

Abstract
The efficacy of tyrosine kinase inhibitors (TKIs) as cancer therapeutics varies amongst individual patients as a result of patient-specific differences in molecular regulation of cancer development and progression, and acquisition of resistance to TKIs during therapy. A sensitive assay that can quantify kinase activity and predict inhibition of that activity from minimally invasive patient tissue samples may aid design of efficacious individualized TKI treatments. A microfluidic format can be useful in reducing limitations in standard protein kinase assays, including sensitivity required and low sample volume available. We present photopatterned macroporous poly(ethylene glycol) diacrylate hydrogel pillars functionalized with kinase substrates within microchannels for quantifying kinase activity in complex cellular lysates. We determined the effect of using a porogen to induce macroporosity in hydrogel pillars and showed that hydrogel poration enhanced the sensitivity of detecting Bcr-Abl activity in cell lysates by an order of magnitude. Bcr-Abl tyrosine kinase activity in K562 cell lysates could be detected from 0.01 μg/μL of cell lysate, corresponding to approximately 500 cells, using GST-Crkl-immobilized in macroporous hydrogels. This device was also capable of quantifying inhibition of Bcr-Abl activity by imatinib mesylate, which demonstrates the potential to predict the biochemical response to drug inhibitors. These results indicate that microfluidic devices containing macroporous hydrogels functionalized with kinase substrates provide a promising platform for sensitive and specific quantification of kinase activity and efficacy of kinase inhibitors in cancer cell lysates.
Keywords: kinase activity, cancer diagnostic, microfluidic device, hydrogels
1. Introduction
Personalized medicine is a rapidly advancing field of medicine that considers a patient's genetic, proteinaceous, and metabolic profile to direct individualized patient therapies against disease. Applications can currently be found in the treatment of cancer. For example, HER-2/neu tyrosine kinase is overexpressed in about 10–34% of patients with metastatic breast cancer (Ross et al. 1998). Trastuzumab (Herceptin) has been developed to treat HER-2+ tumors, but it is not effective in treating HER-2- patients and increases the risk of heart dysfunction. In another example, virtually all cases of chronic myeloid leukemia (CML) are caused by the abnormal expression and constitutive activity of the Bcr-Abl fusion protein tyrosine kinase and can be effectively treated with imatinib mesylate (Gleevec). However, about 15% of patients become resistant, often as a result of the expansion of mutated Bcr-Abl transcripts (Druker et al. 2006, Lewandowski et al. 2009). Additional small molecule kinase inhibitors have been developed against imatinib-resistant CML; however, their efficacy varies among patients depending on each patient's unique mechanism for imatinib mesylate resistance (Weisberg et al. 2007). The development of a sensitive and specific diagnostic to quantitatively profile a patient's kinase activities or potential biochemical responses to tyrosine kinase inhibitor (TKI) therapy may help a physician establish an effective treatment strategy for each patient.
Microfluidics is emerging as a promising platform for medical diagnostics with advantages over traditional diagnostics, including reduced sample size and reagent consumption. A number of microfluidic enzyme assays have been developed to assess kinase activity from cellular lysates, which contain a complex mixture of many proteins and enzymes including phosphorylated proteins, kinases, proteases, and phosphatases (Fang et al. 2010, Lee et al. 2009). Fang et al. used microfluidics to capture Bcr-Abl from cellular lysates and measured activity with a radiometric method (Fang et al. 2010). However, multiplexing analysis of several different kinases from a given sample and increasing throughput is limited with this method. This analysis could be achieved with the use of a microarray format in which substrate immobilization and detection can be spatially controlled.
Assays have been developed that immobilize enzymes or their substrates in hydrogel features within microfluidic devices (Heo et al. 2005, Sung et al. 2009, Yadavalli et al. 2004). Hydrogel arrays maintain a hydrated environment around immobilized substrates which allows them to maintain their native structure and activity (Rubina et al. 2003). Immobilization in a 3-dimensional matrix also permits increased substrate density which may increase assay sensitivity. The majority of hydrogel array assays rely on diffusion of relatively small molecules through the hydrogel mesh. However, analysis of large macromolecules, proteins, and enzymes, including antibodies (150 kDa) and kinases such as Bcr-Abl (190-210 kDa) is limited due to their restricted diffusion into hydrogels (Kim et al. 2008, Lee et al. 2010). Several methods have been used to introduce porosity in hydrogels, including cryogelation and the use of inert porogens such as salt or organic solvents which are leeched from the hydrogel after gelation (Lozinsky et al. 2003, Okay 2000). However, high salt or organic solvent concentrations can denature proteins and thus these methods are usually incompatible for use with most relevant biomolecules. Some porogenic materials that may be compatible with proteins and enzymes include glycerol and PEG (Badiger et al. 1993, Caykara et al. 2006).
Here we report development of sensitive tyrosine kinase assays from cancer cell lysates that utilize kinase substrates immobilized in macroporous poly(ethylene glycol) diacrylate (PEGDA) hydrogels. We investigated the effect of PEG porogen concentration on the porosity of hydrogel pillars formed and their relative performance in quantifying the activity of Bcr-Abl from cell lysates. Photopatterning hydrogel pillars decreased spot-to-spot variation and increased sensitivity compared to our previous assay that measures Bcr-Abl tyrosine kinase activity using spotted arrays (Brueggemeier et al. 2005). Furthermore, macroporous hydrogels containing immobilized GST-Crkl achieved a lower limit of quantitation than nonporous hydrogels. Our results indicate that the diffusion limitation in hydrogel microarrays can be reduced by introducing porosity with PEG porogens. With this improvement, sensitivity and the limit of quantitation can be improved for microfluidic assays that quantify activities of large enzymes from cell lysates.
2. Materials and Methods
2.1. Materials
Isobornyl acrylate, technical grade (IBA); tetraethylene glycol dimethacrylate (TEGDMA); 2,2-dimethoxy-2-phenyl-acetophenone, 99% (DMPA); and poly(ethylene glycol), typical Mn 700 (PEG700DA) were purchased from Sigma-Aldrich (St. Louis, MO). 2-(N-morpholino)ethanesulfonic acid (MES), Brij-35, hydroxylamine hydrochloride, and glass microscope slides (Fisherfinest) were supplied from Fisher Scientific (Waltham, MA). PEG (average Mr 3400) from ICN Biomedicals (Aurora, OH) was dissolved in MES buffer (0.1 M MES, 0.5 M NaCl, pH 5.0) to make a 30% (w/w) solution. 1-[4-(2-Hydroxyethoxy)-phenyl]-2-hydroxy-2-methyl-1-propane-1-one (Irgacure-2959) was obtained from Ciba Specialty Chemicals (Basel, Switzerland) and a stock solution was prepared by dissolving 1 g in ethylene glycol (Fisher) to make 10 mL total solution. 6-((acryloyl)amino)hexanoic acid, succinimidyl ester (Acryloyl-X, SE) was purchased from Invitrogen (Carlsbad, CA), dissolved in anhydrous DMSO to make a 5% (w/v) solution, and aliquots were stored at -80 °C. (3-Acryloxypropyl)-trimethoxysilane was purchased from Gelest (Morrisville, PA). Polycarbonate cartridges (HybriWell) were purchased from Grace Bio-Laboratories (Bend, OR). Transparency masks were printed by Imagesetter (Madison, WI).
Fluorescein isothiocyanate-dextran, average Mr 250,000 (250 kDa dextran-FITC) from Sigma, was dissolved in water, centrifuged with an Amicon Ultra-15 (100,000 MWCO) centrifuge filter (Millipore, Billerica, MA), and repeated until the eluate was colorless (at least five cycles).
GST-GFP fusion protein was produced in Escherichia coli DH5α cells and purified as previously described.(Brueggemeier et al. 2004) GST-Crkl (SH3) fusion protein was produced in Escherichia coli BL21 cells and purified as described by Brueggemeier et al.(Brueggemeier et al. 2005) Imatinib mesylate was purchased from Enzo Life Sciences (Farmingdale, NY). cOmplete Protease Inhibitor Cocktail (Roche, Mannheim, Germany) tablets were dissolved in water according to the manufacturer's instructions to make a 25× stock solution. Halt Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific) was used as supplied. Anti-phosphotyrosine clone 4G10 antibody was purchased from Millipore. All other antibodies were supplied by Invitrogen and all other solvents and reagents were purchased from domestic suppliers and used as received.
2.2. Preparation K562 cell lysate
K562 (ATCC, Manassas, VA) was cultured at 37 °C and 5% CO2 in RPMI-1640 media (Cambrex Bio Science, Walkersville, MD) containing 100 units/mL penicillin, 100 μg/mL streptomycin, and 10% FBS. Cells were lysed by suspending at 5 × 107 cells/mL in cold lysis buffer (42.3 mM HEPES, 126 mM NaCl, 1.27 mM MgCl2, 0.85 mM EDTA, 84.5 mM NaF, 8.45 mM sodium pyrophosphate, 0.169 mM sodium orthovanadate, 1 mM PMSF, Roche cOmplete Protease Inhibitor Cocktail, pH 7.4) and incubated on ice for 20 min. The cell lysate solution was then clarified by spinning at 1500 rpm for 10 min and only the supernatant was retained. Total protein concentration was determined with a bicinchoninic acid assay (Pierce, Rockford, IL), and aliquots of the cell lysate was stored at -80 °C until further use.
2.3. Microchannel fabrication
Microchannels were prepared as previously described.(Beebe et al. 2000, Lee et al. 2010) Briefly, piranha-cleaned glass microscope slides are functionalized with acryloyl groups by incubation with a 95% (v/v) ethanol/water solution containing 2% (v/v) (3-acryloxyproppyl)-trimethoxysilane and dried overnight at room conditions. Polycarbonate cartridges, which have a 150 μm thick adhesive, were affixed to these glass slides and filled with a prepolymer solution containing IBA (1.9 mg), TEGDMA (0.1 mg), and DMPA (0.6 mg). The chamber was covered with a transparency mask and exposed to 320–500 nm light at 5.9 mW/cm2 intensity for 14 s (EXFO OmniCure S1000). The unpolymerized material was removed by thorough flushing with ethanol. The device was re-exposed to the light as described above to ensure complete polymerization. Channel surfaces were then exposed to a poly(dimethylsiloxane)-based solution commercially supplied as “Rain-X” (Illinois Tool Works, Glenview, IL) for 2 minutes, then dried by vacuum suction.
2.4. Photopatterning hydrogel pillars
Hydrogel pillars were photopatterned in a similar fashion as described in the previous section and illustrated in Fig. 1a,b. The PEG prepolymer solution consisted of MES buffer with 5% (v/v) PEG700DA, 0.2 μg/μL Irgacure-2959, and 0.167% (w/v) acryloyl-X, SE. The solution was allowed to flow into the microchannels by capillary flow or gentle syringe suction as needed. A transparency mask was placed over the chamber and held in place with a quartz slide (Fig. 1a). It was then exposed to 320–500 nm light at 16.8 mW/cm2 intensity for 420 s. Macroporous hydrogels were prepared by diluting the 30% PEG3400 in MES buffer stock solution in the PEG prepolymer solution and polymerizing as above for 45 s for solutions containing 20% (w/v) PEG3400 and 180 s for solutions containing 10% (w/v) PEG3400. The addition of PEG porogen affected polymerization kinetics; therefore, polymerization time was varied for each polymer composition in order to keep final pillar diameters similar (0.4 mm) between all polymer compositions. After exposure to UV light, unpolymerized material was flushed from the chamber with cold MES buffer containing 0.05% (w/v) Brij-35.
Fig. 1.
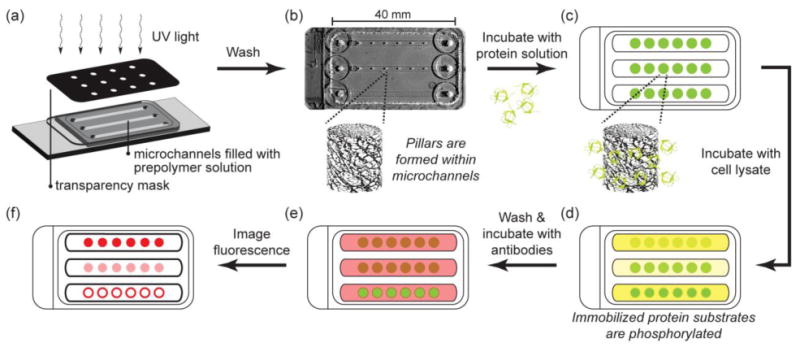
Schematic of hydrogel post fabrication, protein immobilization, kinase assay, and detection of substrate phosphorylation. (a) Hydrogel pillars are photopatterned from PEG prepolymer solutions containing acryloyl-functionalized NHS within microchannels. (b) Photograph of channels with pillars. Unreacted prepolymer solution is washed from the channel leaving polymerized pillars within the microchannels. (c) Protein substrate solution is flowed into the channels and allowed to react with NHS groups in the hydrogel pillars. Unincorporated protein is washed from the channels and (d) a kinase reaction mixture containing cell lysate is flowed into the channels. (e) Phosphorylated substrate is labeled with fluorophore-conjugated antibodies and (f) imaged with epifluorescence microscopy.
2.5. Characterization of porosity
A functional analysis was used to determine the relative effect of PEG3400 porogen concentration in allowing macromolecular diffusion. Microchannels containing hydrogel pillars were filled with 250 kDa dextran-FITC, and a drop of mineral oil was placed over the ports to minimize evaporation. After incubating for 4 h at room temperature, the hydrogel pillars were imaged with an inverted epifluorescence microscope (Olympus IX70) and attached monochrome CCD digital camera (Diagnostic Instruments SPOT RT). Hydrogel pillars appear as circular spots in microscope images (Fig. 1f). The extent of dextran penetration into the hydrogel pillars is described by the relative spot intensity and was calculated by the ratio of the average intensity of the spot to the average intensity of the area surrounding the spot.
2.6. Immobilization of fusion proteins
The inclusion of acryloyl-X, SE in the PEG prepolymer solution allows for the covalent immobilization of proteins to the hydrogel pillars by reaction with their exposed primary amines. Purified protein substrates were diluted in PBSB (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.7 mM KH2PO4, 0.05% (w/v) Brij-35, pH 7.4), flowed into the microchannels containing photopatterned hydrogel pillars, and allowed to incubate in a humid chamber at room temperature for 1 h (Fig 1c). A concentration of 2 μg/μL was used for GST-GFP immobilization. The channels were then washed briefly by flowing PBSB through using gentle syringe suction. Unreacted acryloyl-X, SE was quenched by flowing in 0.5 M hydroxylamine hydrochloride in PBSB adjusted to pH 8 and incubating for 20 minutes at room temperature. After quenching, the channels were washed briefly with TBST (10 mM Tris-HCl, 100 mM NaCl, 0.1% Tween-20, pH 7.5) and blocked with TBST containing 1% bovine serum albumin (BSA) by incubation for 30 minutes.
2.7. Kinase assay with microfluidic channels
BSA blocking buffer was removed from the channels by gentle syringe suction and 1× Abl kinase assay buffer (50 mM Tris-HCl, 10 mM MgCl2, 100 μM EDTA, 1 mM DTT, 0.015% Brij-35, 100 μg/mL BSA, pH 7.5) was flowed into the channels and incubated briefly while the K562 cell lysate reaction mixture was prepared. The K562 cell lysate reaction mixtures contained 3.3 μL of 3× Abl kinase assay buffer, 2 μL of 1 mM ATP, 0–10 μg of K562 cell lysate, 0.1 μL of 100× Halt Phosphatase Inhibitor Cocktail, 0.4 μL of 25× Roche cOmplete Protease Inhibitor (EDTA-free), and water to a total volume of 10 μL. Reaction solutions for experiments with imatinib mesylate inhibitor were prepared by adding K562 cell lysate reaction mixture to serial dilutions of 100 mM imatinib mesylate dissolved in DMSO such that the final inhibitor concentration was 0–100 μM. A 6–7 μL portion of these kinase reaction mixtures was then allowed to flow into the microchannels by capillary forces, and the reactions were allowed to proceed for 60 min at 37 °C in a humid environment (Fig. 1d). Kinetic experiments were performed with reaction mixtures containing 1 μg/μL K562 cell lysate and reaction time was varied from 5 to 120 min.
After the reaction, the microchannels were washed by frequently flowing TBST through the channels over a period of 30 min. The channels were then blocked to reduce nonspecific signal by incubating with blocking buffer for 30 min at room temperature. 2 μg/mL 4G10 anti-phosphotyrosine antibody (Millipore) and 6 μg/mL anti-glutathione S-transferase antibody (Invitrogen) in blocking buffer were then flowed into the channels and incubated for 1 h at room temperature (Fig. 1e). Following a 1 h wash step with TBST, Alexa Fluor 488 goat anti-mouse and Alexa Fluor 594 donkey anti-rabbit secondary antibodies at 10 μg/mL in blocking buffer were flowed into the channels and allowed to incubate overnight at 4 °C, then washed a final time with TBST for 1 h. Fluorescence was imaged with an inverted epifluorescence microscope (Olympus IX70) and attached monochrome CCD digital camera (Diagnostic Instruments SPOT RT). Average fluorescence intensity is quantified by averaging pixel gray values in the images.
2.8. Statistical analysis
Experiments were analyzed using one way analysis of variance (ANOVA). When there was a statistically significant difference in the mean values among the groups (P < 0.05), the Holm-Sidak Test was used to determine significance between groups (P < 0.05).
3. Results and Discussion
3.1. PEG induces porosity in PEGDA hydrogels
Kinase activity can be quantified by incubating kinases with immobilized substrates and subsequently quantifying phosphorylation of the substrate. Hydrogels can be used to immobilize proteins and have been reported to enhance protein microarray sensitivity compared to proteins immobilized directly on surfaces (Zubtsov et al. 2007). However, the pore sizes of hydrogels commonly used for protein microarrays, such as polyacrylamide and PEGDA, are often on the order of a few Å, which is too small to allow the diffusion of many proteins including antibodies and large kinases. The strong dependence of detection signal on the mass transfer of analytes is one of the main limitations on the sensitivity of microspot protein arrays (Kusnezow et al. 2006). To reduce this mass transport limitation and enhance kinase diffusion into hydrogels, we previously investigated the effect of PEG porogen size on PEGDA hydrogel porosity and diffusion of macromolecules, including antibodies (Lee et al. 2010). Of the porogen sizes tested, PEG3400 (Mr 3,400) yielded hydrogels that allowed antibodies the greatest accessibility to hydrogel-immobilized proteins, and thus we used this porogen in this study.
Fig. 2a shows a montage of bright field images of 5% PEG700DA hydrogels formed with 0–20% PEG3400 porogen. The pillar radii remained constant throughout a channel and spot-to-spot variation was low. In the absence of porogen, hydrogels were transparent. However, the addition of PEG porogen caused macroporosity which altered hydrogel opacity (Wu et al. 2010).
Fig. 2.
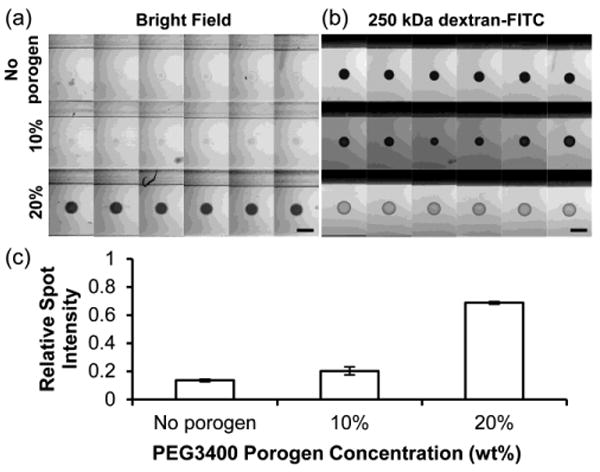
Diffusion of 250kDa dextran-FITC in 5% PEG700DA hydrogels with 0–20% PEG3400 porogen. (a) Bright field image montage of hydrogel pillars (contrast enhanced). (b) Fluorescence microscopy image montage of pillars incubated in 250 kDa dextran-FITC after 4 h. Each row displays six replicate pillars. Scale bars represent 500 μm. (c) Values for spot intensities from images in (a). Relative spot intensity is the ratio of the average gray value (AGV) of the spot with background (AGV outside channels) subtracted to the AGV in an area surrounding the spot with background subtracted. Error bars = SD (n = 6 pillars); P < 0.05 between all pairs.
To investigate the effect of PEG3400 concentration on macromolecular diffusion into PEGDA hydrogels, we incubated 250 kDa dextran-FITC in microchannels containing 5% PEG700DA hydrogels formed with 0% (no porogen), 10%, and 20% PEG3400 for 4 h, during which time a steady-state concentration profile was achieved. Fluorescence microscopy images of the spots were taken to assess dextran penetration into the hydrogel pillars and a montage of the images is shown in Fig. 2b. Dextran of this size has a hydrodynamic radius (RH) of 11.46 nm, which is larger than many proteins including IgG (RH = 6.2 nm; 150 kDa) and fibrinogen (RH = 10.95 nm; 340 kDa) (Armstrong et al. 2004). This dextran is also expected to be larger than Bcr-Abl (185–230 kDa), which has a hydrodynamic radius near 5–10 nm based on comparison with the known radii of several globular proteins of similar molecular mass (Armstrong et al. 2004). 250 kDa dextran-FITC penetration into macroporous PEG hydrogels was over 5 times greater than into nonporous hydrogels formed without porogen (Fig. 2c). Therefore, it is expected that macroporous PEG hydrogel pillars would allow significant diffusion of Bcr-Abl and antibodies used in this study while nonporous PEG hydrogel pillars would limit their diffusion.
3.2. GST-GFP immobilization in PEG hydrogel pillars
In the assessment of kinase and other enzymatic activity, substrate immobilization allows for a robust assay as the enzyme can be quickly removed by washing. Furthermore, proteins are immobilized in hydrogel features in microarrays at a greater density and allow for higher detection sensitivity than if directly immobilized on a surface under the same conditions (Zubtsov et al. 2007). Various strategies have been used to tether biomolecules to a surface including adsorption, entrapment, and biological capture. However, covalent linkage allows greater stability in substrate attachment and subsequent modification and detection. In our group's previous work, we immobilized acryloyl-functionalized kinase substrates by copolymerizing them during hydrogel formation (Brueggemeier et al. 2005, Ghosh et al. 2009, Ghosh et al. 2010). Other labs have immobilized proteins by first activating the surface or copolymerizing the hydrogel with N-hydroxysuccinimide (NHS) based molecules which react with primary amines of proteins (Kim et al. 2008, Schnaar et al. 1975). In this study, we avoided PEG-mediated protein precipitation by adopting the latter method and incubating hydrogel pillars containing copolymerized NHS functionality with protein substrate solutions.
To investigate the ability of protein substrates to penetrate and covalently attach to PEG700DA hydrogels formed with and without porogen, we used GST-GFP as a model protein (Mr = 54 kDa). Fig. 3 shows the fluorescence of GST-GFP immobilized in PEG hydrogels of different porogen concentrations after incubating with a 2 μg/μL solution of GST-GFP and washing to remove non-covalently bound protein. Unreacted acryloyl-X was quenched by incubation with hydroxylamine, which removes the NHS functionality. This quench step was important to reduce nonspecific protein attachment during subsequent exposure to antibodies, and is demonstrated by low GST-GFP and anti-GST signal in the pillars that were quenched before exposure to GST-GFP as shown in Fig. 3b,c (last columns). Interestingly, greater GST-GFP fluorescence was observed in hydrogels without porogen compared with hydrogels formed with porogen (Fig. 3b,d). This may be due to a lower polymer volume fraction and fewer accessible acryloyl-X crosslinkers within the structure of macroporous PEG hydrogels compared to nonporous hydrogels. Fig. 3e shows the fluorescence intensity profile of immobilized GST-GFP along a diameter of typical hydrogel pillars. In all of the hydrogels, GST-GFP was able to fully diffuse to the interior of the pillar. However, as porogen concentration decreased, mass transport limitations become increasingly important and immobilization of GST-GFP at the center of the hydrogel became lower relative to the periphery.
Fig. 3.
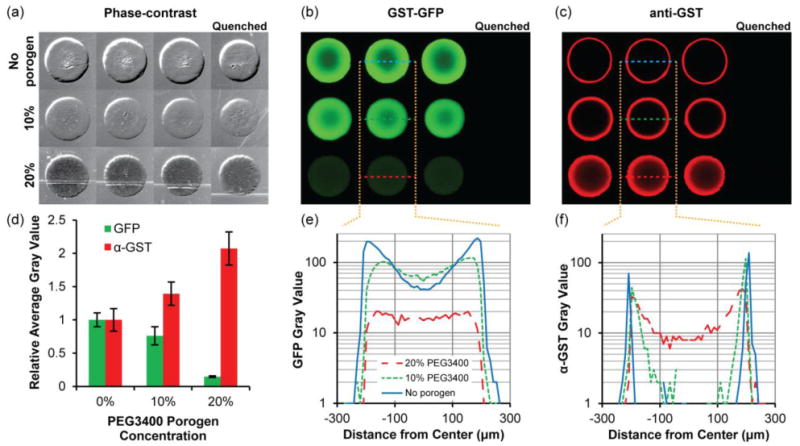
Immobilization of GST-GFP in 5% PEG700DA hydrogels with 0–20% PEG3400 porogen. (a) Phase-contrast images of hydrogel pillars. Pillars were incubated with 2 μg/μL GST-GFP and subsequently incubated with anti-GST and Alexa Fluor 594-conjugated secondary antibody. Fluorescence images showing (b) GST-GFP and (c) anti-GST with Alexa Fluor 594-conjugated secondary antibody localization throughout the pillars. The first three columns are triplicates and pillars shown in the last column were quenched with 0.5 M hydroxylamine prior to exposure to GST-GFP. Contrast is enhanced in image to emphasize low-signal areas. (d) Fluorescence intensity of GST-GFP and anti-GST relative to those detected in hydrogels lacking porogen. Error bars = SD (n ≥ 4 pillars). (e) Typical GST-GFP and (f) anti-GST fluorescence intensity profiles along a diameter of the hydrogels pillars. Gray value range = 0–255.
Having identified the immobilization profiles of the model GST-GFP protein, we then investigated accessibility of immobilized protein for detection with antibodies by incubating GST-GFP-immobilized pillars with anti-GST antibody followed by an Alexa Fluor 594-conjugated secondary antibody (Fig 3c). The fluorescence intensity profile (Fig. 3f) of anti-GST in the macroporous hydrogel shows that antibodies are able to fully penetrate and bind to protein immobilized in the interior of these macroporous pillars in comparison to nonporous hydrogels which limited antibody binding to only GST-GFP immobilized near the pillar periphery. Antibody signal in macroporous hydrogels was twice as intense as nonporous hydrogels (Fig. 3d), which indicates the potential for greater signal sensitivity in detecting immobilized proteins in macroporous hydrogels.
3.3. GST-Crkl immobilization and phosphorylation in hydrogel pillars
Having demonstrated hydrogel pillar formation and subsequent protein immobilization and detection with low spot-to-spot variability using the model protein GST-GFP, we next immobilized GST-Crkl (SH3) to assess the ability to detect the activity of Bcr-Abl. The Crkl fragment used comprises only the SH3 domains of full-length Crkl that flank the tyrosine residue that is phosphorylated by Bcr-Abl. This GST-Crkl (SH3) fusion construct is 50 kDa, which is smaller than the GST-GFP fusion protein (54 kDa) immobilized in the previous section (Brueggemeier et al. 2005). Therefore, it is expected that GST-Crkl would penetrate and exhibit a similar immobilization profile as demonstrated by GST-GFP. GST-Crkl (SH3) fusion protein was purified from BL21 E. coli cell cultures, immobilized in hydrogel pillars formed with 0– 20% PEG3400 porogen, and detected using anti-GST and Alexa Fluor 594-conjugated secondary antibody to verify immobilization. The quantity of GST-Crkl immobilized and detectable by anti-GST in these pillars was consistent between independent experiments (Fig. 4a). Consistent with the trend observed with antibody detection of GST-GFP, GST-Crkl was more accessible in hydrogels formed with 20% PEG3400 porogen than without porogen (P < 0.05).
Fig. 4.
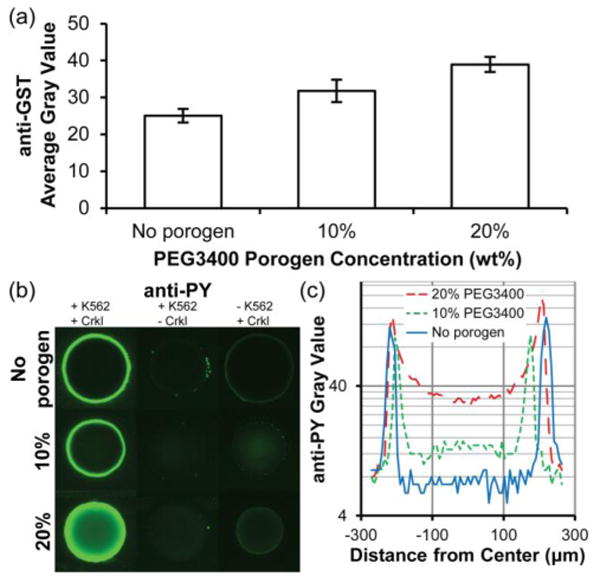
Immobilization of GST-Crkl and detection of K562 cell lysate-mediated phosphorylation in 5% PEG700DA hydrogels with 0–20% PEG3400 porogen. (a) Average gray values for anti-GST detection of GST-Crkl after incubating pillars in a solution of 4 μg/μL GST-Crkl and exposure to K562 cell lysate. Error bars = SE (n ≥ 3 experiments). (b) Images of anti-phosphotyrosine signal (overexposed to emphasize low signal areas) in these pillars. Pillars shown in the middle and last column were negative controls and any observable signal suggests nonspecific cell lysate adsorption (middle column, no GST-Crkl immobilized) or nonspecific antibody adsorption (last column, no K562 cell lysate). (c) Typical fluorescence intensity profiles of anti-phosphotyrosine along the diameters of the representative pillars shown in (b), first column. Gray value range = 0–255.
To demonstrate that hydrogel pillars containing immobilized GST-Crkl can be used to quantify kinase activity from cellular extracts, we incubated K562 cell lysates in microfluidic chambers containing GST-Crkl-immobilized hydrogel pillars. Extracts from mammalian cells contain an abundance of phosphorylated proteins, including endogenous Crkl. Therefore, it is crucial to minimize nonspecific binding and subsequent antibody detection of these phosphoproteins. Preliminary experiments were plagued by high background signal caused by nonspecific binding of cell lysate proteins to both the glass and polycarbonate channel surfaces and antibodies to the polymer hydrogels (data not shown). We determined that exposing channel surfaces to a poly(dimethylsiloxane)-based coating and in conjunction with using a nonionic surfactant, Brij-35, during protein immobilization, drastically reduced nonspecific anti-phosphotyrosine signal on channel surfaces. In addition, low signal was achieved in control pillars without immobilized GST-Crkl that were incubated with K562 cell lysate (Fig. 4b, middle column). Some anti-phosphotyrosine signal observed in control spots with immobilized GST-Crkl but not exposed to K562 cell lysate (Fig. 4b, column 3) may be the result of nonspecific anti-phosphotyrosine antibody interactions with immobilized protein. Nonetheless, this signal is low in comparison to pillars containing immobilized GST-Crkl and exposed to 1 μg/μL K562 cell lysate (Fig. 4b).
The effect of porogen concentration on phosphorylation of immobilized GST-Crkl by Bcr-Abl present in K562 cell lysate is shown in Fig. 4b,c. Consistent with previous anti-GST signal profiles, there was a greater observable anti-phosphotyrosine signal in the interior of hydrogel pillars formed with 20% porogen than in pillars without porogen. This indicates that kinases as large as Bcr-Abl (210 kDa) were able to diffuse into these macroporous hydrogels and phosphorylate substrate while only the periphery was accessible in nonporous pillars.
Overall signal intensity can be controlled by adjusting either the concentration of acryloyl-X in the prepolymer mixture or the concentration of GST-Crkl in the immobilization solution. Modifying acryloyl-X concentration in the prepolymer mixture will alter the mechanical and structural properties of the resulting hydrogel; therefore, we varied substrate immobilization to maximize signal. Fig. 5 shows anti-phosphotyrosine and anti-GST levels in macroporous hydrogel pillars that have been incubated with 0–10 μg/μL GST-Crkl and reacted with 1 μg/μL K562 cell lysate. Saturation of NHS reactive sites in the hydrogel pillar was reached near 4 μg/μL GST-Crkl and higher concentrations did not yield significantly greater anti-phosphotyrosine or anti-GST signal. Signal due to phosphorylation is dependent on the anti-GST signal (Fig. 5b), which suggests that the antibody-accessible GST-Crkl is also accessible for phosphorylation by Bcr-Abl. To maximize phosphotyrosine signal without consuming excessive amounts of GST-Crkl substrate, a concentration of 4 μg/μL was subsequently used to assess Bcr-Abl activity in this study.
Fig. 5.

Phosphorylation of 0–10 μg/μL GST-Crkl immobilized in 5% PEG700DA/20% PEG3400 hydrogel pillars by K562 cell lysates. (a) Anti-phosphotyrosine and anti-GST signal as a function of the concentration of GST-Crkl used for immobilization after incubating with 1 μg/μL K562 cell lysate for 1 h and labeling with antibodies. (b) Anti-phosphotyrosine signal is dependent on the anti-GST signal. Error bars = SD (n = 5 spots).
3.5. Effect of PEG porogen concentration on reaction time for GST-Crkl phosphorylation
Immobilizing protein substrates on surfaces alters enzyme kinetics compared to solution-phase reactions due to considerations of surface adsorption kinetics in addition to Michaelis-Menten enzyme kinetics (Gaspers et al. 1995, Gaspers et al. 1994, Kim et al. 2002, Lee et al. 2005). For the overall reaction, enzyme surface diffusion, diffusion through a porous medium, and substrate linker size all introduce complexity and affect the reaction time necessary for saturation (Deere et al. 2008, Gaspers et al. 1994). Here we examined the effect of polymer porosity on the overall rate of reaction. Time course data for the phosphorylation of GST-Crkl in 5% PEG700DA hydrogels fabricated with 0–20% PEG3400 porogen are shown in Fig. 6. Similar to our previous studies of GST-Crkl phosphorylation using spotted nonporous polyacrylamide arrays, the reaction appeared to be nearly complete after 30 minutes with nonporous PEG hydrogel pillars (Brueggemeier et al. 2005). However, with increasing concentrations of porogen used, the time to saturation increased. At reaction times less than 30 minutes, the difference in phosphorylation signals between nonporous and macroporous hydrogels was not significant (P > 0.05). However, after 30 minutes, phosphorylation signal continued to rise for porous hydrogels while steady-state was reached in nonporous hydrogels. In addition, it is expected that large molecules like Bcr-Abl require at least 40 minutes to diffuse into macroporous hydrogel pillars (Lee et al. 2010). These observations suggest that although a greater maximum signal can be achieved with macroporous hydrogels in this microchannel-based assay, reaching this maximum signal is more diffusion limited than in nonporous hydrogels. Based on these results, a 60 minute reaction time was chosen for subsequent experiments.
Fig. 6.
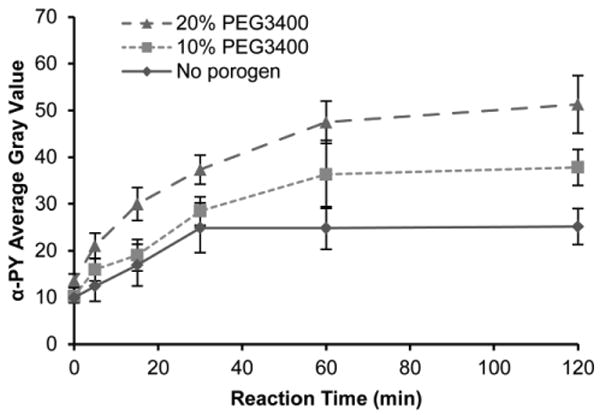
K562 cell lysate-mediated phosphorylation in 5% PEG700DA hydrogel pillars incubated with 4 μg/μL GST-Crkl. Average gray values of anti-phosphotyrosine signal in pillars formed with 20% PEG3400 (▴), 10% PEG3400 (▪) and without porogen (◆). Error bars = SE (n ≥ 5 experiments).
Because macroporous hydrogel pillars were diffusion limited, the reaction time could be reduced by modifying the device design. For instance, convective flow can decrease reaction times by increasing transport of kinases through the hydrogel, and there are several methods to introduce flow or mixing in microfluidic channels including the use of active external mixers and passive pumping (Hart et al. 2010, Sigurdson et al. 2005, Walker et al. 2002). In addition, a decrease in the pillar diameter would decrease diffusion lengths and reduce reaction time to saturation. Further studies to miniaturize this system will investigate the formation of smaller-diameter hydrogel pillars and the corresponding effect on kinase assay reaction time.
3.6. Microchannel-based assay for Bcr-Abl activity in K562 cell lysates
Having determined reaction parameters to maximize kinase assay signal, we investigated the relative performance of macroporous hydrogels and nonporous hydrogels containing immobilized GST-Crkl in assessing Bcr-Abl activity in cell lysates. Fig. 7a shows anti-phosphotyrosine signal in these hydrogels after reaction with 0–1 μg/μL K562 cell lysate relative to the maximum signal obtained in nonporous hydrogels. Hydrogels that were formed with 20% PEG3400 had greater signal at ≥ 0.3 μg/μL K562 cell lysate than hydrogels formed in the absence of porogen (P < 0.05). Consequently, K562 activity was detected over a greater dynamic range of lysates concentration in macroporous hydrogels than in nonporous hydrogels. The data in Fig. 7a were obtained from images taken at 2 s exposure times such that the images of pillars exposed to 1 μg/μL K562 cell lysate were not overexposed. However, at this exposure time the microscope and attached digital camera were not able to discriminate differences at low fluorescence intensity (< 0.3 μg/μL K562 cell lysate). To resolve differences at low K562 concentrations, images taken at a longer exposure time (5 s) are shown in Fig. 7b. Images of hydrogel pillars incubated with greater than 0.1 μg/μL K562 cell lysate were overexposed and are not included. We defined the limit of quantitation (LOQ) as the lowest K562 cell lysate concentration tested that has a significant (P < 0.05) difference compared to the control hydrogels which were not exposed to K562 cell lysate. For hydrogels formed without porogen, the LOQ was 0.1 μg/μL K562 cell lysate, which was similar to the 0.225 μg/μL K562 cell lysate LOQ in our previous studies with nonporous polyacrylamide hydrogel spot arrays (Brueggemeier et al. 2005). The LOQ for hydrogels formed with 10% and 20% PEG3400 porogen was 0.01 μg/μL K562 cell lysate, an order of magnitude improvement compared to nonporous hydrogels.
Fig. 7.
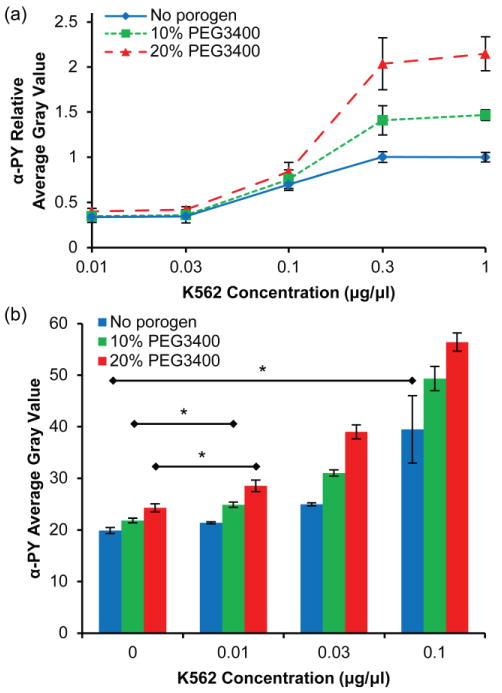
Effect of porogen concentration on detection sensitivity of the microchannel kinase assay. (a) Relative anti-phosphotyrosine signal (exposure time = 2 s) as a function of K562 cell lysate dilution for PEG hydrogels with 20% PEG3400 (▴), 10% PEG3400 (▪) and without porogen (◆). Data are relative to the maximum average gray value for the hydrogel formed without porogen. Error bars = SE (n > 6 experiments). (b) Average gray value of anti-phosphotyrosine signal (exposure time = 5 s) for hydrogels with and without porogen at low K562 concentrations. Error bars = SD (n > 3 spots). * P < 0.05 indicates limit of quantitation for each hydrogel.
A microfluidic format minimizes the quantity of cell lysate required compared to traditional solution or array-based methods. It is estimated that a cubic centimeter of tissue contains about 109 cells while core needle biopsies or cell aspirates could contain fewer than 105 cells (Liotta et al. 2003). Because tissues are highly heterogeneous, perhaps only a few thousand cancer cells may be present in a core biopsy. In our previous studies using nonporous polyacrylamide arrays (Brueggemeier et al. 2005), about 56 μg of K562 cell lysate (about 330,000 cells) was required at the LOQ. In this study, phosphorylation was detected from as few as 500 cells—a reduction in sample size by almost 3 orders of magnitude. Sample and reagent volumes could be further reduced by fabricating smaller channels and increasing the spot density within a channel.
Sensitivity, in this study, is defined as the ability to discriminate differences in anti-phosphotyrosine signal due to small changes in K562 concentration. Practically, it is defined as the slope of the plot of anti-phosphotyrosine signal vs. K562 concentration. The relative sensitivities of the hydrogels normalized against the value from hydrogels formed without porogen are tabulated in Table 1. Hydrogels formed with 20% PEG3400 porogen have almost 60% greater sensitivity at detecting Bcr-Abl activity than nonporous hydrogels under the conditions used in this study. This is due to the increase in accessible GST-Crkl substrate to both Bcr-Abl and antibodies used for detection. Additional improvements in sensitivity may be achieved through the use of high performance tools for fluorescence detection such as lasers for fluorophore excitation and photomultiplier tubes which raise signal above the background electronic noise.
Table 1.
Sensitivity of microchannel kinase assay using hydrogels formed with 0–20% PEG3400.
| PEG3400 Porogen Concentration | Relative Sensitivitya |
|---|---|
| No porogen | 1.00 (0.03) |
| 10% | 1.38 (0.03) |
| 20% | 1.58 (0.19) |
Slope of the curve obtained by plotting anti-phosphotyrosine average gray value against K562 concentration; value normalized to no porogen hydrogel. Parentheses indicate standard error of the slope.
3.7. Detection of imatinib mesylate inhibitor activity
A sensitive assay that can screen the effect of TKIs on patient cell lysates may be a useful diagnostic tool for the determination of patient-specific drug therapies. The microchannel-based kinase assay with macroporous (20% PEG3400) hydrogels was assessed for its ability to quantify the effect of inhibitor concentration. 1 μg/μL K562 cell lysate was mixed with serial dilutions of imatinib mesylate immediately prior to incubation in microfluidic channels containing hydrogels with immobilized GST-Crkl. The resultant anti-phosphotyrosine average gray values are shown in Fig. 8. Under these conditions, the half-maximal inhibitory concentration (IC50) of imatinib mesylate against Bcr-Abl was close to 5 μM. This is lower than the IC50 (20 μM) determined in our previous studies with nonporous polyacrylamide arrays (Brueggemeier et al. 2005). White et al. found that the IC50 of imatinib mesylate inhibition of endogendous Crkl phosphorylation was variable amongst patients with values up to 1.8 μM (White et al. 2005). Other studies have determined IC50 for autophosphorylation of Abl to be about 20-30 nM (Corbin et al. 2003, Hochhaus et al. 2002). These are lower than our study and can partially be explained by our addition of ATP in the reaction mixture, which would increase the concentration of the competitive inhibitor needed. Despite these differences in observed biochemical inhibition of Bcr-Abl compared to other studies, inhibited K562 activity was significant between each dilution of inhibitor used (P < 0.05). This indicates the potential of this device to assess inhibitor activity against kinases in cell lysates.
Fig. 8.

Inhibition of Bcr-Abl in K562 cell lysates by imatinib mesylate. K562 cell lysate was added to serial dilutions (0–100 μM) of imatinib mesylate and incubated in microchannels containing 5% PEG700DA/20% PEG3400 macroporous hydrogel pillars with immobilized GST-Crkl, and detected with anti-phosphotyrosine. Control pillars (No K562) were not exposed to K562 cell extracts or imatinib mesylate. Error bars = SD (n ≥ 6 spots). P < 0.05 between all inhibitor concentrations.
4. Conclusions
Macroporous PEGDA hydrogels can be formed by incorporating inert PEG porogens in the prepolymer mixture. Photopatterning allows size and spatial control when forming hydrogel pillars within microfluidic channels, which yields low spot-to-spot variability. These hydrogels can be used to immobilize protein substrates in order to access enzymatic activity from solutions, which is limited in current hydrogel-based assays due to mass transport limitations. We suggest that the use of macroporous hydrogels provides rapid formation of porous features within microfluidic devices which achieve a greater sensitivity than nonporous hydrogels in applications requiring diffusion of large molecules, such as quantifying kinase activity and assessing inhibition in cell lysates. Finally, we provide a proof-of-principle that the activity of specific tyrosine kinases can be determined from minute sample sizes as small as those obtained from core needle biopsies, and with a heterogeneous mixture of proteins, suggesting clinical relevance for hydrogel-based kinase assays in a microfluidic format.
Acknowledgments
We thank Alicia Powers for helpful discussion and revisions in preparing this manuscript. This work was supported by the NIH/NIGMS Grant R01GM074691.
References
- Armstrong JK, Wenby RB, Meiselman HJ, Fisher TC. Biophys J. 2004;87:4259. doi: 10.1529/biophysj.104.047746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badiger MV, Mcneill ME, Graham NB. Biomaterials. 1993;14:1059. doi: 10.1016/0142-9612(93)90206-h. [DOI] [PubMed] [Google Scholar]
- Beebe DJ, Moore JS, Yu Q, Liu RH, Kraft ML, Jo BH, Devadoss C. Proc Natl Acad Sci USA. 2000;97:13488. doi: 10.1073/pnas.250273097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueggemeier SB, Kron SJ, Palecek SP. Anal Biochem. 2004;329:180. doi: 10.1016/j.ab.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Brueggemeier SB, Wu D, Kron SJ, Palecek SP. Biomacromolecules. 2005;6:2765. doi: 10.1021/bm050257v. [DOI] [PubMed] [Google Scholar]
- Caykara T, Bulut M, Dilsiz N, Akyuz Y, Macromol J. Sci Part A-Pure Appl Chem. 2006;43:889. [Google Scholar]
- Corbin AS, La Rosee P, Stoffregen EP, Druker BJ, Deininger MW. Blood. 2003;101:4611. doi: 10.1182/blood-2002-12-3659. [DOI] [PubMed] [Google Scholar]
- Deere J, De Oliveira RF, Tomaszewski B, Millar S, Lalaouni A, Solares LF, Flitsch SL, Halling PJ. Langmuir. 2008;24:11762. doi: 10.1021/la801932f. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, Cervantes F, Hochhaus A, Powell BL, Gabrilove JL, Rousselot P, Reiffers J, Cornelissen JJ, Hughes T, Agis H, Fischer T, Verhoef G, Shepherd J, Saglio G, Gratwohl A, Nielsen JL, Radich JP, Simonsson B, Taylor K, Baccarani M, So C, Letvak L, Larson RA. The New England journal of medicine. 2006;355:2408. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- Fang C, Wang YJ, Vu NT, Lin WY, Hsieh YT, Rubbi L, Phelps ME, Muschen M, Kim YM, Chatziioannou AF, Tseng HR, Graeber TG. Cancer Research. 2010;70:8299. doi: 10.1158/0008-5472.CAN-10-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspers PB, Gast AP, Robertson CR. J Colloid Interface Sci. 1995;172:518. doi: 10.1006/jcis.1999.6109. [DOI] [PubMed] [Google Scholar]
- Gaspers PB, Robertson CR, Gast AP. Langmuir. 1994;10:2699. [Google Scholar]
- Ghosh G, Lee AG, Palecek SP. Anal Biochem. 2009;393:205. doi: 10.1016/j.ab.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh G, Yan X, Lee AG, Kron SJ, Palecek SP. Biosens Bioelectron. 2010;26:424. doi: 10.1016/j.bios.2010.07.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart R, Lec R, Noh H. Sens Actuator B-Chem. 2010;147:366. [Google Scholar]
- Heo J, Crooks RM. Anal Chem. 2005;77:6843. doi: 10.1021/ac0507993. [DOI] [PubMed] [Google Scholar]
- Hochhaus A, Kreil S, Corbin AS, La Rosee P, Muller MC, Lahaye T, Hanfstein B, Schoch C, Cross NC, Berger U, Gschaidmeier H, Druker BJ, Hehlmann R. Leukemia. 2002;16:2190. doi: 10.1038/sj.leu.2402741. [DOI] [PubMed] [Google Scholar]
- Kim DN, Lee W, Koh WG. Analytica Chimica Acta. 2008;609:59. doi: 10.1016/j.aca.2007.12.024. [DOI] [PubMed] [Google Scholar]
- Kim JH, Roy S, Kellis JT, Poulose AJ, Gast AP, Robertson CR. Langmuir. 2002;18:6312. [Google Scholar]
- Kusnezow W, Syagailo YV, Goychuk I, Hoheisel JD, Wild DG. Expert Rev Mol Diagn. 2006;6:111. doi: 10.1586/14737159.6.1.111. [DOI] [PubMed] [Google Scholar]
- Lee AG, Arena CP, Beebe DJ, Palecek SP. Biomacromolecules. 2010;11:3316. doi: 10.1021/bm100792y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Wark AW, Goodrich TT, Fang SP, Corn RM. Langmuir. 2005;21:4050. doi: 10.1021/la046822h. [DOI] [PubMed] [Google Scholar]
- Lee JH, Cosgrove BD, Lauffenburger DA, Han J. Journal of the American Chemical Society. 2009;131:10340. doi: 10.1021/ja902594f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski K, Warzocha K, Hellmann A, Skotnicki A, Prejzner W, Foryciarz K, Sacha T, Gniot M, Majewski M, Solarska I, Nowak G, Wasag B, Kobelski M, Scibiorski C, Siemiatkowski M, Lewandowska M, Komarnicki M. Pol Arch Med Wewn. 2009;119:789. [PubMed] [Google Scholar]
- Liotta LA, Espina V, Mehta AI, Calvert V, Rosenblatt K, Geho D, Munson PJ, Young L, Wulfkuhle J, Petricoin EF. Cancer Cell. 2003;3:317. doi: 10.1016/s1535-6108(03)00086-2. [DOI] [PubMed] [Google Scholar]
- Lozinsky VI, Galaev IY, Plieva FM, Savinal IN, Jungvid H, Mattiasson B. Trends Biotechnol. 2003;21:445. doi: 10.1016/j.tibtech.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Okay O. Prog Polym Sci. 2000;25:711. [Google Scholar]
- Ross JS, Fletcher JA. The oncologist. 1998;3:237. [PubMed] [Google Scholar]
- Rubina AY, Dementieva EI, Stomakhin AA, Darii EL, Pan'kov SV, Barsky VE, Ivanov SM, Konovalova EV, Mirzabekov AD. Biotechniques. 2003;34:1008. doi: 10.2144/03345rr01. [DOI] [PubMed] [Google Scholar]
- Schnaar RL, Lee YC. Biochemistry. 1975;14:1535. doi: 10.1021/bi00678a030. [DOI] [PubMed] [Google Scholar]
- Sigurdson M, Wang D, Meinhart CD. Lab Chip. 2005;5:1366. doi: 10.1039/b508224b. [DOI] [PubMed] [Google Scholar]
- Sung WC, Chen HH, Makamba H, Chen SH. Anal Chem. 2009;81:7967. doi: 10.1021/ac901138w. [DOI] [PubMed] [Google Scholar]
- Walker GM, Beebe DJ. Lab Chip. 2002;2:131. doi: 10.1039/b202473j. [DOI] [PubMed] [Google Scholar]
- Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Nat Rev Cancer. 2007;7:345. doi: 10.1038/nrc2126. [DOI] [PubMed] [Google Scholar]
- White D, Saunders V, Lyons AB, Branford S, Grigg A, To LB, Hughes T. Blood. 2005;106:2520. doi: 10.1182/blood-2005-03-1103. [DOI] [PubMed] [Google Scholar]
- Wu YH, Park HB, Kai T, Freeman BD, Kalika DS. J Membr Sci. 2010;347:197. [Google Scholar]
- Yadavalli VK, Koh WG, Lazur GJ, Pishko MV. Sens Actuator B-Chem. 2004;97:290. [Google Scholar]
- Zubtsov DA, Savvateeva EN, Rubina AY, Pan'kov SV, Konovalova EV, Moiseeva OV, Chechetkin VR, Zasedatelev AS. Anal Biochem. 2007;368:205. doi: 10.1016/j.ab.2007.04.040. [DOI] [PubMed] [Google Scholar]