

Abstract
Signs or symptoms of impaired autonomic regulation of the circulation often attend Parkinson disease (PD). This review covers biomarkers and mechanisms of autonomic cardiovascular abnormalities in PD and related alpha-synucleinopathies. The clearest clinical laboratory correlate of dysautonomia in PD is loss of myocardial noradrenergic innervation, detected by cardiac sympathetic neuroimaging. About 30–40% of PD patients have orthostatic hypotension (OH), defined as a persistent, consistent fall in systolic blood pressure of at least 20 mm Hg or diastolic blood pressure of at least 10 mm Hg within three minutes of change in position from supine to standing. Neuroimaging evidence of cardiac sympathetic denervation is universal in PD with OH (PD+OH). In PD without OH about half the patients have diffuse left ventricular myocardial sympathetic denervation, a substantial minority have partial denervation confined to the inferolateral or apical walls, and a small number have normal innervation. Among patients with partial denervation the neuronal loss invariably progresses over time, and in those with normal innervation at least some loss eventually becomes evident. Thus, cardiac sympathetic denervation in PD occurs independently of the movement disorder. PD+OH also entails extra-cardiac noradrenergic denervation, but this is not as severe as in pure autonomic failure. PD+OH patients have failure of both the parasympathetic and sympathetic components of the arterial baroreflex. OH in PD therefore seems to reflect a “triple whammy” of cardiac and extra-cardiac noradrenergic denervation and baroreflex failure. In contrast, most patients with multiple system atrophy, which can resemble PD+OH clinically, do not have evidence for cardiac or extra-cardiac noradrenergic denervation. Catecholamines in the neuronal cytoplasm are potentially toxic, via spontaneous and enzyme-catalyzed oxidation. Normally cytoplasmic catecholamines are efficiently taken up into vesicles via the vesicular monoamine transporter. The recent finding of decreased vesicular uptake in Lewy body diseases therefore suggests a pathogenetic mechanism for loss of catecholaminergic neurons in the periphery and brain.
Parkinson disease (PD) is one of the most common chronic neurodegenerative diseases of the elderly, and it is likely that as populations age PD will become even more prevalent and more of a public health burden.
Severe depletion of dopaminergic neurons of the nigrostriatal system characterizes and likely produces the movement disorder (rest tremor, slowness of movement, rigid muscle tone, and postural instability) in PD. Over the past two decades, compelling evidence has accrued that PD also involves loss of noradrenergic neurons in the heart. This finding supports the view that loss of catecholaminergic neurons, both in the nigrostriatal system and the heart, is fundamental in PD.
By the time PD manifests clinically, most of the nigrostriatal dopaminergic neurons are already lost. Identifying laboratory measures—biomarkers of the—disease process is therefore crucial for advances in treatment and prevention.
Deposition of the protein, alpha-synuclein, in the form of Lewy bodies in catecholaminergic neurons is a pathologic hallmark of PD. Alpha-synucleinopathy in autonomic neurons may occur early in the pathogenetic process. The timing of cardiac noradrenergic denervation in PD is therefore a key issue.
This review updates the field of autonomic cardiovascular abnormalities in PD and related disorders, with emphasis on relationships among striatal dopamine depletion, sympathetic noradrenergic denervation, and alpha-synucleinopathy.
Overview of Cardiovascular Autonomic Abnormalities in PD
Alterations in autonomic functions, known as dysautonomias, adversely affect health. Before considering cardiovascular manifestations of dysautonomia in PD it is important to recognize that the autonomic nervous system has multiple components (Figure 1), which seem to be involved differentially in PD.
Figure 1.
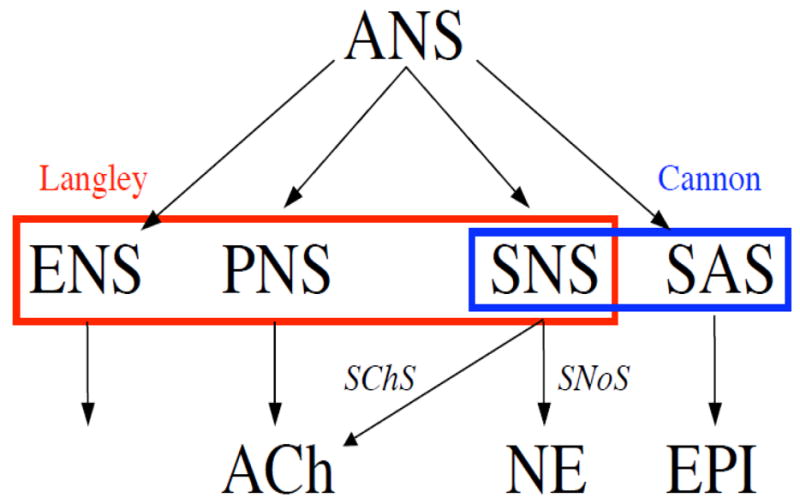
Components of the autonomic nervous system (ANS). Langley taught the ANS has three components: the sympathetic nervous system (SNS), parasympathetic nervous system (PNS), and enteric nervous system (ENS). Cannon added a hormonal component, the sympathetic adrenomedullary system (SAS), in which epinephrine (EPI) is the chemical messenger. Acetylcholine (ACh) is the neurotransmitter of the sympathetic cholinergic system (SChS) and norepinephrine (NE) the neurotransmitter of the sympathetic noradrenergic system (SNoS). Cannon taught that the sympathoadrenal system is a functional unit.
Langley coined the term autonomic nervous system (ANS) and recognized three components, the sympathetic nervous system (SNS), parasympathetic nervous system (another phrased he introduced, PNS), and enteric nervous system (ENS). The sympathetic nervous system is composed of two subsystems based on their main chemical messengers norepinephrine (NE), epinephrine (also known as adrenaline) and acetylcholine. The sympathetic noradrenergic system (SNaS) is the SNS component responsible for reflexive constriction of blood vessels and stimulation of the heart. The sympathetic cholinergic (SChS) system mediates sweating. The parasympathetic nervous system is responsible for a constellation of phenomena including respiratory sinus arrhythmia, gastrointestinal and urinary bladder tone, salivation, lacrimation, and pupillary constriction in response to light. Parasympathetic cholinergic failure therefore results in dry mouth and eyes, constipation, urinary retention, and photophobia. The sympathetic adrenomedullary system (SAS) uses the hormone epinephrine as the chemical effector. Epinephrine is one of the three main hormones regulating serum glucose, the others being insulin and glucagon. It is difficult if not impossible clinically to distinguish parasympathetic cholinergic denervation, enteric denervation, and disruption of reflexive modulation of autonomic outflows as determinants of symptoms such as constipation, abdominal bloating, and esophageal reflux.
At least three pathophysiologic mechanisms underie the cardiovascular autonomic abnormalities attending PD. The first is loss of cardiac sympathetic noradrenergic nerves. As discussed below, cardiac sympathetic denervation occurs virtually universally in PD, in a manner that seems surprisingly independent of the movement disorder in individual patients. The second is extra-cardiac noradrenergic denervation. For unknown reasons, loss of extra-cardiac noradrenergic innervation in PD is less extensive than is loss of cardiac innervation. The third determinant is arterial baroreflex failure. Severely decreased function of both the parasympathetic and sympathetic components of the arterial baroreflex is characteristic in PD+OH.
These three determinants together result in orthostatic hypotension (OH). Conversely, OH is a key manifestation of cardiovascular dysautonomia in PD.
Cardiac Sympathetic Denervation in PD: Scientific and Clinical Implications
More than a movement disorder, more than a brain disease
The discovery about 15 years ago of neuroimaging evidence for cardiac sympathetic denervation in PD (Goldstein et al., 1997) was a watershed for scientific and clinical understanding of the disease (Figure 2). Until then, although it had been well known that PD patients often have symptoms or signs of autonomic failure, pathophysiologic bases for these complaints had been mysterious. The findings from cardiac sympathetic neuroimaging indicated that at least one mechanism of dysautonomia in PD is loss of post-ganglionic noradrenergic nerves.
Figure 2.
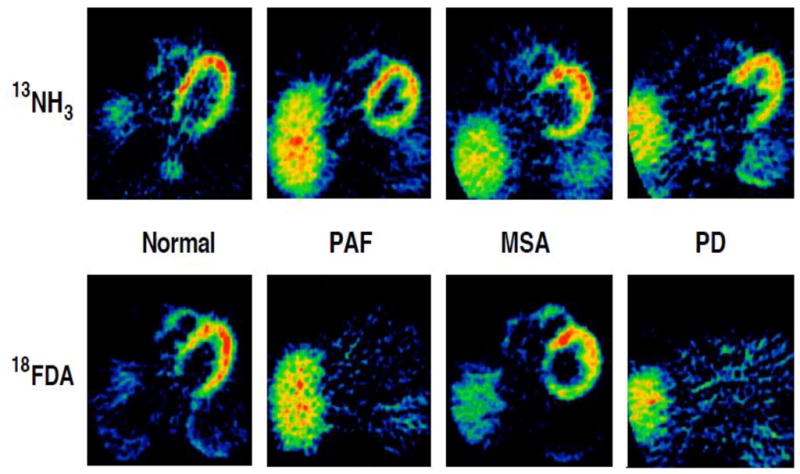
Typical 13NH3 and 18F-dopamine (18FDA) positron emission tomographic scans in a control subject, a patient with pure autonomic failure (PAF), a patient with multiple system atrophy (MSA), and a patient with Parkinson disease (PD). Note absence of discernible myocardial 18FDA-derived radioactivity in the PAF and PD patients and normal radioactivity in the MSA patient.
More than 50 studies since then have agreed with perfect unanimity that PD involves decreased myocardial innervation as assessed by a variety of sympathetic neuroimaging agents, including 123I-metaiodobenzylguanidine, 18F-dopamine, 11C-epinephrine, and 18F-metaraminol. Subsequent neuropathologic studies based on tyrosine hydroxylase immunostaining in epicardial nerves have consistently confirmed profound cardiac sympathetic denervation in PD (Amino et al., 2005; Orimo et al., 2002).
Sympathetic noradrenergic innervation of the heart derives from ganglia. The fact that dopamine is the immediate chemical precursor of NE reinforces the view that loss of catecholaminergic neurons is fundamental in PD.
The discovery and confirmation of cardiac noradrenergic denervation in PD has led to two key questions. First, when in the course of the disease does cardiac sympathetic denervation occur? If it occurred early on, then loss of cardiac noradrenergic innervation could represent a long-sought biomarker of pre-motor PD. Tracking cardiac innervation quantitatively might then enable testing the efficacy of putative neuroprotective or neurorescue agents. Second, what are the mechanisms for the loss of cardiac noradrenergic neurons in PD? The answer to this question could help elucidate the pathogenesis of PD.
Differential involvement of components of the autonomic nervous system in PD
PD involves relatively selective dysfunction the sympathetic noradrenergic component of the autonomic nervous system. Although some PD patients complain of decreased sweating, which normally is mediated mainly by the sympathetic cholinergic system, other patients have increased sweating. Thus, considered as a single group, PD patients have approximately normal results of the quantitative sudomotor axon reflex test (QSART), a measure of sympathetic cholinergic innervation (Sharabi et al., 2003). Constipation is common in PD; however, the extent to which this reflects loss of parasympathetic cholinergic or enteric neurons, as opposed to decreased afferent information to the brain, pathology in brainstem centers mediating neuronal outflows, disruption of descending pathways in the spinal cord, or loss of pre-ganglionic parasympathetic neurons is unknown.
Among PD patients there is greater loss of noradrenergic innervation in the heart than in the body as a whole. Plasma levels of both NE and of dihydroxyphenylglycol (DHPG), the main neuronal metabolite of norepinephrine, are normal in PD considered as a single group. Some clues about bases for cardioselectivity of sympathetic noradrenergic denervation in PD may be that the myocardium contains a high tissue concentration of NE, implying relatively dense innervation; cardiac sympathetic nerves avidly remove circulating catecholamines from the coronary arterial blood; and there is greater production of DHPG, a product of oxidation of NE, than other organs, suggesting a high rate of production of potentially toxic quinones, aldehydes, and other oxidation products.
Relationship to other non-motor manifestations
It is becoming increasingly recognized that PD involves a variety of non-motor manifestations, which sometimes dominate the clinical picture and can precede the movement disorder, even by decades. These include constipation, depression, cognitive dysfunction, anosmia, REM behavior disorder, and orthostatic hypotension (OH). The latter three non-motor manifestations are associated with cardiac sympathetic denervation (Goldstein et al., 2002b; Goldstein et al., 2009; Kashihara et al., 2010).
Almost all patients with PD have at least some olfactory dysfunction, and a substantial minority have absent sense of smell—anosmia. Olfactory function can be quantified by the University of Pennsylvania Smell Identification Test (UPSIT). Among patients with alpha-synucleinopathies such as PD, UPSIT scores are positively correlated with myocardial noradrenergic innervation as assessed by cardiac sympathetic neuroimaging (Goldstein et al., 2008). Perhaps surprisingly, UPSIT scores are unrelated to either the severity of the movement disorder, assessed by the Uniform Parkinson’s Disease Rating Scale, or with the putamen:occipital cortex ratio of 18F-DOPA-derived radioactivity, a measure of striatal dopaminergic innervation.
Several sleep disorders are associated with PD; however, REM behavior disorder stands out. In REM behavior disorder (RBD) the patients act out their dreams (“dream enactment behavior”) and thrash about in bed. Polysomnography shows absence of the atonia that normally accompanies REM sleep. Patients with RBD can injure themselves or their bed-mates, while they are asleep. RBD patients have substantial myocardial noradrenergic denervation as indicated by cardiac sympathetic neuroimaging. Indeed, heart:mediastum ratios of 123I-MIBG can be even more severely decreased in RBD than in PD (Kashihara et al., 2010).
Most PD patients eventually develop dementia. In some patients dementia precedes the movement disorder or dominates the clinical picture, or the presence of visual hallucinations and fluctuating cognition leads to a diagnosis of Lewy Body Dementia (LBD) without sufficient Parkinsonism to diagnose PD. LBD features neuroimaging evidence of substantial cardiac sympathetic denervation. In this regard LBD differs from Alzheimer’s disease, in which cardiac sympathetic innervation is thought to be generally intact. Because of this distinction, cardiac sympathetic neuroimaging has been proposed as a means to diagnose LBD differentially from Alzheimer’s disease (Yoshita et al., 2006).
PD+OH: A Distinctive Pathophysiologic Entity?
About 30–40% of PD patients have OH (Goldstein, 2003; Velseboer et al., 2011). The prevalence of OH varies substantially across studies, probably reflecting differences in referral patterns in different centers. If one relies on symptoms to diagnose PD+OH, then OH is quite infrequent, because patients with OH can have surprisingly low blood pressure and not notice symptoms.
The “Triple Whammy”
Three determinants—cardiac noradrenergic denervation, extra-cardiac noradrenergic denervation, and arterial baroreflex failure—seem to work together in a “triple whammy,” resulting in not only OH but a syndrome that also includes post-prandial hypotension, blood pressure lability, supine hypertension, and possibly fatigue and exercise intolerance.
All patients with PD+OH have markedly reduced sympathetic noradrenergic innervation of the left ventricular myocardium (Goldstein and Orimo, 2009). This abnormality is so characteristic that in patients with central neurodegeneration and OH, neuroimaging evidence for normal myocardial noradrenergic innervation excludes PD.
As will be discussed in more detail later, about one-half of patients with PD and no OH (PD No OH) have neuroimaging evidence of cardiac noradrenergic denervation (Li et al., 2002). Of the remaining half, a substantial minority have partial denervation, with sparing of the anterobasal septum, and a small minority have intact innervation.
Plasma levels both of NE and DHPG are lower in PD+OH than in PD No OH, indicating a relatively smaller overall complement of sympathetic nerves in PD+OH (Goldstein et al., 2003; Senard et al., 1993); however, patients with pure autonomic failure (PAF), a rare Lewy body disease in which the patients always have OH without Parkinsonism, have even lower NE and DHPG levels than do PD+OH patients (Goldstein et al., 2011a), indicating more extensive extra-cardiac noradrenergic denervation in PAF than in PD+OH.
Arterial baroreflex failure in PD+OH involves both the cardiovagal and sympathoneural limbs. Thus, during the Valsalva maneuver, the extent of decrease in the time between heartbeats (interbeat interval), expressed as a function of the decrease in systolic blood pressure (a measure of parasympathetic gain), is remarkably low, and during orthostasis the proportionate increment in plasma NE (a measure sympathetic gain) is blunted. These abnormalities are present regardless of levodopa treatment (Goldstein et al., 2005). Most PD No OH patients have more subtle baroreflex abnormalities.
Differential Diagnosis of PD vs. MSA
The cardiac neuroimaging findings in PD+OH contrast with those in multiple system atrophy (MSA), which can be difficult to distinguish from PD+OH, because MSA usually entails intact sympathetic innervation of the heart.
A minority of MSA patients have neuroimaging evidence of cardiac sympathetic denervation (Orimo et al., 2007). Therefore, in a patient with central neurodegeneration and OH, the finding of myocardial noradrenergic denervation by neuroimaging does not exclude MSA. Post-mortem studies show that these unusual MSA patients have Lewy bodies or Lewy neurites in sympathetic neurons, suggesting a “hybrid” disease involving alpha-synuclein deposition both in glial cells and in catecholaminergic neurons.
Which Autonomic Function Tests for Cardiovascular Dysautonomia are Sensitive and Which Specific in PD?
There are several clinical laboratory tests available for evaluating involvement of different components of the autonomic nervous system in PD. Some are approved and widely used, while others are considered research tools. This section discusses tests that seem most relevant to cardiovascular dysautonomia in PD.
In our view the most important single test in this evaluation is beat-to-beat hemodynamics associated with performance of the Valsalva maneuver (Figure 3). The combination of a progressive fall in blood pressure early in the maneuver (Phase II), absence of overshoot above baseline later in the maneuver (Phase IV), and prolonged pressure recovery time in Phase IV indicates failure of reflexive vasoconstriction mediated by the sympathetic noradrenergic system (Goldstein and Tack, 2000; Schrezenmaier et al., 2007).
Figure 3.

Continuous recordings of heart rate and blood pressure in a control subject and a patient with neurogenic orthostatic hypotension associated with performance of the Valsalva maneuver. Note that the patient has a progressive decrease in pressure in Phase II, no pressure overshoot in Phase IV, and blunted heart rate responses.
During Phase II, heart rate normally increases, and in response to the pressure overshoot in Phase IV, heart rate normally decreases rapidly to baseline. Attenuation of the increase in heart rate, taking into consideration the fall in blood pressure, in Phase II indicates decreased baroreflex-cardiovagal (parasympathetic) gain. As noted above, patients with PD+OH virtually always have baroreflex-sympathetic neurocirculatory and baroreflex-cardiovagal failure.
We routinely also monitor beat-to-beat hemodynamics after 15 minutes of supine rest and then up to 5 minutes of 90 degrees upright posture using a motorized tilt table. This provides a means to examine orthostatic blood pressure and heart rate and detect OH, a cardinal manifestation of sympathetic neurocirculatory failure. Measurement of forearm blood flow by impedance plethysmography or Doppler ultrasound, coupled with measurement of blood pressure, enables assessment of reflexive increases in forearm vascular resistance, which are blunted in baroreflex-sympathetic neurocirculatory failure. Skin electrical conductance, a measure of sympathetic cholinergic function, increases abruptly with tilting (unpublished observations) and can be assessed simultaneously.
In conjunction with brief upright tilting, sampling of arm venous blood for assays of plasma levels of catechols provides a neurochemical assessment that can be quite informative. Plasma NE levels normally approximately double by 5 minutes of orthostasis. Plasma DHPG provides a better overall measure of sympathetic noradrenergic innervation than does plasma NE (Goldstein et al., 1988). Plasma DOPA and dihydroxyphenylacetic acid (DOPAC) can determine whether the patient has taken levodopa/carbidopa recently; this can be important, because dopamine generated from therapeutic levodopa acts as a vasodilator and can artifactually decreased blood pressure during standing.
Power spectral analysis of heart rate variability (HRV) has been proposed as a means to assess cardiac sympathetic “tone” or “cardiovagal balance.” In the context of assessing possible autonomic failure in PD, this approach has limited value and validity. On the other hand, the log of low frequency power (LF) is strongly positively associated with the log of baroreflex-cardiovagal gain (Moak et al., 2009; Rahman et al., 2011). This suggests that measuring LF power can detect baroreflex failure in patients who cannot perform a technically adequate Valsalva maneuver or cannot tolerate being upright for 5 minutes.
A variety of pharmacologic tests have been used to examine cardiovascular autonomic function. Test drugs include sympathomimetics such as yohimbine, tyramine, desipramine, glucagon, and atomoxetine as well as isoproterenol (which blocks both sympathetic and parasympathetic signaling) and edrophonium (a parasympathomimetic) (Imrich et al., 2009a; Polinsky et al., 1981; Shannon et al., 2000). Concurrent measurement of plasma catechols improves the sensitivity of these tests. Because of baroreflex failure and denervation supersensitivity, blood pressure responses can be normal, despite clear abnormalities in plasma NE and DHPG responses (Sharabi et al., 2006).
Generally available tests of cardiovascular autonomic functions in PD can be sensitive but offer little or no specificity in distinguishing PD+OH from MSA. For diagnosing PD+OH from MSA (and Lewy body dementia from Alzheimer’s disease), cardiac sympathetic neuroimaging is most helpful.
In our experience, all patients with PD+OH have neuroimaging evidence of myocardial noradrenergic denervation. In contrast, most patients with MSA have intact innervation. Irrespective of diagnosis, cardiac sympathetic neuroimaging can provide important information relative to treatment. Analysis has identified a bi-phasic distribution of 18F-dopamine-derived radioactivity among patients with primary neurogenic OH, regardless of clinical diagnosis, with one group having cardiac sympathetic denervation and the other normal innervation (Goldstein and Sharabi, 2009).
Patients with neuroimaging evidence of cardiac noradrenergic denervation have evidence also of loss of noradrenergic innervation in the body as a whole, corresponding abnormalities in neurochemical responses to test drugs, and evidence of compensatory up-regulation of adrenoceptors (Senard et al., 1990). Patients with central neurodegeneration and neuroimaging evidence of intact cardiac noradrenergic innervation have neurochemical abnormalities suggesting central NE deficiency. Collectively, the neurochemical, neuroimaging, and neuropharmacologic results complement each other.
Considering the many positive intercorrelations, across a variety of assessment modalities, a classification scheme has been proposed for patients with neurogenic OH, based on evidence of peripheral noradrenergic denervation (Goldstein and Sharabi, 2009). An algorithm for clinical evaluation of orthostatic hypotension therefore emphasizes identifying OH as neurogenic and then peripheral noradrenergic deficiency (Figure 4).
Figure 4.

Algorithm for evaluation of orthostatic hypotension. The algorithm asks if orthostatic hypotension is persistent and consistent; if it is neurogenic; and if it is associated with post-ganglionic, sympathetic, noradrenergic denervation. Abbreviations: AAG=autoimmune autonomic ganglionopathy; BP=blood pressure; CNS=central nervous system; LBD=Lewy body dementia; MSA=multiple system atrophy; NE=norepinephrine; PAF=pure autonomic failure; PD+NOH=Parkinson disease with neurogenic orthostatic hypotension.
This classification schema has implications for the clinical management of patients with neurogenic OH, both in terms of diagnosis and predicted responses to treatment. In patients with neurogenic OH who have no clinical evidence of central neurodegeneration, the finding of normal peripheral noradrenergic innervation casts doubt on the diagnosis of pure autonomic failure. Testing for a circulating antibody to the neuronal nicotinic receptor (nAChR) is appropriate in this situation (Goldstein et al., 2002a; Imrich et al., 2009b; Vernino et al., 2000), because of the potential for improvement of autoimmune autonomic ganglionopathy by total plasma exchange or immunosuppressive therapy. In patients with neurogenic OH and central neurodegeneration, the finding of normal cardiac noradrenergic innervation excludes PD+OH and favors a diagnosis of MSA.
Once the patient has been placed into one of the two categories, therapy may be tailored according to the pathophysiologic classification. Patients with neurogenic OH and intact peripheral noradrenergic innervation might benefit from oral yohimbine (Shannon et al., 2000), whereas those with noradrenergic denervation might not (Senard et al., 1993). Yohimbine blocks alpha-2 adrenoceptors, which when occurring pre-synaptically leads to increased release of NE and dopamine. One must exercise caution because of the likelihood of oral yohimbine worsening supine hypertension. An indirectly acting sympathomimetic amine such as tyramine given with a monoamine oxidase inhibitor might also be tried (Nanda et al., 1976), but with the same precaution.
Patients with neurogenic OH have baroreflex failure and therefore have exaggerated responses to vasoactive drugs. For the same severity of baroreflex failure, patients with peripheral noradrenergic denervation have larger responses to drugs that increase occupation of adrenoceptors and smaller responses to drugs that work by releasing NE (Sharabi et al., 2006). In neurogenic OH with peripheral noradrenergic denervation, considering evidence for adrenoceptor up-regulation, a directly acting adrenoceptor agonist would seem appropriate. Midodrine is the only orally acting alpha-adrenoceptor agonist approved for marketing in the US. Norepinephrine precursor treatment with L-DOPS might play a role here in the future (Kaufmann et al., 2003), because conversion of L-DOPS to NE does not require intact noradrenergic nerves. Patients with noradrenergic denervation would be less likely to respond well to yohimbine or to an indirectly acting sympathomimetic amine, but they may still do so because of baroreflex failure.
Is Cardiovascular Dysautonomia a Biomarker of Pre-motor PD?
Progression and timing of partial cardiac denervation in PD No OH
Although patients with PD+OH invariably have loss of cardiac sympathetic nerves diffusely in the left ventricular myocardium, PD patients without OH (PD No OH) can have normal innervation or have denervation that is heterogeneous within the left ventricular myocardium. The apical or inferolateral wall is denervated, while the anterobasal septal innervation is still intact. The 18F-dopamine scans in Figure 5 exemplify this phenomenon (Li et al., 2002). Over the course of relatively few years, the loss of sympathetic nerves progresses to diffuse denervation. Since PD No OH patients already have the characteristic movement disorder, one would expect from the Braak schema (Braak et al., 2004; Braak et al., 2003) that they would already have cardiac sympathetic denervation.
Figure 5.

Serial 18F-dopamine scans in a patient with Parkinson disease who did not have orthostatic hypotension. Note initially normal septal radioactivity, with progressive loss of radioactivity.
Although evidence of cardiac denervation can be a pre-motor finding in PD, it may also occur after the movement disorder is already overt. We previously reported the case of a patient who had neuroimaging evidence of cardiac sympathetic denervation four years before motor onset of PD (Goldstein et al., 2007). We also noted cardiac and overall sympathetic denervation in patients with neurogenic orthostatic hypotension who subsequently developed parkinsonism (Goldstein, 2006). Another patient had neurogenic OH and neuroimaging and neurochemical evidence of cardiac and extra-cardiac noradrenergic denervation without parkinsonism upon initial evaluation (Goldstein et al., 2011 (in press)-a). Two years later he had a subtle decrease in putamen 6-[18F]DOPA-derived radioactivity and reported visual hallucinations, and at four years he had clearly decreased 6-[18F]DOPA-derived radioactivity and incipient dementia, still without parkinsonism (Figure 6). These cases illustrate cardiac sympathetic noradrenergic denervation years before development of parkinsonism or neuroimaging evidence of a striatal dopaminergic lesion. On the other hand, the data in Figure 7 are from a patient with clinically classic PD who had neuroimaging evidence of normal cardiac sympathetic innervation for about a decade, despite low striatal 18F-DOPA-derived radioactivity. Decreased 18F-dopamine-derived radioactivity was first noted in the inferolateral wall at 8 years, followed two years later by generalized denervation (Goldstein et al., 2011 (in press)-b). The findings suggest that in this case the disease process began with the more distal nerves and progressed proximally.
Figure 6.

Serial 18F-DOPA, 18F-dopamine, and 13NH3 scans in a patient with pure autonomic failure. Note initially normal striatal 18F-DOPA-derived radioactivity and low myocardial 18F-dopamine-derived radioactivity, with progressive loss of striatal radioactivity.
Figure 7.
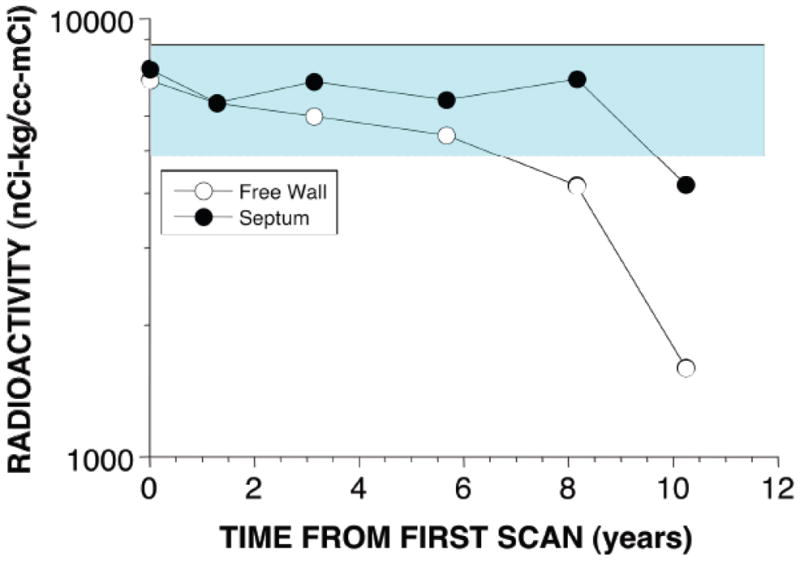
Cardiac septal and free wall 18F-dopamine-derived radioactivity as a function of years of follow-up in a patient with Parkinson disease who did not have orthostatic hypotension. Note that the patient had normal radioactivity for several years.
Independence of cardiovascular dysautonomia from striatal dopamine depletion
Across individual patients neither the severity of cardiac sympathetic denervation nor values for measures of other non-motor manifestations seem related to the severity of loss of nigrostriatal dopaminergic neurons (Goldstein et al., 2008; Goldstein et al., 2011 (in press)-b). In other words, autonomic dysfunction, at least as reflected by sympathetic noradrenergic denervation, seems to occur independently of the dopaminergic lesion that produces the movement disorder in PD. Because of this independence, although cardiac sympathetic denervation invariably progresses over time in PD and can come on years before the motor disorder, the denervation can also be a late finding.
As for cardiac sympathetic denervation, other non-motor manifestations, such as olfactory dysfunction, are more closely associated with cardiac noradrenergic than with striatal dopaminergic denervation in PD (Goldstein et al., 2008).
PD and pure autonomic failure feature not only decreased neuronal catecholamine uptake but also accelerated intra-neuronal catecholamine loss, which seems to reflect decreased vesicular uptake via the vesicular monoamine transporter (Figure 8)(Goldstein et al., 2011a). Multiple system atrophy patients have normal values for these parameters. The potential relevance of these findings to the pathogenesis of Lewy body diseases is discussed in the next section.
Figure 8.
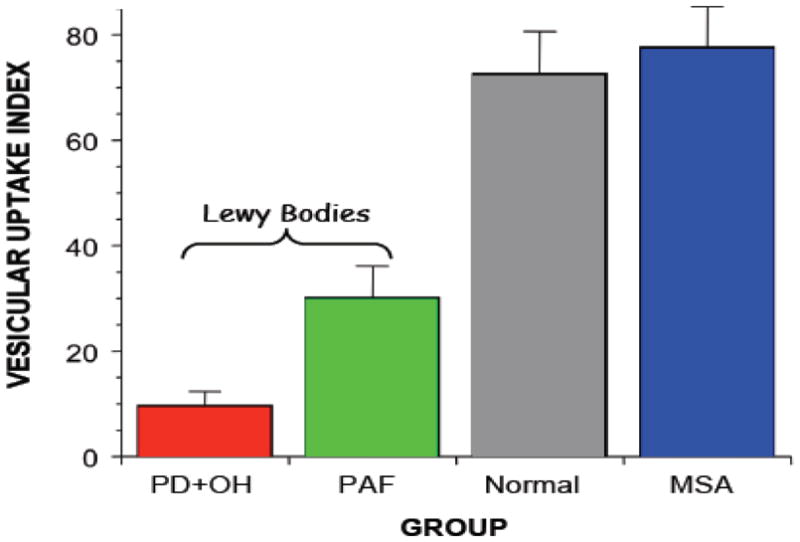
Mean (±SEM) values for a vesicular uptake index in patient groups with Parkinson disease and orthostatic hypotension (PD+OH), pure autonomic failure (PAF), or multiple system atrophy (MSA) and normal control subjects. Note decreased vesicular uptake indices in the Lewy body diseases.
Pathogenetic Insights
Impaired vesicular uptake of intraneuronal catecholamines in Lewy body diseases
Mechanisms of striatal and cardiac catecholaminergic denervation in Lewy body diseases such as PD have been poorly understood. A recent study examined the uptake, loss, and intra-neuronal vesicular sequestration of catecholamines, by assessing myocardial 6-[18F]dopamine- (18F-DA)-derived radioactivity and arterial plasma 18F-dihydroxyphenylacetic acid (18F-DOPAC) in PD with or without OH, pure autonomic failure (a Lewy body disease without parkinsonism), multiple system atrophy (a non-Lewy body synucleinopathy), and normal controls (Goldstein et al., 2011a). Vesicular uptake was decreased in PD+OH and pure autonomic failure compared to the multiple system atrophy and control groups and correlated positively with the rate of loss of radioactivity. Taken together, the results indicate that in Lewy body diseases sympathetic denervation is associated with accelerated neuronal catecholamine loss and decreased vesicular uptake.
Significance of decreased vesicular uptake
If catecholaminergic denervation in Lewy body diseases were related to decreased VMAT activity, this could be relevant to disease pathogenesis (Caudle et al., 2007). In the setting of ongoing catecholamine biosynthesis and vesicular leakage, decreased vesicular sequestration builds up cytosolic catecholamines, and cytosolic catecholamines are toxic, by spontaneous auto-oxidation to quinones or enzymatic conversion to catecholaldehydes (Marchitti et al., 2007). Moreover, cytosolic DA interacts with calcium and alpha-synuclein to destroy substantia nigra neurons (Mosharov et al., 2009).
Dihydroxyphenylacetaldehyde, the catecholaldehyde produced from the action of monoamine oxidase on dopamine, potently oligomerizes and aggregates alpha-synuclein (Burke et al., 2008), and lipid peroxidation products interfere with detoxification of dihydroxyphenylacetaldehyde by inhibiting aldehyde dehydrogenase and aldehyde reductase (Jinsmaa et al., 2009). These abnormalities could initiate deadly positive feedback loops.
Although exogenous DOPAL is toxic to dopaminergic neurons (Panneton et al., 2010), whether endogenously produced DOPAL plays a pathogenetic role remains unclear (Legros et al., 2004). Consistent with such a role, a recent post-mortem study noted a high ratio of dihydroxyphenylacetaldehyde:DA in the putamen of patients with end-stage PD (Goldstein et al., 2011b). Moreover, increased production of 18F-DOPAC from cytosolic 18F-DA implies increased production of the catecholaldehyde 18F-fluoroDOPAL, the obligate intermediate metabolite.
Reserpine, which inhibits vesicular sequestration by blocking the monoamine transporter, depletes catecholamine stores in the brain and heart, produces decreased locomotion and other findings reminiscent of PD, and evokes apoptosis of striatal neurons by a glutamate-dependent process (Mitchell et al., 1994); and mice with genetically determined very low activity of the type 2 VMAT have motor and non-motor manifestations similar to those in PD (Caudle et al., 2007; Taylor et al., 2009). Inhibition of vesicular sequestration augments the toxicity of a variety of agents such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 6-hydroxydopamine. Conversely, the pesticide and complex 1 inhibitor rotenone inhibits vesicular uptake, increases production of DOPAL, and evokes apoptosis via oxidation of cytosolic dopamine (Lamensdorf et al., 2000a; Lamensdorf et al., 2000b). Thus, by several routes involving multiple interactions among genetic predispositions, environmental exposures, synucleinopathy, and time, decreased VMAT activity may contribute to the catecholamine depletion that characterizes and produces major clinical manifestations of Lewy body diseases.
Summary
Cardiac and extra-cardiac noradrenergic denervation and baroreflex failure characterize and probably produce OH in PD. These abnormalities occur independently of striatal dopamine depletion. Cardiovascular autonomic variables may provide biomarkers of pre-motor PD in some patients. Understanding mechanisms of cardiac sympathetic dysfunction and denervation in PD may also help elucidate bases of central catecholaminergic lesions in a variety of neurodegenerative diseases. In particular, Lewy body forms of alpha-synucleinopathy seem to be associated with decreased vesicular sequestration of cytosolic catecholamines, and since cytosolic catecholamines are toxic, decreased vesicular recycling may be part of a pathogenetic pathway in the catecholaminergic denervation that characterizes these diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Samay Jain, Email: jains@upmc.edu.
David S. Goldstein, Email: goldsteind@ninds.nih.gov.
References
- Amino T, et al. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Path. 2005;15:29–34. doi: 10.1111/j.1750-3639.2005.tb00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, et al. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- Braak H, et al. Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003;110:517–36. doi: 10.1007/s00702-002-0808-2. [DOI] [PubMed] [Google Scholar]
- Burke WJ, et al. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 2008;115:193–203. doi: 10.1007/s00401-007-0303-9. [DOI] [PubMed] [Google Scholar]
- Caudle WM, et al. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci. 2007;27:8138–48. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS. Dysautonomia in Parkinson's disease: neurocardiological abnormalities. Lancet Neurol. 2003;2:669–676. doi: 10.1016/s1474-4422(03)00555-6. [DOI] [PubMed] [Google Scholar]
- Goldstein DS. Orthostatic hypotension as an early finding in Parkinson disease. Clin Auton Res. 2006;16:46–64. doi: 10.1007/s10286-006-0317-8. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, et al. Plasma dihydroxyphenylglycol and the intraneuronal disposition of norepinephrine in humans. J Clin Invest. 1988;81:213–220. doi: 10.1172/JCI113298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, et al. Neurocirculatory abnormalities in Parkinson disease with orthostatic hypotension: independence from levodopa treatment. Hypertension. 2005;46:1333–9. doi: 10.1161/01.HYP.0000188052.69549.e4. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, et al. Biomarkers to detect central dopamine deficiency and distinguish Parkinson disease from multiple system atrophy. Parkinsonism Relat Disord. 2008;14:600–7. doi: 10.1016/j.parkreldis.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, et al. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med. 1997;336:696–702. doi: 10.1056/NEJM199703063361004. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, et al. Pandysautonomia associated with impaired ganglionic neurotransmission and circulating antibody to the neuronal nicotinic receptor. Clin Auton Res. 2002a;12:281–5. doi: 10.1007/s10286-002-0020-3. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, et al. Intra-neuronal vesicular uptake of catecholamines is decreased in patients with Lewy body diseases. J Clin Inv. 2011a;121:3320–3330. doi: 10.1172/JCI45803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, et al. Sympathetic noradrenergic before striatal dopaminergic denervation: Relevance to Braak staging of synucleinopathy. Clin Auton Res. 2011 doi: 10.1007/s10286-011-0136-4. (in press)-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, et al. Plasma levels of catechols and metanephrines in neurogenic orthostatic hypotension. Neurology. 2003;60:1327–32. doi: 10.1212/01.wnl.0000058766.46428.f3. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, et al. Orthostatic hypotension from sympathetic denervation in Parkinson's disease. Neurology. 2002b;58:1247–55. doi: 10.1212/wnl.58.8.1247. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Orimo S. Cardiac sympathetic neuroimaging: summary of the First International Symposium. Clin Auton Res. 2009;19:133–136. doi: 10.1007/s10286-009-0002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, et al. Association of anosmia with autonomic failure in Parkinson disease. Neurology. 2009;74:245–251. doi: 10.1212/WNL.0b013e3181ca014c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, et al. Autonomic dysfunction in PD: A window to early detection? J Neurol Sci. 2011 doi: 10.1016/j.jns.2011.04.011. (in press)-b. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Sharabi Y. Neurogenic orthostatic hypotension: a pathophysiological approach. Circulation. 2009;119:139–46. doi: 10.1161/CIRCULATIONAHA.108.805887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, et al. Cardiac sympathetic denervation preceding motor signs in Parkinson disease. Clin Auton Res. 2007;17:118–21. doi: 10.1007/s10286-007-0396-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, et al. Catechols in post-mortem brain of patients with Parkinson disease. Eur J Neurol. 2011b;18:703–710. doi: 10.1111/j.1468-1331.2010.03246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Tack C. Non-invasive detection of sympathetic neurocirculatory failure. Clin Auton Res. 2000;10:285–291. doi: 10.1007/BF02281111. [DOI] [PubMed] [Google Scholar]
- Imrich R, et al. Functional effects of cardiac sympathetic denervation in neurogenic orthostatic hypotension. Parkinsonism Relat Disord. 2009a;15:122–7. doi: 10.1016/j.parkreldis.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imrich R, et al. Autoimmune autonomic ganglionopathy: treatment by plasma exchanges and rituximab. Clin Auton Res. 2009b;19:259–62. doi: 10.1007/s10286-009-0012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinsmaa Y, et al. Products of oxidative stress inhibit aldehyde oxidation and reduction pathways in dopamine catabolism yielding elevated levels of a reactive intermediate. Chem Res Toxicol. 2009;22:835–41. doi: 10.1021/tx800405v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashihara K, et al. Cardiac 123I-MIBG uptake is reduced more markedly in patients with REM sleep behavior disorder than in those with early stage Parkinson's disease. Parkinsonism Relat Disord. 2010;16:252–5. doi: 10.1016/j.parkreldis.2009.12.010. [DOI] [PubMed] [Google Scholar]
- Kaufmann H, et al. Norepinephrine precursor therapy in neurogenic orthostatic hypotension. Circulation. 2003;108:724–8. doi: 10.1161/01.CIR.0000083721.49847.D7. [DOI] [PubMed] [Google Scholar]
- Lamensdorf I, et al. Metabolic stress in PC12 cells induces the formation of the endogenous dopaminergic neurotoxin, 3,4-dihydroxyphenylacetaldehyde. J Neurosci Res. 2000a;60:552–8. doi: 10.1002/(SICI)1097-4547(20000515)60:4<552::AID-JNR14>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Lamensdorf I, et al. 3,4-Dihydroxyphenylacetaldehyde potentiates the toxic effects of metabolic stress in PC12 cells. Brain Res. 2000b;868:191–201. doi: 10.1016/s0006-8993(00)02309-x. [DOI] [PubMed] [Google Scholar]
- Legros H, et al. Semi-chronic increase in striatal level of 3,4-dihydroxyphenylacetaldehyde does not result in alteration of nigrostriatal dopaminergic neurones. J Neurosci Res. 2004;75:429–35. doi: 10.1002/jnr.10880. [DOI] [PubMed] [Google Scholar]
- Li ST, et al. Progressive loss of cardiac sympathetic innervation in Parkinson's disease. Ann Neurol. 2002;52:220–3. doi: 10.1002/ana.10236. [DOI] [PubMed] [Google Scholar]
- Marchitti SA, et al. Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol Rev. 2007;59:125–150. doi: 10.1124/pr.59.2.1. [DOI] [PubMed] [Google Scholar]
- Mitchell IJ, et al. Glutamate-induced apoptosis results in a loss of striatal neurons in the parkinsonian rat. Neuroscience. 1994;63:1–5. doi: 10.1016/0306-4522(94)90002-7. [DOI] [PubMed] [Google Scholar]
- Moak JP, et al. Supine low-frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Cleve Clin J Med. 2009;76(Suppl 2):S51–9. doi: 10.3949/ccjm.76.s2.11. [DOI] [PubMed] [Google Scholar]
- Mosharov EV, et al. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–29. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda RN, et al. Treatment of neurogenic orthostatic hypotension with a monoamine oxidase inhibitor and tyramine. Lancet. 1976;2:1164–7. doi: 10.1016/s0140-6736(76)91681-0. [DOI] [PubMed] [Google Scholar]
- Orimo S, et al. Degeneration of cardiac sympathetic nerve can occur in multiple system atrophy. Acta Neuropathol (Berl) 2007;113:81–86. doi: 10.1007/s00401-006-0160-y. [DOI] [PubMed] [Google Scholar]
- Orimo S, et al. Sympathetic cardiac denervation in Parkinson's disease and pure autonomic failure but not in multiple system atrophy. J Neurol Neurosurg Psychiatry. 2002;73:776–777. doi: 10.1136/jnnp.73.6.776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panneton WM, et al. The neurotoxicity of DOPAL: behavioral and stereological evidence for its role in Parkinson disease pathogenesis. PLoS One. 2010;5:e15251. doi: 10.1371/journal.pone.0015251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polinsky RJ, et al. Pharmacologic distinction of different orthostatic hypotension syndromes. Neurology. 1981;31:1–7. doi: 10.1212/wnl.31.1.1. [DOI] [PubMed] [Google Scholar]
- Rahman F, et al. Low frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Clin Auton Res. 2011;21:133–141. doi: 10.1007/s10286-010-0098-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrezenmaier C, et al. Adrenergic and vagal baroreflex sensitivity in autonomic failure. Arch Neurol. 2007;64:381–386. doi: 10.1001/archneur.64.3.381. [DOI] [PubMed] [Google Scholar]
- Senard JM, et al. Effects of yohimbine on plasma catecholamine levels in orthostatic hypotension related to Parkinson disease or multiple system atrophy. Clin Neuropharmacol. 1993;16:70–76. doi: 10.1097/00002826-199302000-00008. [DOI] [PubMed] [Google Scholar]
- Senard JM, et al. Adrenergic supersensitivity in parkinsonians with orthostatic hypotension. Eur J Clin Invest. 1990;20:613–619. doi: 10.1111/j.1365-2362.1990.tb01909.x. [DOI] [PubMed] [Google Scholar]
- Shannon JR, et al. Sympathetically mediated hypertension in autonomic failure. Circulation. 2000;101:2710–5. doi: 10.1161/01.cir.101.23.2710. [DOI] [PubMed] [Google Scholar]
- Sharabi Y, et al. Neuropharmacologic distinction of neurogenic orthostatic hypotension syndromes. Clin Neuropharmacol. 2006;29:97–105. doi: 10.1097/01.WNF.0000220822.80640.0D. [DOI] [PubMed] [Google Scholar]
- Sharabi Y, et al. Neurotransmitter specificity of sympathetic denervation in Parkinson's disease. Neurology. 2003;60:1036–9. doi: 10.1212/01.wnl.0000052690.91036.ff. [DOI] [PubMed] [Google Scholar]
- Taylor TN, et al. Nonmotor symptoms of Parkinson's disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci. 2009;29:8103–8113. doi: 10.1523/JNEUROSCI.1495-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velseboer DC, et al. Prevalence of orthostatic hypotension in Parkinson's disease: A systematic review and meta-analysis. Parkinsonism Relat Disord. 2011 doi: 10.1016/j.parkreldis.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernino S, et al. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343:847–55. doi: 10.1056/NEJM200009213431204. [DOI] [PubMed] [Google Scholar]
- Yoshita M, et al. Value of 123I-MIBG radioactivity in the differential diagnosis of DLB from AD. Neurology. 2006;66:1850–4. doi: 10.1212/01.wnl.0000219640.59984.a7. [DOI] [PubMed] [Google Scholar]