation
DNA fragmentation in NaF-exposed mESCs was assayed using a Cell Death Detection ELISA kit (Roche Diagnostics, Mannheim, Germany) and all procedures were conducted according to the manufacturer's instructions.
Measurement of mitochondrial membrane potential (MMP)
The mESCs (2 × 106 cells) were washed two times with 1 ml PBS and then stained with 50 nM 3,3′-dihexyloxacarbocyanine iodide (DiOC6; Invitrogen, Carlsbad, CA) for 20 min at 37 °C. Fluorescence related to MMP was measured using a FACS Calibur® system (Becton Dickinson), and the change in MMP level was determined using the Window Multiple Document Interface (WinMDI) 2.9 Software (PUCL, West Lafayette, IN).
Measurement of intracellular ROS
A stock solution of 2’,7’-dichlorodihydrofluorescein-diacetate (DCFH-DA) (50 mM; Calbiochem, Darmstadt, Germany) was prepared in DMSO and stored at −20 °C in the dark. The mESCs exposed to NaF were incubated with 25 μM DCFH-DA for 20 min. The green fluorescence of 2’,7’-dichlorofluorescein (DCF) was recorded at 515 nm (FL 1) using a FACS Vantage® system (Becton Dickinson), and 10,000 events were counted per sample.
Electron spin resonance (ESR) assay
All ESR measurements were conducted using a Bruker EMX spectrometer (Bruker Instruments, Billerica, MA) and a flat cell assembly as described previously (Son et al., 2011). A 5,5-dimethyl-1-pyrroline-1-oxide (DMPO) spin trap was charcoal purified and distilled to remove all ESR detectable impurities prior to use. Hyperfine couplings were measured (to 0.1 G) directly from magnetic field separation using potassium tetraperoxochromate (K3CrO8) and 1,1-diphenyl-2-picrylhydrazyl as reference standards. The Acquisit program was used for data acquisition and analysis (Bruker Instruments, Madison, MI). Reactants were mixed to a final volume of 0.5 ml and the reaction mixture was then transferred to a flat cell for ESR measurement. Experiments were performed at room temperature and under ambient air conditions.
Measurement of caspase activity
Caspase activity was assessed using the luminescent Caspase-Glo® 3/7 Assay system (Promega, Madison, WI) according to the manufacturer’s instructions. In brief, mESCs were treated with various NaF concentrations (0–3 mM) for 24 h and then 100 μl Caspase-Glo® 3/7 Reagent was added to each well of the 96-multiwell plates. The plates were incubated at room temperature for 1 h before measuring luminescence using a GlomaxTM 96 microplate luminometer (Promega). In this assay, N-(4-hydroxyphenyl) retinamide (4-HPR) was used as a caspase-dependent positive control.
Preparation of cell fractions
Whole cell lysates were made in NP-40 lysis buffer (30 mM Tris-Cl, pH 7.5, 1 mM EDTA, 150 mM NaCl, 1% NP-40, 1 mM PMSF, and a protease inhibitor mixture containing μ1 g/ml aprotinin and leupeptin). Mitochondrial and cytosolic fractions were prepared using a Mitochondria Isolation Kit for Cultured Cells (Thermo Fisher Scientific, Inc.) according to the manufacturer’s protocols. Protein samples were analyzed by western blotting after determining protein concentrations using a BCA protein assay kit.
Western blot analysis
After quantifying protein concentrations, protein lysates (30 g/sample) were analyzed by SDS-PAGE (12–15% gels) and blotted onto polyvinyl difluoride membranes. The blots were probed with primary antibodies and incubated with horseradish peroxidase-conjugated anti-IgG in blocking buffer for 1 h. After washing, the blots were developed with enhanced chemiluminescence (Santa Cruz Biotechnology Inc., Santa Cruz, CA) and exposed to X-ray film (Eastman-Kodak, Rochester, NY). Unless specified otherwise, all antibodies used in this study were obtained from Santa Cruz Biotechnology, Inc.
Statistical analysis
Unless otherwise indicated, all data are expressed as mean ± standard deviation (S.D.) from at least triplicate experiments. One-way ANOVA was used for multiple comparisons using SPSS version 18.0 software. A p value < 0.05 was considered statistically significant.
Results
NaF reduces viability and induces cell cycle arrest in mESCs in a time- and dose-dependent manner
This study initially examined how NaF influences the viability of mESCs. Untreated control cells showed a time-dependent increase in viability during experimental periods, which was not affected by the addition of 1 mM NaF until 24 h of co-incubation (Fig. 1A). In contrast, cells exposed to 2 mM NaF did not show such an increase; rather, they showed a time-dependent reduction in their viability. To verify the effects of NaF on viability, cells were either treated with various concentrations of NaF for 24 h (Fig. 1B) or with 2 mM for various incubation times (Fig. 1C). As shown in the figures, NaF-mediated reduction of viability occurred at 2 mM NaF after 24 h incubation compared to the untreated control cells. Almost complete inhibition of viability was observed when the cells were exposed to more than 4 mM NaF for 24 h or 2 mM NaF for 72 h.
Fig. 1. NaF reduces the viability of mESCs in a dose- and time-dependent manner.

(A) Cells were incubated in the presence and absence of 1 or 2 mM NaF and at various incubation times. Viability was determined by the WST-8 assay. In addition, mESCs were treated with the increasing concentrations (0–5 mM) of NaF for 24 h (B) or with 2 mM for various incubation times (0–72 h) (C) and then processed for the WST-8 assay. ***p < 0.001 vs. the untreated controls.
NaF inhibited DNA synthesis in a dose-dependent manner (Fig. 2A). Treating the cells with 3 and 5 mM NaF for 24 h decreased TdR uptake levels by 81 ± 3% and 44 ± 6%, respectively, compared to the non-treated control. Cell cycle analysis revealed that NaF treatment led to cell population migration into the sub-G1 and G2/M phases with a concomitant decrease of cells in the S phase (Figs. 2B and C). Subsequently, the levels of cyclin-dependent kinase 2 (CDK2), cyclin E, and proliferating cell nuclear antigen (PCNA) were analyzed by western blot analysis. NaF treatment did not affect CDK2 and PCNA protein levels but it markedly decreased cyclin E levels (Figs. 2D and E).
Fig. 2. NaF inhibits DNA synthesis and induces cell cycle arrest in the G2/M phase in mESCs.

(A) Cells were exposed to various concentrations (0–5 mM) of NaF for 24 h and then processed for the tritium incorporation assay. (B) Cells were incubated in the presence of NaF (0–3 mM). After 24 h incubation, cell cycle progression was analyzed by flow cytometery after PI staining. (C) The results represent the mean ± S.D. from three independent experiments. (D) mESCs were incubated at the indicated doses of NaF for 24 h and processed for western blot analysis. In panel E, the cyclin E level was calculated from triplicate experiments after normalizing the bands to β-actin. *p < 0.04, **p < 0.01, and ***p < 0.001 vs. the untreated controls.
NaF treatment causes cell death in mESCs mainly via apoptosis
Flow cytometric analysis after PI staining showed that the cell population in the sub-G1 phase of cell cycle progression, which indicates apoptotic cell death, increased after treatment with NaF in a dose-dependent manner (data not shown). FITC-annexin V/PI staining experiments also revealed that cell populations showing low-PI and high-FITC and high-PI and high-FITC signals increased to 17.5% and 24.6%, respectively, after exposing the cells to 5 mM NaF for 24 h as compared to the untreated control level of 2.0% (Fig. 3A). Figure 3B shows a significant increase in the number of apoptotic cells according to NaF concentration, although there was also a mild increase in necrotic cells as indicated by the high-PI and low-FITC signals. NaF-mediated apoptosis was supported by results from ELISA-based TUNEL assays, where NaF treatment induced a dose-dependent increase in DNA strand breaks (Fig. 3C). In addition, exposure of mESCs to NaF resulted in a marked decrease of Akt1 protein levels and an increase of poly (ADP-ribose) polymerase (PARP) cleavage (Figs. 3D and E).
Fig. 3. NaF induces cell death of mESCs mainly by apoptosis.

(A) Cells were exposed to increasing concentrations (0–5 mM) of NaF for 24 h and assessed using flow cytometry with FITC-annexin V/PI double staining. In panel B, filled and open bars represent the early and late apoptotic cells and necrotic cells, respectively (n = 5), after calculation using the WinMDI 2.9 program. (C) ELISA assay of DNA fragmentation, or (D) western blot analysis was performed as described in the Materials and Methods. In panel E, the results represent the relative fold intensity of Akt1 and cleaved-PARP after normalizing the bands to β-actin (n = 5). *p < 0.05, **p < 0.01, and ***p < 0.001 vs. untreated control values.
ROS are related to NaF-induced reduction in cell viability
Since the accumulation of intracellular ROS is related to cell death induced by toxic heavy metals, this study investigated whether NaF induced intracellular ROS accumulation in mESCs. Flow cytometric analysis revealed that NaF treatment increased ROS levels within the cells in a dose-dependent manner (Fig. 4A). This finding was supported by ESR signals showing the dose-dependent increase of hydroxyl radicals in NaF-treated mESCs (Fig. 4B). Subsequently, the effects of superoxide dismutase (SOD), catalase (CAT), N-acetyl cysteine (NAC), and apocynin (APO) antioxidants on viability in NaF-exposed mESCs were determined. Pre-treatment with 2,500 U/ml CAT, but not with other antioxidants, showed a significant inhibition in the NaF-mediated reduction of cell viability (Fig. 4C). To better understand the effects of CAT, mESCs were exposed to various concentrations of NaF in the presence and absence of 500 and 2,500 U/ml CAT for 24 h. As shown in Figure 4D, treating cells with 500 U/ml CAT showed mild protection against NaF-induced toxicity only when the cells were exposed to 2 mM NaF, whereas treatment with 2,500 U/ml markedly inhibited the NaF-mediated decrease in cell viability at the exposed NaF concentrations.
Fig. 4. Intracellular ROS is not directly related to NaF-induced reduction of mESC viability.

Cells were exposed to increasing concentrations of NaF (0–5 mM) for 24 h and then processed for flow cytometric analysis after (A) DCFH-DA staining and (B) ESR measurements. (C) Cells were incubated with 5 mM NaF in the presence and absence of various antioxidants for 24 h and then processed for the WST-8 assay. (D) Cells were incubated in the presence of NaF (0–5 mM) with and without 500 U/ml CAT or 2,500 U/ml CAT 24 h before determination of viability. *p < 0.05, **p < 0.01, and ***p < 0.001 vs. the untreated controls. ###p < 0.001 vs. 5 mM NaF treatment alone.
MAPK-mediated and caspase-dependent pathways are in part related to NaF-mediated cell death
We next explored the effects of MAPKs on NaF-mediated cell death because the activation of MAPKs tightly regulates cellular events such as proliferation, survival, and apoptosis. Pretreatment of cells with an extracellular signal-regulated kinase (ERK) inhibitor (2.5 μM PD98059) or a p38 MAPK inhibitor (2.5 μM SB203580) for 2 h did not reduce the NaF-mediated decrease in cell viability to a significant level (Suppl. 1A). In contrast, a JNK inhibitor (2.5 μM SP600125) suppressed the decrease in cells exposed to 2 or 3 mM, but not 5 mM, NaF. Western blot analysis revealed that NaF treatment increased the phosphorylated levels of JNK (p-JNK) in a dose-dependent manner (Fig. 5A), and the phosphorylation was blocked by treatment with 2,500 U/ml CAT (Fig. 5B). However, the NaF-mediated increase in p-JNK levels was not diminished by 5 μM pifithrin-α (PFT-α ) (data not shown). Similarly, pre-treatment of the cells with 5 μM PFT-α did not inhibit the NaF-mediated increase of JNK activity as determined by ELISA-based assay (Fig. 5C). NaF treatment appeared to induce the activation of caspase-3 and -9 in that the band at a molecular weight of 17 kDa, which is the active form corresponding to these caspases, was slightly increased after exposure to 2 mM NaF (Suppl. 1B). The results of enzymatic analysis also showed that NaF treatment resulted in a mild increase in caspase 3/7 activities in mESCs (Fig. 5D). Treating the cells with the pan-caspase inhibitor, z-VAD-fmk significantly inhibited the NaF-mediated caspase activation. Further, pretreatment of the cells with 2.5 μM z-VAD-fmk for 1 h before the addition of 2 or 3 mM NaF significantly inhibited the NaF-induced reduction in cell viability (Fig. 5E).
Fig. 5. JNK and caspase activation is at least in part associated with NaF-induced toxicity in mESCs.

(A) Cells were exposed to the increasing doses (0–5 mM) of NaF for 12 h and then processed for western analysis. Cells were also exposed to the indicated concentrations of NaF in the presence and absence of 2,500 U/ml CAT or 5 μM PFT-α for 12 h and then the (B) levels of p-JNK and total JNK protein, and (C) activity of JNK were detected using western blot analysis and enzyme immunometric assays. In addition, cells were exposed to the indicated concentrations of NaF in the presence and absence of 2.5 μM z-VAD-fmk or 10 μM 4-HPR for 24 h, and (D) caspase 3/7 activities in the lysates or (E) cell viability was analyzed. *p < 0.05 and **p < 0.01 vs. the experiments. In panel D, *p < 0.05 and #p < 0.05 indicate significant differences from the untreated control cells and the 2 mM NaF treatment alone, respectively.
NaF induces mitochondrial stress in mESCs, where intracellular calcium chelation accelerates NaF-mediated reduction of cell viability
Analysis of DiOC6-specific fluorescence intensity using flow cytometry revealed that NaF treatment induced a mild reduction in cellular MMP levels at doses higher than 2 mM (data not shown). A 7% and 14% reduction in MMP level was seen in cells when they were treated with 3 and 5 mM NaF for 24 h as compared to the control (Fig. 6A). NaF treatment at 3 mM resulted in a decrease in mitochondrial Bcl-2 (Fig. 6B). A mild relocation of cytochrome c into the cytoplasm from the mitochondria was found in cells exposed to more than 1 mM NaF for 24 h (Fig. 6C). However, NaF treatment did not induce an alteration of apoptosis-inducing factor (AIF) protein level both in the mitochondria and cytoplasm as determined by western blot analysis. We subsequently examined the effects of sodium and calcium channel blockers in NaF-exposed mESCs, where combined treatment of the cells with 10 μM NFD or 10 μM TTX did not diminish the NaF-mediated reduction of viability in mESCs (data not shown). Treatment of cells with BAPTA-AM, an intracellular free calcium chelator, facilitated the NaF-mediated toxicity in the cells in a dose-dependent manner (Fig. 6D). The addition of 5 μM BAPTA-AM into mESCs exposed to 2 mM NaF did not affect the NaF-induced increase in p-JNK levels, whereas the increased p-JNK levels were almost completely inhibited by the addition of 2,500 U/ml CAT (Fig. 6E).
Fig. 6. Association of mitochondrial stress and intracellular free calcium ions in NaF-mediated reduction of cell viability.

(A) The relative percentage of NaF-induced MMP loss was calculated by monitoring the DiOC6 fluorescence intensity shift between the experimental and control values using WinMDI 2.9 (n = 4). (B) Mitochondrial and (C) cytosolic fractions were prepared from cells exposed to the indicated NaF concentrations for 24 h and then analyzed by western blotting. Cells were also incubated with 2 mM NaF at the indicated concentrations of BAPTA-AM (0–10 μM) or 2,500 U/ml CAT, and then (D) cell viability and (E) JNK activity were determined at 24 h and 12 h after the incubation, respectively. *p < 0.05 and **p < 0.01 vs. the untreated controls.
ROS act as upstream effectors of JNK- and p53-mediated signaling in NaF-exposed mESCs
NaF treatment significantly increased growth arrest and DNA damage-inducible protein 45α (GADD45α) levels in a dose- and time-dependent manner (Fig. 7A). The NaF-mediated GADD45α increase was inhibited by pre-treating cells with 2.5 μM SP600125, but not with 5 μM PFT-α (Fig. 7B). Combined treatment with PFT-α significantly attenuated the NaF-mediated MMP loss in mESCs and this was further verified by the addition of CAT (Fig. 7C). In contrast, a JNK inhibitor, SP600125, did not show a significant reduction in MMP loss. Further, flow cytometric analysis showed that the NaF-mediated increase in ROS levels was suppressed by treating the cells with CAT, but not with SP600125 or PFT-α (Fig. 7D).
Fig. 7. ROS act upstream of p53- and JNK-mediated signaling in NaF-exposed cells.

(A) Cells were exposed to the indicated concentrations (0–5 mM) of NaF for various times (0–24 h), and then GADD45α protein levels were determined by western blotting. A representative result from triplicate experiments is shown. Cells were also incubated with 5 mM NaF in the presence of the indicated concentrations of SP600125, PFT-α, or CAT for 24 h and then processed for (B) western blotting, (C) MMP determination, and (D) flow cytometric analysis. In panel B, the results represent the mean ± S.D. from triplicate experiments and are expressed as relative expression (fold) to the control value after normalizing the bands to that of β-actin. ***p < 0.001 vs. the untreated control. #p < 0.05 and ###p < 0.05 vs. the 5 mM NaF treatment alone.
Discussion
Numerous studies have been focused on the elucidation of the exact influences of fluoride on cells and tissues. It is generally accepted that NaF at concentrations greater than 1 mM causes growth arrest and cell death either by necrosis or apoptosis, although the deleterious effects of NaF differ according to the exposed concentrations and the types of cells examined (Chien et al., 2006; Lee et al., 2008; Qu et al., 2008; Wurtz et al., 2008; Yan et al., 2007). In the present study, we for the first time show that 1 mM NaF did not affect the proliferation and survival of mESCs, but at higher doses NaF reduced cell viability in a dose-dependent manner. NaF at high doses induced G2/M growth arrest with a concomitant reduction in cells in the S phase of the cell cycle progression. NaF also led to apoptotic cell death, as shown by the migration of many cell populations into the sub-G1 phase, the increase of annexin V/PI stained cells, and the formation of DNA fragments.
The mitochondria-mediated and death receptor-mediated pathways are considered to be involved in apoptosis induced by fluoride (Barbier et al., 2010). Mitochondria play central roles in both caspase-dependent and caspase-independent death pathways (Tsujimoto, 2003). An important mitochondrial event during apoptosis is the reduction of MMP, which is accompanied by the alteration of Bcl-2 family proteins (Kroemer and Reed, 2000). MMP loss causes the cytoplasmic release of pro-apoptotic molecules such as AIF and cytochrome c from the mitochondria (Kook et al., 2007; Newmeyer and Ferguson-Miller, 2003). Accumulated evidence has suggested that apoptotic cell death mediated by toxic heavy metals is related to mitochondrial stress followed by MMP reduction (Son et al., 2010; 2011). This sequence is believed to be involved in the metal-mediated increase in intracellular ROS. We observed mild reductions in the levels of MMP and mitochondrial Bcl-2 proteins. The cytoplasmic levels of cytochrome c were also increased after treatment with NaF at 2 mM, and this increase was in parallel with the pattern of caspase activities.
In addition, the present results revealed that CAT, but not SOD, NAC, and APO, diminished the NaF-mediated reduction in cell viability and inhibited the MMP loss caused by NaF. This suggests that ROS are a mediator of NaF-mediated cell death, where mitochondrial stress is at least in part related to cell death. This is similar to previous studies showing that NaF induces apoptosis by elevating oxidative stress-mediated lipid peroxidation, eventually leading to mitochondrial dysfunction with the activation of downstream pathways (Barbier et al., 2010; Karube et al., 2009; Wang et al., 2010). The current findings also indicate that hydroxyl radicals are the direct mediator of NaF-mediated cell death, as evidenced by the dose-dependent increase in ESR signal and DCF fluorescence and the CAT-mediated prevention of cell toxicity in NaF-treated mESCs. These data are also consistent with previous findings, in which hydroxyl radicals were shown to be the main toxic radicals in mycotoxin or heavy metal-exposed cells (Barbouti et al., 2002; Son et al., 2009).
Cytoplasmic release of cytochrome c and its complex formation with Apaf-1 and procaspase-9 activates executive caspase-3 (Li et al., 1997). In the current study, NaF induced a marked cleavage of PARP in mESCs. NaF-mediated reduction in cell viability was also suppressed by treatment with a pan-caspase inhibitor. These results support strongly the involvement of the caspase-mediated pathway in NaF-mediated apoptosis in mESCs. Moreover, our results suggest that the decrease in Akt levels is related to a NaF-mediated reduction of cell viability, although more detailed experiments to clarify the role of Akt in NaF-exposed mESCs will be required. Collectively, the mitochondrial- and caspase-mediated signaling accompanied by intracellular ROS accumulation appears to be involved in NaF-mediated apoptosis.
A few reports have suggested the involvement of the JNK pathway in fluoride-induced apoptosis. Fluoride exposure at 2 to 10 mM induced prolonged phosphorylation of JNK in MDPC-23 odontoblast-like cells (Karube et al., 2009). Chronic fluorosis increased p-JNK levels in rat brains, which is similar to the results of SH-SY5Y cells treated with excessive fluoride (Liu et al., 2011). These reports suggest that over-exposure to excessive fluoride could activate the JNK pathway. There is also considerable evidence that GADD45α has an important role in the induction of apoptosis (Hildesheim et al., 2002), in which its transcription and function are controlled either by JNK1 or JNK2 (Zhang et al., 2006). In a previous study, cadmium increased the production of GADD45α in JB6 Cl41 cells and this was suppressed by its pharmacological inhibitor or si-JNK transfection (Son et al., 2010). In parallel with this report, NaF treatment increased the induction of GADD45α in a dose- and time-dependent manner and a JNK specific inhibitor prevented this effect. In contrast, NaF-mediated MMP loss was inhibited by PFT-α or CAT, but not by SP600125. Further, NaF-mediated ROS accumulation was inhibited only by CAT rather than by JNK or p53 inhibitors. These results suggest that JNK-GADD45α- and p53-mediated signaling is critical for NaF-mediated apoptosis in mESCs, where ROS act as the most important upstream mediator (Fig. 8).
Fig. 8.
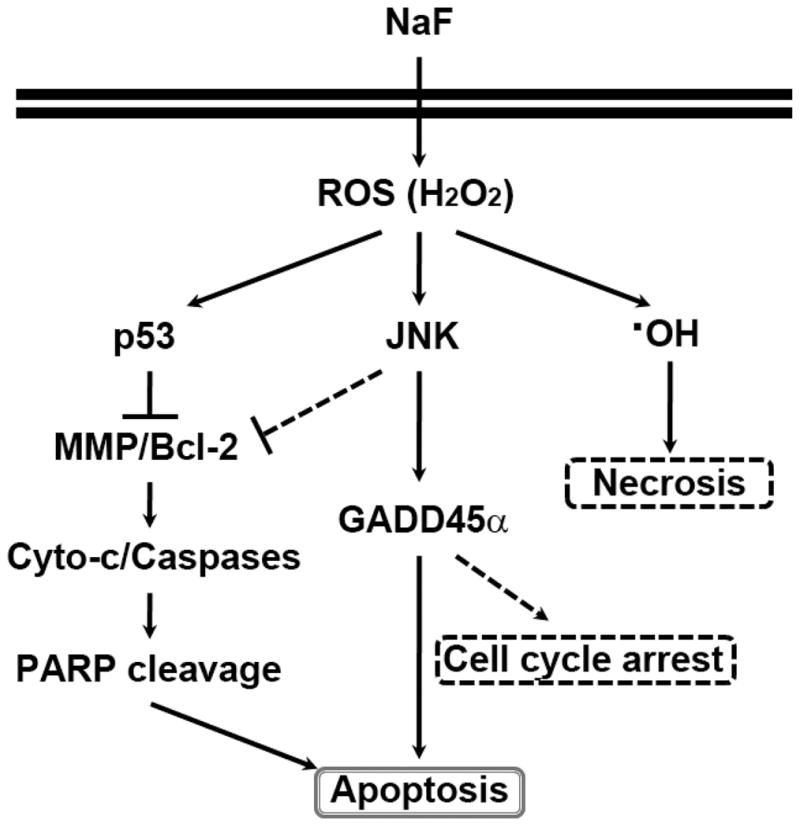
Proposed signaling pathways involved in NaF-induced apoptosis in mESCs. NaF induces intracellular ROS accumulation, especially H2O2, and this stimulates both p53- and JNK-mediated pathways eventually leading to cell death mainly by apoptosis through the induction of mitochondrial stress, caspase activation, and the increase of GADD45α . Necrotic cell death and cell cycle progression arrest in the G2/M phase also occurred at least in part in the NaF-exposed cells.
Intracellular calcium ions can play critical roles in fluoride-induced apoptosis (Dominguez et al., 1991; Kubota et al., 2005). Intracellular calcium homeostasis is also critical for maintaining cellular functions in response to extra- and/or endogenous stimuli. Similarly, cadmium increased intracellular calcium levels and then mediated apoptosis (Kim and Sharma, 2004; Shih et al., 2005; Wang et al., 2008). However, the present study revealed the opposite result, in that treatment with calcium channel blockers did not inhibit NaF-mediated reduction in cell viability; rather BAPTA-AM facilitated the NaF-mediated toxic effects. In addition, BAPTA-AM did not attenuate the activity of JNK in NaF-exposed mESCs. There are reports emphasizing the relationship between intracellular calcium and ROS during fluoride-induced cytotoxicity. In fact, treatment with BAPTA-AM reduced the fluoride-induced increase in calcium as well as ROS and lactate dehydrogenase leakage levels (Xu et al., 2011). Changes in calcium concentrations in fluoride-exposed cells were also observed (Bourgoin et al., 1996). Moreover, endoplasmic reticulum (ER) stress is an important mediator of NaF-mediated apoptosis (Kubota et al., 2005; Matsuo et al., 1996; Sharma et al., 2008). ER stress causes an overall reduction in protein synthesis so that cells can cope with the existing unfolded or misfolded proteins (Niederreiter and Kaser, 2011). This indicates the possibility of cytoplasmic release of calcium ions accompanied by ER stress in NaF-treated cells. Interestingly, Chien et al. (2006) reported that NaF-mediated cytotoxicity in PLFs was reduced by calcium treatment, whereas it was augmented by the removal of calcium from the culture medium. More detailed experiments to clarify the relationship between intracellular calcium ions, ER stress, and apoptotic cell death in NaF-exposed cells are required.
In summary, our findings demonstrate that NaF influences the viability and survival of mESCs according to the exposed concentrations. In high doses (from 2 mM), NaF induces cell death mainly by apoptosis through mitochondrial stress and caspase- and JNK-mediated pathways, where ROS play important roles as upstream effectors. It is also believed that hydroxyl radicals generated by H2O2 might cause acute damage to cellular macromolecules in NaF-exposed cells, especially DNA, thereby leading to necrotic cell death. It is considered that fluoride uptake by water fluoridation or by treating osteoporosis does not result in severe problems which can occur by an acute and high-concentration exposure, mainly by inhalation in occupational settings (Fordyce, 2011). However, the present findings suggest that fluoride above a threshold concentration exert toxic effects sensitively on stem cells and thus the younger should pay the more caution before its treatment.
Supplementary Material
Effects of NaF on the activation of MAPK and caspase in mESCs. (A) Cells were incubated in the presence of NaF (0–5 mM) with and without 2.5 μM PD98059, SB203580, or SP600125. At 24 h of the incubation, cell viability was determined by WST-8 assay. *p < 0.05 and **p < 0.01 vs. the experiments. (B) Cellular proteins were prepared from the cells exposed to 2 mM NaF for the indicated times (3–24 h) and then adjusted for Western blot analysis. A representative data from triplicate experiments was shown, where all experiments showed similar results.
Acknowledgments
This research was supported by Basic Science Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2011-0009123). Part of this research was supported by NIH grants (R01ES015518-04, R01CA116697-05, and R01CA119028-05S1). Dr. J.-C. Lee was supported by the Chonbuk National University for his sabbatical leave of absence at the University of Kentucky to perform part of the experiments for this work.
Abbreviations
- AIF
apoptosis-inducing factor
- CAT
catalase
- DCF-DA
2′,7′-dichlorodihydrofluorescein diacetate
- DiOC6
3,3′-dihexyloxacarbocyanine iodide
- ESR
electron spin resonance
- 4-HPR
N-(4-hydroxyphenyl) retinamide
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- mESCs
mouse embryonic stem cells
- MMP
mitochondrial membrane potential
- NaF
sodium fluoride
- NFD
nifedipine
- PARP
poly (ADP-ribose) polymerase
- pifithrin- α
PFT- α
- PI
propidium iodide
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- TTX
tetrodotoxin
- WST
water soluble tetrazolium
Footnotes
Conflict of interest
The authors declare that there is no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barbier O, Arreola-Mendoza L, Del Razo LM. Molecular mechanisms of fluoride toxicity. Chem Biol Interact. 2010;188:319–333. doi: 10.1016/j.cbi.2010.07.011. [DOI] [PubMed] [Google Scholar]
- Barbouti A, Doulias PT, Nousis L, Tenopoulou M, Galaris D. DNA damage and apoptosis in hydrogen peroxide-exposed Jurkat cells: bolus addition versus continuous generation. Free Radic Biol Med. 2002;33:691–702. doi: 10.1016/s0891-5849(02)00967-x. [DOI] [PubMed] [Google Scholar]
- Ba Y, Zhu JY, Yang YJ, Yu B, Huang H, Wang C, Ren LJ, Cheng XM, Cui LX, Zhang YW. Serum calciotropic hormone levels, and dental fluorisis in children exposed to different concentrations of fluoride and iodine in dringking water. Chin Med J. 2010;123:675–679. [PubMed] [Google Scholar]
- Bourgoin SG, Harbour D, Poubelle PE. Role of protein kinase C alpha, Arf, and cytoplasmic calcium transients in phospholipase D activation by sodium fluoride in osteoblast-like cells. J Bone Miner Res. 1996;11:1655–1665. doi: 10.1002/jbmr.5650111109. [DOI] [PubMed] [Google Scholar]
- Buzalaf MA, Levy SM. Fluoride intake of children: considerations for dental caries and dental fluorosis. Monogr Oral Sci. 2011;22:1–19. doi: 10.1159/000325101. [DOI] [PubMed] [Google Scholar]
- Caverzasio J, Palmer G, Bonjour JP. Fluoride: mode of action. Bone. 1998;22:585–589. doi: 10.1016/s8756-3282(98)00058-1. [DOI] [PubMed] [Google Scholar]
- Chien CH, Otsuki S, Chowdhury SA, Kobayashi M, Takahashi K, Kanda Y, Kunii S, Sakagami H, Kanegae H. Enhancement of cytotoxic activity of sodium fluoride against human periodontal ligament fibroblasts by water pressure. In Vivo. 2006;20:849–856. [PubMed] [Google Scholar]
- Dominguez JH, Garcia JG, Rothrock JK, English D, Mann C. Fluoride mobilizes intracellular calcium and promotes Ca2+ influx in rat proximal tubules. Am J Physiol. 1991;261:F318–327. doi: 10.1152/ajprenal.1991.261.2.F318. [DOI] [PubMed] [Google Scholar]
- Everett ET. Fluoride’s effects on the formation of teeth and bones, and the influence of genetics. J Dent Res. 2011;90:552–560. doi: 10.1177/0022034510384626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordyce FM. Fluoride: Human health risks. Encyclopedia of Environmental Health. 2011;5:776–785. [Google Scholar]
- Gazzano E, Bergandi L, Riganti C, Aldieri E, Doublier S, Costamagna C, Bosia A, Ghigo D. Fluoride effects: the two faces of Janus. Curr Med Chem. 2010;17:2431–2441. doi: 10.2174/092986710791698503. [DOI] [PubMed] [Google Scholar]
- Hildesheim J, Bulavin DV, Anver MR, Alvord WG, Hollander MC, Vardanian L, Fornace AJ., Jr Gadd45α protects against UV irradiation-induced skin tumors, and promotes apoptosis and stress signaling via MAPK and p53. Cancer Res. 2002;62:7305–7315. [PubMed] [Google Scholar]
- Karube H, Nishitai G, Inageda K, Kurosu H, Matsuoka M. NaF activates MAPKs and induces apoptosis in odontoblast-like cells. J Dent Res. 2009;88:461–465. doi: 10.1177/0022034509334771. [DOI] [PubMed] [Google Scholar]
- Kim J, Sharma RP. Calcium-mediated activation of c-Jun NH2-terminal kinase (JNK) and apoptosis in response to cadmium in murine macrophages. Toxicol Sci. 2004;81:518–527. doi: 10.1093/toxsci/kfh221. [DOI] [PubMed] [Google Scholar]
- Klaunig JE, Wang Z, Pu X, Zhou S. Oxidative stress and oxidative damage in chemical carcinogenesis. Toxicol Appl Pharmacol. 2011;254:86–99. doi: 10.1016/j.taap.2009.11.028. [DOI] [PubMed] [Google Scholar]
- Kook SH, Son YO, Chung SW, Lee SA, Kim JG, Jeon YM, Lee JC. Caspase-independent death of human osteosarcoma cells by flavonoids is driven by p53-mediated mitochondrial stress and nuclear translocation of AIF and endonuclease G. Apoptosis. 2007;12:1289–1298. doi: 10.1007/s10495-007-0056-x. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Kubota K, Lee DH, Tsuchiya M, Young CS, Everett ET, Martinez-Mier EA, Snead ML, Nguyen L, Urano F, Bartlett JD. Fluoride induces endoplasmic reticulum stress in ameloblasts responsible for dental enamel formation. J Biol Chem. 2005;280:23194–231202. doi: 10.1074/jbc.M503288200. [DOI] [PubMed] [Google Scholar]
- Lee JH, Jung JY, Jeong YJ, Park JH, Yang KH, Choi NK, Kim SH, Kim WJ. Involvement of both mitochondrial- and death receptor-dependent apoptotic pathways regulated by Bcl-2 family in sodium fluoride-induced apoptosis of the human gingival fibroblasts. Toxicology. 2008;243:340–347. doi: 10.1016/j.tox.2007.10.026. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Liu YJ, Guan ZZ, Gao Q, Pei JJ. Increased level of apoptosis in rat brains and SH-SY5Y cells exposed to excessive fluoride-a mechanism connected with activating JNK phosphorylation. Toxicol Lett. 2011;204:183–189. doi: 10.1016/j.toxlet.2011.04.030. [DOI] [PubMed] [Google Scholar]
- Matsuo S, Inai T, Kurisu K, Kiyomiya K, Kurebe M. Influence of fluoride on secretory pathway of the secretory ameloblast in rat incisor tooth germs exposed to sodium fluoride. Arch Toxicol. 1996;70:420–429. doi: 10.1007/s002040050294. [DOI] [PubMed] [Google Scholar]
- Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–490. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- Niederreiter L, Kaser A. Endoplasmic reticulum stress and inflammatory bowel disease. Acta Gastroenterol Belg. 2011;74:330–333. [PubMed] [Google Scholar]
- Qu WJ, Zhong DB, Wu PF, Wang JF, Han B. Sodium fluoride modulates caprine osteoblast proliferation and differentiation. J Bone Miner Metab. 2008;26:328–334. doi: 10.1007/s00774-007-0832-2. [DOI] [PubMed] [Google Scholar]
- Ren G, Ferreri M, Wang Z, Su Y, Han B, Su J. Sodium fluoride affects proliferation and apoptosis through insulin-like growth factor I receptor in primary cultured mouse osteoblasts. Biol Trace Elem Res. 2011;144:914–923. doi: 10.1007/s12011-011-9059-0. [DOI] [PubMed] [Google Scholar]
- Shih YL, Lin CJ, Hsu SW, Wang SH, Chen WL, Lee MT, Wei YH, Shih CM. Cadmium toxicity toward caspaseindependent apoptosis through the mitochondria-calcium pathway in mtDNA-depleted cells. Ann N Y Acad Sci. 2005;1042:497–505. doi: 10.1196/annals.1338.043. [DOI] [PubMed] [Google Scholar]
- Sharma R, Tsuchiya M, Bartlett JD. Fluoride induces endoplasmic reticulum stress and inhibits protein synthesis and secretion. Environ Health Perspect. 2008;116:1142–1146. doi: 10.1289/ehp.11375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son YO, Jang YS, Heo JS, Chung WT, Choi KC, Lee JC. Apoptosis-inducing factor plays a critical role in caspase-independent, pyknotic cell death in hydrogen peroxide-exposed cells. Apoptosis. 2009;14:796–808. doi: 10.1007/s10495-009-0353-7. [DOI] [PubMed] [Google Scholar]
- Son YO, Lee JC, Hitron JA, Pan J, Zhang Z, Shi X. Cadmium induces intracellular Ca2+- and H2O2-dependent apoptosis through JNK- and p53-mediated pathways in skin epidermal cell line. Toxicol Sci. 2010;113:127–137. doi: 10.1093/toxsci/kfp259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son YO, Wang X, Hitron JA, Zhang Z, Cheng S, Budhraja A, Ding S, Lee JC, Shi X. Cadmium induces autophagy through ROS-dependent activation of the LKB1-AMPK signaling in skin epidermal cells. Toxicol Appl Pharmacol. 2011;255:287–296. doi: 10.1016/j.taap.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrane EV, Refsnes M, Thoresen GH, Låg M, Schwarze PE. Fluoride-induced apoptosis in epithelial lung cells involves activation of MAP kinases p38 and possibly JNK. Toxicol Sci. 2001;61:83–91. doi: 10.1093/toxsci/61.1.83. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y. Cell death regulation by the Bcl-2 protein family in the mitochondria. J Cell Physiol. 2003;195:158–167. doi: 10.1002/jcp.10254. [DOI] [PubMed] [Google Scholar]
- Wang SH, Shih YL, Ko WC, Wei YH, Shih CM. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci. 2008;65:3640–3652. doi: 10.1007/s00018-008-8383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZH, Li XL, Yang ZQ, Xu M. Fluorine-induced apoptosis and lipid peroxidation in human hair follicles in vitro. Biol Trace Elem Res. 2010;137:280–288. doi: 10.1007/s12011-009-8592-6. [DOI] [PubMed] [Google Scholar]
- Wang Z, Yang X, Yang S, Ren G, Ferreri M, Su Y, Chen L, Han B. Sodium fluoride suppress proliferation and induce apoptosis through decreased insulin-like growth factor-I expression and oxidative stress in primary cultured mouse osteoblasts. Arch Toxicol. 2011;85:1407–1417. doi: 10.1007/s00204-011-0697-y. [DOI] [PubMed] [Google Scholar]
- Wurtz T, Houari S, Mauro N, MacDougall M, Peters H, Berdal A. Fluoride at non-toxic dose affects odontoblast gene expression in vitro. Toxicology. 2008;249:26–34. doi: 10.1016/j.tox.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Xu Z, Xu B, Xia T, He W, Gao P, Guo L, Wang Z, Niu Q, Wang A. Relationship between intracellular Ca2+ and ROS during fluoride-induced injury in SH-SY5Y cells. Environ Toxicol. 2011 doi: 10.1002/tox.20721. In press. [DOI] [PubMed] [Google Scholar]
- Yang S, Wang Z, Farquharson C, Alkasir R, Zahra M, Ren G, Han B. Sodium fluoride induces apoptosis and alters bcl-2 family protein expression in MC3T3-E1 osteoblastic cells. Biochem Biophys Res Commun. 2011;410:910–915. doi: 10.1016/j.bbrc.2011.06.094. [DOI] [PubMed] [Google Scholar]
- Yan X, Feng C, Chen Q, Li W, Wang H, Lv L, Smith GW, Wang J. Effects of sodium fluoride treatment in vitro on cell proliferation, apoptosis and caspase-3 and caspase-9 mRNA expression by neonatal rat osteoblasts. Arch Toxicol. 2009;83:451–458. doi: 10.1007/s00204-008-0365-z. [DOI] [PubMed] [Google Scholar]
- Zhang D, Song L, Li J, Wu K, Huang C. Coordination of JNK1 and JNK2 is critical for GADD45alpha induction and its mediated cell apoptosis in arsenite responses. J Biol Chem. 2006;281:34113–34123. doi: 10.1074/jbc.M602821200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects of NaF on the activation of MAPK and caspase in mESCs. (A) Cells were incubated in the presence of NaF (0–5 mM) with and without 2.5 μM PD98059, SB203580, or SP600125. At 24 h of the incubation, cell viability was determined by WST-8 assay. *p < 0.05 and **p < 0.01 vs. the experiments. (B) Cellular proteins were prepared from the cells exposed to 2 mM NaF for the indicated times (3–24 h) and then adjusted for Western blot analysis. A representative data from triplicate experiments was shown, where all experiments showed similar results.