

Abstract
Pten, the potent tumor suppressor, is a lipid phosphatase that is best known as a regulator of cell proliferation and cell survival. Here we show that mouse embryos that lack Pten have a striking set of morphogenetic defects, including the failure to correctly specify the anterior-posterior body axis, that are not caused by changes in proliferation or cell death. The majority of Pten null embryos express markers of the primitive streak at ectopic locations around the embryonic circumference, rather than at a single site at the posterior of the embryo. Epiblast-specific deletion shows that Pten is not required in the cells of the primitive streak; instead, Pten is required for normal migration of cells of the Anterior Visceral Endoderm (AVE), an extraembryonic organizer that controls the position of the streak. Cells of the wild-type AVE migrate within the visceral endoderm epithelium from the distal tip of the embryo to a position adjacent to the extraembryonic region. In all Pten null mutants, AVE cells move a reduced distance and disperse in random directions, instead of moving as a coordinated group to the anterior of the embryo. Aberrant AVE migration is associated with the formation of ectopic F-actin foci, which indicates absence of Pten disrupts the actin-based migration of these cells. After the initiation of gastrulation, embryos that lack Pten in the epiblast show defects in the migration of mesoderm and/or endoderm. The findings suggest that Pten has an essential and general role in the control of mammalian collective cell migration.
Introduction
Phosphoinositides are important regulators of membrane localization of proteins, trafficking, polarity and signaling, whose roles in development are only beginning to be understood (Skwarek and Boulianne, 2009). Pten (phosphatase and tensin homologue on chromosome 10) is an important regulator of phosphoinositides that converts phosphoinositol-3,4,5 tri-phosphate (PIP3) into phosphatidylinositol (4,5) bisphosphate (PIP2). PIP3 anchors a number of important signaling proteins to the plasma membrane to promote proliferation, cell survival, increased cell size and epithelial polarity (Manning and Cantley, 2007). Pten is a classic tumor suppressor: individuals that inherit one mutant allele of Pten show spontaneous benign tumors and a predisposition to malignant tumors, along with developmental defects that include macrocephaly (Waite and Eng, 2002). After p53, somatic mutations in Pten are the second most common genetic lesion in human cancers (Yin and Shen, 2008; Parsons, 2004; Chalhoub and Baker, 2009). The majority of studies on Pten in cancer have focused on its role in the Akt-mTor-S6K pathway, which regulates translation and cell growth and is an important target for tumor therapy (Manning and Cantley, 2007; Sabatini, 2006). Most studies on the roles of Pten in development in Drosophila and C. elegans have focused on its roles in the insulin receptor/Akt pathway to control cell size, dauer formation and longevity (Ogg and Ruvkun, 1998; Stocker and Hafen, 2000).
Pten also has other cellular functions that are likely to play important roles in development and tumorigenesis. Studies in Dictyostelium amoebae defined the importance of enrichment of PIP3 at the leading edge for the directional movement of individual migrating cells. PIP3 recruits WASP, WAVE and several PH-domain proteins to the leading edge of the cell (Myers et al., 2005; Meili et al., 1999; Oikawa et al., 2004; Padrick and Rosen, 2010). Pten, which degrades PIP3, becomes localized to the trailing edge of these cells; this enhances the gradient of PIP3 within the cell and is required for directional migration (Iijima and Devreotes, 2002). Pten appears to have similar functions in mammalian hematopoietic cells: Pten is localized to the trailing edge of migrating mammalian neutrophils (Wu et al., 2004; Li et al., 2005), and loss of Pten in neutrophils and B cells disrupts polarized migration and the ability to respond to chemoattractants (Heit et al., 2008; Anzelon et al., 2003). Conditional deletion experiments in the mouse have revealed complex roles for Pten in the developing brain, including providing structural support for neuronal migration in the developing cerebellum (Yue et al., 2005; Endersby and Baker, 2008).
Null mutations in Pten cause embryonic lethality in the mouse, but the cellular and developmental bases of lethality have not been defined. Embryos homozygous for two different targeted null mutations were reported to die prior to e7.5 with defects in epiblast organization and differentiation (Di Cristofano et al., 1998; Podsypanina et al., 1999), whereas embryos homozygous for a third null allele in a different genetic background survived to e9.5, with defects in chorio-allantoic fusion and formation of the cranial neural folds (Stambolic et al., 1998; Suzuki et al., 1998). Subsequent studies showed that the differences between the phenotypes were due to differences in genetic background and that the outbred strain background CD1 allowed longer survival (Cully et al., 2004; Freeman et al., 2006). Here we use null and conditional alleles in the CD1 background to define the function of Pten in early embryogenesis. Pten mutants display an array of morphogenetic defects, including broad neural folds, cardia bifida and aberrant mesoderm migration. In contrast to expectation, we find that cell proliferation and cell death are normal in embryos that lack Pten, indicating that the developmental phenotypes must reflect other functions of Pten.
The most dramatic defect in Pten mutant embryos is disruption of the anterior-posterior body axis in the majority of mutant embryos. We trace this phenotype to defects in the anterior visceral endoderm (AVE), a group of extra-embryonic cells that is required for the establishment of the anterior-posterior axis. AVE cells originate at the distal tip of the e5.5 embryo and migrate along one side of the embryo to the edge of the extraembryonic region between e5.5 and e6.0 (Rivera-Perez et al., 2003; Srinivas et al., 2004; Migeotte et al., 2010; Takaoka et al., 2011). Cells of the AVE express inhibitors of Wnt and Nodal signaling that are required to restrict the primitive streak to a single site on the opposite, posterior side of the embryo, thereby establishing the anterior-posterior body axis (Perea-Gomez et al., 2002; Yamamoto et al., 2004). High-resolution imaging showed that the leading wild-type AVE cells extend long lamellar protrusions that span several cell diameters and are polarized in the direction of cell movement (Srinivas et al., 2004; Migeotte et al., 2010). AVE cells move in an actin-mediated process that requires the WAVE complex and Rac1 (Rakeman and Anderson, 2006; Migeotte et al., 2010). Wild-type AVE cells display hallmarks of collective cell migration: they retain tight and adherens junctions as they migrate and exchange neighbors within the plane of the visceral endoderm epithelium (Migeotte et al., 2010; Trichas et al., 2011; Takaoka et al., 2011).
We find that the defect in axis specification in Pten mutant embryos is caused by abnormal behavior of AVE cells. Pten mutant AVE cells initiate movement, but fail to migrate as far as wild-type cells. In addition, Pten−/− AVE cells disperse in random directions rather than migrating as a group towards the future anterior of the embryo, and the abnormal final position of the AVE cells accounts for the defects in axis specification. We identify additional defects in cell migration after gastrulation, which suggest Pten has a general role in collective cell migration in embryonic tissues.
Materials and Methods
Mouse strains and genotyping
Mice carrying the targeted Ptentm1PPP null mutation (Di Cristofano et al., 1998), the floxed allele of Pten (Ptenflox) (Trotman et al., 2003), the ENU-induced point mutation of Pten (PtenM1un) (García-García et al., 2005), and the BAT-gal (Maretto et al., 2003), Hex-GFP (Rodriguez et al., 2001) and Sox2-Cre (Hayashi et al., 2002) transgenes were crossed into the outbred CD1 strain background (>8 generations). The KJB5 (TCGCCTTCTTGACGAGTTCT) and KJB6 (GCTCCAATCCTTCCATTCAA) primer pairs were used to detect the targeted Pten null allele. The 93F (CTGGGATTATCTTTTTGCAACAGT) and 405R (ACAGCTTACCTTTTTGTCTCTGGT) primers detect the wild-type allele of Pten but not the targeted allele. PCR amplification using primers 83F (TCTTGACCAATGGCTAAGTG) and M1unBgl-IIR (GCCCCGATGCAATAAATATGCACAGATC) create a Bgl-II restriction site in the PtenM1un allele. PCR amplification using primers M1unAse1F (TCACTGTAAAGCTGGAAAGGGACGGACTGGATTAA) and 405R creates an Ase1 restriction site in the wild-type allele of Pten, but not the PtenM1un allele. Primers for detecting the floxed allele of Pten (Ptenflox) and the conditionally deleted (Ptencko) were previously published (Trotman et al., 2003). Primers and genotyping protocols to detect DNA containing Cre, LacZ and GFP sequences were obtained from the Jackson Laboratories (www.jax.org).
Phenotypic analysis
Standard procedures were used for in situ hybridizations, LacZ staining, and immunofluorescent staining of cryosections (Eggenschwiler and Anderson, 2000). Embryos for SEM were fixed in 2.5% glutaraldehyde overnight at 4°C, processed using standard procedures and imaged with a Zeiss Supra 25 Field Emission Scanning Electron Microscope. 1mg BrdU per 10g of mouse body weight was injected into pregnant females, and after a one-hour incubation, embryos were dissected and processed for anti-BrdU staining.
Immunohistochemistry
The antibodies used were: rabbit or chicken anti-GFP (Molecular probes, 1:500); rabbit anti-laminin (Sigma, 1:200); rat anti-E-cadherin (Sigma, 1:500); rabbit anti-active caspase-3 (Abcam, 1:500); rabbit anti-phospho-histone H3 (Ser28) (Cell signaling, 1:300); rabbit anti-phospho-Akt (Ser473) (Cell signaling, 1:50); mouse anti-Pten (Cascade, clone 6H2.1; 1:500); and mouse anti-BrdU (Sigma, 1:10). Alexa-conjugated secondary antibodies were obtained from Invitrogen. An anti-mouse biotinylated, streptavidin-Alexa-488 (Vector, cat. No BA-9200) amplification step was used to visualize BrdU localization. F-actin was visualized using TRITC-Phalloidin (Molecular probes, 1:50–1:200). Nuclei were visualized using DAPI (Sigma, 1mg/ml).
Imaging and Image analysis
Whole mount embryos after in situ hybridization or LacZ staining were imaged using a Zeiss Axiocam HRC digital camera on a Leica MZFLIII microscope. Immunofluorescence images (whole mount and sections) were captured with an inverted Leica TCS SP2 confocal microscope. Confocal stacks were taken at a 1024×1024 resolution with 20x and 63x magnification. Identical camera and microscope settings were used to image wild-type and mutant embryos. Confocal stacks were reconstructed with Volocity software (Improvision).
The software program Amira (Mercury Computer Systems) was used to reconstruct confocal stacks and count the number of Hex-GFP cells in wild-type and Pten mutant embryos. The software program ImageJ were used to count number of cells and determine the position of AVE cells along the circumference and along the proximal-distal axis of the embryo. Snapshots of 3D reconstructions from confocal stacks (Volocity, Improvision) were taken as the reconstruction was rotated 90 degrees around the proximal-distal axis. Using the cell counter plug-in (K. De Bos) within ImageJ, the position of Hex-GFP cells was manually marked in wild-type and Pten mutant embryos. Reference points along the proximal-distal axis were marked at the most distal and proximal positions on the embryo. The measure function within the cell counter plug-in was used to extract coordinates (X, Y) of the Hex-GFP cells and reference points. These numbers were imported into Excel (Microsoft) and used to determine the vertical distance Hex-GFP cells had migrated from the most distal point in the embryo, relative to the overall length of the embryo. Because each image represented a quadrant of the embryo, the number of Hex-GFP cells in each quadrant was also extracted from these data and used along with the position of each cell along the X-axis to determine the distribution of cells around the circumference of the embryo. Bar graphs and polar plots were created in excel (Microsoft). For the polar plots (θ r), θ represents the position of individual Hex-GFP+ cells around the circumference and r represents relative vertical distance the Hex-GFP+ cell traveled.
Statistical analysis was carried out using an unpaired two-tailed Students’s t-test with unequal variance.
Results
Pten null embryos die at midgestation with a set of defects in embryonic patterning and morphogenesis
We identified a missense allele of Pten, PtenM1un, in an ENU mutagenesis screen, based on interesting defects in morphogenesis in homozygous mutant embryos (García-García et al., 2005). We therefore decided to define the embryonic phenotype caused by complete absence of Pten. In the original 129/C57BL/6 mixed genetic background, the null mutation Ptentm1PPP caused recessive peri-implantation lethality (Di Cristofano et al., 1998), but we found that homozygous null mutants survived to midgestation in the CD1 background, when they showed clear morphological defects. The most striking aspects of the null phenotype were the abnormal headfolds (Fig. 1B, E), as previously noted (Stambolic et al., 1998; Suzuki et al., 1998), and incompletely penetrant duplication of the anterior-posterior body axis (Fig. 1C, F; see below). These phenotypes were also seen in PtenM1un homozygotes, although at lower penetrance (Fig. S1).
Figure 1. Morphological phenotypes of e8.5 Pten mutant embryos.
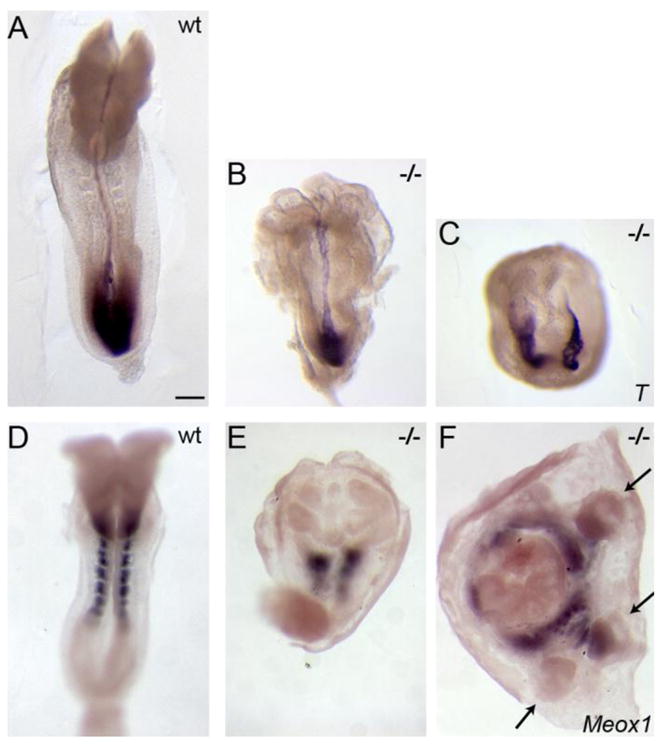
(A–C) In situ hybridization of e8.5 embryos to show the pattern of expression of Brachyury (T), which marks the primitive streak and midline, and (D–F) Meox1, which is expressed in the paraxial mesoderm. The majority of Pten null embryos show ruffled, accordion-like neural folds (B) and abnormal paraxial mesoderm (E). (C, F). At this stage, ~20% of Pten null embryos display an ectopic anterior-posterior axis; arrows in F indicate ectopic allantoides, suggesting a triplication of the posterior body axis. Anterior is up. Scale bar = 275μm.
Proliferation and cell death occur at normal rates in Pten mutant embryos
Previous studies using conditional alleles to delete Pten in specific mouse tissues have been able to account for many of the observed phenotypes on the basis of increased proliferation, increased cell size or decreased cell death (Knobbe et al., 2008). In the early embryo, it was suggested that increased proliferation in Pten null mutant embryos could be responsible for the abnormal morphology of the headfolds (Stambolic et al., 1998). At e5.75 when the anterior-posterior asymmetry is initiated, we did not observe a significant difference in the number of mitotic cells in wild type and null mutants, as assayed by staining for phospho-histone H3 (Ser28) (pHH3) (Fig. 2A, D, Table S1). At later stages, we compared wild-type and Pten-epiblast deleted embryos, where Pten was removed specifically in derivatives of the epiblast, and found no difference at e7.5 or e8.5 in either the fraction of mitotic cells (Fig. 2B, E; Fig. S2A, B) or the number of BrdU labeled cells after a one hour pulse (total number of cells scored >4000 for each genotype) (Fig. S2C, D; Table S1). As an independent method to assess whether there was a difference in proliferation or death in the Pten mutants that could account for the changes in neural morphology, we counted the total number of Sox2+ neuroepithelial cells in the cranial neural folds of two wild-type and two Pten−/− mutant embryos at the 5 somite stage, when the phenotype was clear and embryos were healthy. We found indistinguishable numbers of nuclei in the cranial neuroepithelium of wild-type and Pten−/− embryos between the posterior of the heart and the anterior end of the embryo (wild type: 12,156 and 13,799; Pten−/− 10,940 and 13,142 nuclei).
Fig. 2. Proliferation and cell death in early Pten mutant embryos.

Confocal slices of immunostained wild-type (A–C) and Pten mutant embryos (D–F), using anti-GFP (green) to mark the location of the Hex-GFP transgene. Wild-type (A) and Pten−/− (D) e5.75 embryos stained for anti-phospho-histoneH3 (magenta) to highlight cells in mitosis; phalloidin (red) and DAPI (blue) mark F-actin and nuclei. Wild-type (B) and Pten epiblast deleted (D) e7.5 embryos stained for anti-phospho-histoneH3 (magenta); phalloidin (red) and DAPI (blue). Wild-type (B) and Pten−/− (E) e6.0 embryos stained for active-caspase3 antibody (red) to label cells undergoing apoptosis. Scale bars = 20 μm.
Loss of Pten has been shown to cause inappropriate cell survival in many contexts; for example, we observed that the cell death in Rac1 mutant embryos was largely rescued by reducing the gene dosage of Pten (Migeotte et al., 2011). We assayed cell death in wild-type, Pten null e6.0 embryos, and Pten-epiblast deleted e8.5 embryos by staining for activated capase-3 (Fig. 2C, F; Fig. S2E, F). There is very little cell death in wild-type embryos at these early stages, and no decrease in cell death was detected in the Pten mutants. Increase in the size of cells and nuclei has been seen in Pten−/− cells in the brain (Kwon et al., 2001), but we did not see a significant difference in nuclear size in Pten mutant embryos (J. G.-B. and J. B. unpublished). Thus, Pten is not required for normal proliferation or cell death through midgestation of the mouse embryo and the phenotypes of Pten mutants instead reflect other essential functions of Pten.
Pten is required for normal specification of the anterior-posterior body axis
To define the functions of Pten in the early embryo, we investigated the earliest and most dramatic phenotype observed in Pten−/− embryos, the duplication of the anterior-posterior body axis in a fraction of the mutant embryos (Fig. 1C,F), which was apparent from the morphology of the mutants and by expression of molecular markers. Brachyury, a marker of the primitive streak at the posterior of the embryo and of the midline, was ectopically expressed in more than half of e7.5 Pten−/− and PtenM1un/M1un mutant embryos examined (8/14; Fig. 3A, B). At e8.5, only ~20% of Pten−/− mutant embryos (4/21) displayed an ectopic primitive streak (Fig. 1C). A similar fraction of e8.5 embryos showed ectopic expression of the paraxial mesoderm marker Meox1 (Fig. 1E, F). The lower penetrance of the axis specification phenotype at e8.5 than at e7.5 did not appear to be due to embryo death, as the ratio of e8.5 Pten−/− mutants to total embryos (21/66, 30%) was close to the expected Mendelian ratio. Instead, it is likely that regulatory mechanisms partially corrected the early defects in axis specification, as has been described for other mutations (e.g. Otx2; Perea-Gomez et al., 2001). The axis defects in e8.5 Pten−/− embryos ranged from a small ectopic spot of Brachyury expression, in addition to a wild-type primitive streak, to a complete duplication or triplication of posterior primitive streak derivatives such as the allantois (Fig. 1F, arrows) and embryos with two parallel primitive streaks (Fig. 1C).
Figure 3. Pten controls specification of the anterior-posterior body axis.

(A–B) In situ hybridization for Brachyury (T), which marks the primitive streak; viewed from the posterior. 50% of e7.5 Pten−/− embryos (B) show ectopic expression of T. (C, D) In situ hybridization for the expression of Wnt3 at e6.5, lateral view, indicates that Wnt3 is expressed ectopically in Pten−/− embryos. Arrow indicates position of a cluster of mis-positioned anterior visceral endoderm (AVE) cells near the distal tip of this Pten−/− embryo at e6.5. (E, F) Expression of the BAT-gal reporter of canonical Wnt signaling at e7.5, lateral view, is mislocalized in Pten−/− embryos (4/8). In situ hybridization for Foxa2 (G. H) and Lefty2 (I, J), posterior views, show that Lefty2 and Foxa2 are also expressed ectopically in Pten−/− embryos (3/6 and 3/6 respectively). Bmp4 expression is unchanged in e6.5 Pten−/− embryos (7/7) (K, L). Scale bar = 250 μm.
Other primitive streak markers were also expressed ectopically in the mutant embryos. Wnt3 is the earliest marker of the primitive streak (Liu et al., 1999; Rivera-Perez et al., 2003). Wnt3 was expressed in a ring around proximal portion of the majority of Pten−/− mutant embryos at e6.5 (n=2/3), instead of being correctly restricted to the proximal posterior side of the embryo (Fig. 3C, D). The BAT-gal transgene is a reporter for canonical Wnt signaling that is expressed in the primitive streak (Maretto et al., 2003). BAT-gal activity was found in a proximal ring in half of the e7.5 Pten−/− embryos examined (4/8), rather than the wild-type pattern along the posterior side of the embryo (Fig. 3E, F). Additional streak markers, Lefty2 and Foxa2, were expressed in a ring around the proximal region of ~50% of e7.0–7.5 Pten−/− mutant embryos (Lefty2 (3/6); Foxa2 (3/6)) (Fig. 3G–J). These results demonstrate that the activity of Pten is important for specification of a single primitive streak.
Pten appears to act in extraembryonic cells to control axis specification
To determine whether Pten acts in embryonic or in extra-embryonic tissues to control axis specification, we deleted a conditional Pten allele using the Sox2-Cre transgene, which deletes the floxed wild-type allele in all derivatives of the epiblast, but not in the extraembryonic lineages (Hayashi et al., 2002). Both the Sox2-Cre transgene and a conditional allele of Pten (Trotman et al., 2003) were made congenic in the CD1 background. We then carried out crosses to generate Sox2-Cre; Ptenflox/− embryos, which we refer as Pten epiblast deleted embryos. Pten epiblast deleted embryos arrested at approximately e9.0, when all Pten epiblast deleted embryos had abnormal headfolds (Fig. 4B) and cardia bifida (Fig. 4C). These embryos showed normal expression of primitive streak markers at the posterior of the embryo, normal migration of the AVE, as assayed by the anterior position of Cer1 expression (Fig. 4A) and always had a single anterior-posterior body axis (Fig. 4B). Thus Pten is required in extraembryonic, rather than embryonic, tissues for axis specification.
Figure 4. Conditional deletion of Pten in the epiblast shows that Pten acts in extraembryonic cells to control axis specification.

(A) The primitive streak marker T was expressed correctly in a single primitive streak in all Pten-epiblast deleted (Δepi) embryos examined (13/13), as was the anterior streak marker FoxA2. Scale bars = 250 μm. (B) The morphology of e8.5 Pten-epiblast deleted embryos, viewed from the dorsal side. All embryos of this genotype form a single anterior-posterior body axis (n>50). Like the wild-type embryo on the left, the three mutants (on the right) form somites and close the neural tube in the trunk, but have headfolds that fold in irregular patterns. Scale bar =250 μm. (C) SEM views of the ventral side of wild-type and Pten-epiblast deleted e8.5 embryos. In contrast to the single, looping heart tube in wild type, the mutant heart is composed of two tubes that have not fused on the midline (cardia bifida). Scale bar = 100 μm. (D) Pten protein localization in e6.5 wild-type and Pten-epiblast deleted embryos. Arrows indicate the epiblast; arrowheads indicate extraembryonic ectoderm. Pten is detectable in the epiblast, visceral endoderm and extraembryonic ectoderm of wild-type embryos (8/8). Pten protein is not detectable in the epiblast of Pten-epiblast deleted embryos (3/3), although it is still detectable in the extraembryonic ectoderm and visceral endoderm. Scale bar = 50μm.
Two extraembryonic cell types, the visceral endoderm and the extraembryonic ectoderm, play a role of AVE migration (Rivera-Perez et al., 2003; Srinivas et al., 2004; Rodriguez et al., 2005) and we find that Pten is expressed in both these cell types in wild-type embryos (Fig. 4D). Phosphorylated-Akt (S473), a target of the Pten phosphatase, was present high levels in both mutant visceral endoderm and extraembryonic ectoderm of mutant embryos (Fig. 5K–M). The only factor known to be important in the extraembryonic ectoderm for proper AVE specification and migration is Bmp4 (Rodriguez et al., 2005; Soares et al., 2008). Bmp4 was expressed normally in Pten−/− mutant embryos (Fig. 3K, L), which suggested that Pten is required autonomously in the other extraembryonic lineage, the visceral endoderm, for migration of the AVE. Consistent the action of Pten in the visceral endoderm, Phosphorylated-Akt is tethered to the membrane by a PIP3-binding PH domain and was associated with the membrane on all faces of all visceral endoderm cells in Pten−/− mutant embryos (Fig. 5K–M), demonstrating that Pten is required in the visceral endoderm to prevent high levels of membrane PIP3.
Figure 5. AVE cells remain distal or are evenly dispersed over the embryonic region in Pten null embryos.

(A–D) e6.5 embryos, anterior to the left. Cer1, a marker of the AVE, is expressed on the anterior of the wild-type embryo (A). In 37% of Pten−/−mutant embryos (11/30) (B), most Cer1-expressing cells remain near the distal tip of the embryo. In an additional 13% of Pten−/−mutant embryos (4/30) Cer1 expression appears to cover most of the embryonic region (C). Cer1 is expressed in the anterior of Pten-epiblast deleted embryos (D; 4/4 embryos). (E–G) Hex-GFP expression (stained with anti-GFP antibody (green); DAPI is blue) in e6.5 embryos, anterior to the left. In wild-type (E), Hex-expressing cells form a contiguous group on the anterior, proximal side of the embryo, while Hex-expressing cells remain distal in Pten−/− mutants (F) or are scattered over the embryonic region (G), similar to the Cer1 expression pattern. (H–M) Staining for phospho-Akt (S473) (magenta) in wild-type (H–J) and Pten−/− (K–M) e6.0 embryos. Phospho-Akt was detected at a high level at the membrane of visceral endoderm cells in Pten−/− embryos, but was not detected above background in the membrane of visceral endoderm cells in the embryonic region of wild-type embryos. The Hex-GFP+ cells are near the distal tip of the mutant embryo, but have migrated toward the anterior in wild type at this stage. (I–J, L–M) p-Akt signal only; high magnification views in J and M. Scale bars in A–G, H–I and K–L = 100μm.
Pten is required for the normal migration of AVE cells
In addition to its roles in the control of cell proliferation, death and size, Pten is important in polarized migration of Dictyostelium amoebae and in mammalian hematopoietic cells (Iijima and Devreotes, 2002; Wu et al., 2004; Li et al., 2005). We therefore asked whether Pten was important for the migration of the anterior visceral endoderm (AVE), the extra-embryonic organizer that defines the anterior-posterior axis of the animal (Perea-Gomez et al., 2002). Cer1, a marker of the AVE, is normally expressed along the anterior side of the embryo at e6.5, up to the embryonic/extraembryonic boundary (Fig. 5A). In contrast, Cer1 was expressed at the distal tip or around the circumference of the visceral endoderm in half the Pten−/−mutant embryos (Fig. 5B, C).
The transcription factor Hex is expressed in AVE cells, and a Hex-GFP transgene marks AVE cells with cellular resolution (Srinivas et al., 2004). There were ~50 Hex-GFP+ cells at e5.75 in both Pten−/− and wild-type embryos (n=2 for both wild-type and mutant), demonstrating that loss of Pten did not affect the specification or number of AVE cells. Hex-GFP expressing cells form a coherent group on the anterior side of the wild-type embryo (Fig. 5E). In contrast, Hex-GFP cells were abnormally distributed in all Pten−/− embryos. In some embryos, the Hex-expressing cells were located near the distal tip (Fig. 5F); in other embryos the Hex+ cells were distributed across the embryonic region (Fig. 5G). Thus Pten is required for the normal migration of the cells of the AVE.
Pten mutant AVE cells disperse in nearly random directions from their distal origin
To define how Pten affected behavior of the AVE cells, we carried out quantitative analysis of the position of Hex-GFP+ cells from 3D confocal reconstructions of whole mount wild-type, heterozygous and homozygous mutants at e5.75 and e6.5 (e.g. Fig. 6A–D). Pten−/− Hex-GFP+ cells did not migrate as far from the distal tip of the embryo as wild-type cells. Graphical representations of the position of the AVE cells, shown in Fig. 6E–H, indicate that while wild-type cells moved away from the distal tip of the embryo (the center of the polar plot), many mutant cells at both e5.75 (Fig. 6F) and e6.5 (Fig. 6H) remained near their origin (the center of the plot). At both e5.75 and e6.5, Hex-GFP expressing cells in Pten−/− embryos had moved, on average, only about 60% as far away from the distal tip as in wild-type embryos (Table S2; Fig. S3).
Figure 6. Pten mutant Hex+ AVE cells are dispersed around the embryo.

(A–B) 3D reconstructions of e5.75 embryos during AVE migration, showing expression of Hex-GFP (green, staining with anti-GFP antibody) and F-actin (phalloidin, red). White line marks the embryonic/extraembryonic boundary. (A) Wild type, a left/anterior view. Migration is in progress and Hex+ cells have not yet reached the boundary of the embryonic region. (B) The left and right sides a Pten−/− embryo. AVE cells are more dispersed in the mutant embryo. The leading cells have arrived at the embryonic/extraembryonic border, but many cells remain distally located. Scale bar for A and B = 40μm. (C, D) 3D reconstructions of e6.5 embryos after AVE migration, showing expression of Hex-GFP (green, staining with anti-GFP antibody) and E-cadherin (magenta). (C) Anterior view of wild-type; there are no AVE cells in the back (not shown) or at the distal tip of the embryo. (D) Two views of a Pten−/−embryo; posterior view to the left and anterior view on the right. AVE cells are present all sides of the embryo, and numerous cells remain distally located. (C) Scale bar for C and D = 50μm. (E–H) Polar plots representing the distribution of AVE cells in embryos like those in (A–D). Each dot represents the position of one Hex+ cell. Plots are oriented so the most populated quadrant (the presumptive anterior) is oriented to the left. The position of each cell is indicated both with respect to its position around the circumference of the embryo (angle) and distance migrated along the proximal-distal axis (proximity to center of the plot). Data from 4 wild-type embryos (+/+) at e5.75 (E) and 4 wild-type embryos (+/+) at e6.5 (G) reveal that most Hex+ cells are in the 3 anterior quadrants and have moved away from the distal tip. Data from 3 Pten−/− mutant embryos at e5.75 (F) and 5 Pten−/− mutant embryos at e6.5 (H) indicate that cells are more evenly distributed among the quadrants and many more remain near the distal tip. Heterozygous embryos were excluded from this analysis, as they appear to have an intermediate behavior between +/+ and Pten−/− embryos (Supp. Table S2).
In addition to incomplete migration, Hex-GFP expressing cells were distributed more evenly around the circumference of Pten−/− embryos compared to the restriction to the anterior seen in wild type (Fig. 6A–D). To quantitate the change in distribution of AVE cells, we divided 3D images of whole mount embryos into quadrants and counted the Hex-GFP expressing cells in each quadrant (Fig. 6E–H; Table S2). At both e5.75 and e6.5, only 4–5% of Hex-GFP+ cells were in the least-populated (posterior) quadrant of wild type. In contrast, 16–17% of the Hex-GFP+ cells were in the least-populated quadrant of Pten−/− mutants; this was close to the 25% expected if the cells had dispersed randomly, in contrast to the oriented migration of wild-type AVE cells. Consistent with the haploinsufficiency of Pten in other contexts (e.g. Trotman et al., 2003; Blando et al., 2009), the behaviors of Pten−/+ heterozygous AVE cells were intermediate between wild-type and homozygous mutant cells (Table S2).
Ectopic apical actin accumulates in cells of the Pten visceral endoderm
Because active rearrangements of the actin cytoskeleton are required for AVE migration (Rakeman and Anderson, 2006), we examined the organization of F-actin in the 3D reconstructions of early embryos. In the visceral endoderm of e6.5 wild-type embryos, F-actin was enriched at cell-cell boundaries and at vertices where multiple cells met (Fig. 7C). In Pten−/− mutant embryos, there was an increase in the staining of F-actin in the middle of apical surface in both AVE cells and in other cells of the visceral endoderm (Figure 7A, D, E). Ectopic puncta of apical actin were already visible in visceral endoderm at the time of AVE migration in Pten−/− embryos (Fig. S4; Fig. 6A–B). It is likely that this defect in actin organization in the visceral endoderm could disrupt the migration of Pten−/− AVE cells. Our results are consistent with the ectopic F-actin seen in zebrafish ptenb morphants (Yeh et al., 2011), although the cellular and developmental defects seen in zebrafish embryos are different from those described here in the mouse.
Figure 7. Abnormal distribution of F-actin in the visceral endoderm of mutant embryos.
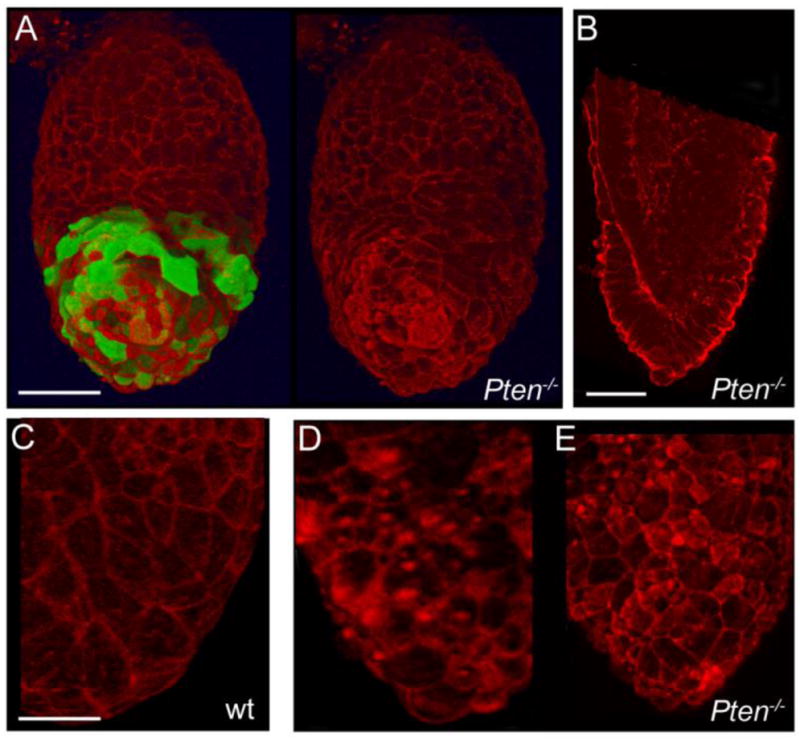
e6.5 embryos stained with rhodamine-conjugated phalloidin (red) to reveal the distribution of F-actin. (A) Anterior view of a 3D reconstruction of a Pten−/− embryo, with and without the green channel for Hex-GFP. Dense F-actin is present at the apical surface of some VE cells, including both Hex+ and Hex− cells. Scale bar = 50μm. (B) An optical section through another embryo shows the columnar morphology of cells in the distal region of the visceral endoderm and strong apical F-actin staining. Scale bar = 50μm. (C–E) High magnification images from 3D reconstructions, showing the difference in the F-actin distribution in wild-type (C) and two representative mutant embryos (D, E). Many mutant VE cells show a high level of apical F-actin, including strong foci of F-actin. Scale bar in C–E = 100μm.
Pten-epiblast deleted embryos show additional defects in cell migration
After establishment of the position of the primitive streak, cells from the wild-type epiblast undergo an epithelial-to-mesenchymal transition (EMT) at the streak to generate the mesoderm and definitive endoderm. In about one-third of e7.5 Pten−/− and Pten-epiblast deleted mutant embryos, a group of cells from the primitive streak protruded into the amniotic cavity (5/15, Fig. 8A–C), similar to the phenotype of mutants with defects in the EMT or mesoderm migration (García-García and Anderson, 2003; Rakeman and Anderson, 2006; Sun et al., 1999). Transverse sections of Pten−/− and Pten epiblast deleted embryos at e7.5 stained with anti-E-cadherin and anti-Laminin (Fig. 8D–F) showed that E-cadherin was properly down regulated in the mutant mesoderm wings, indicating that this aspect of the EMT was not disrupted in Pten mutant embryos. After ingression through the primitive streak, nascent mesoderm cells migrate around the circumference of the embryo. Pten epiblast deleted embryos developed somites (Fig. 4B), demonstrating that mesoderm migration and somitogenesis could proceed in the absence of Pten. However, sections of those Pten epiblast deleted embryos in which cells accumulated in the amniotic cavity showed an accumulation of cells in the mesodermal wings near the primitive streak (Fig. 8D–F). Thus Pten was important for efficient migration of mesoderm cells away from the primitive streak. Additionally, most Pten epiblast deleted embryos showed clear cardia bifida (8/10; Fig. 4C), the failure of the two laterally positioned anlage of the heart to move to the ventral midline. Defects in either migration of the definitive foregut endoderm or the nascent cardiac mesoderm can lead to cardia bifida (Constam and Robertson, 2000; Saga et al., 1999). These findings suggest that Pten has a general role in cell migration during dynamic tissue rearrangements.
Figure 8. Mesoderm migration defects in some Pten epiblast-deleted embryos.

(AC) Bright field views of e7.5 embryos. Cells appear to accumulate in the amniotic cavity in some Pten−/− (B) and Pten-epiblast deleted (C) embryos. Scale bar in A–C = 240μm. (D–F) Transverse sections of e7.5 wild-type (D), Pten−/− (E) and Pten epiblast-deleted (F) embryos, stained for laminin (green), E- cadherin (red), and DAPI (blue). Arrowheads indicate the location of the primitive streak. E-cadherin down-regulation is normal, indicating that the EMT is normal. Arrows point to mesodermal cells; in contrast to the thin mesodermal wings that extend around the circumference of the embryo in wild-type, mesodermal cells accumulate near the primitive streak in the mutants, indicating that a defect in mesoderm migration is responsible for the accumulation of cells seen in (B and C). Scale bar in D–F = 40μm.
Discussion
Pten was originally studied for its roles in promoting proliferation and preventing apoptosis, which are important for its function as a tumor suppressor. In contrast, we find that Pten null mouse embryos show a syndrome of developmental defects that are independent of the roles of Pten in proliferation and apoptosis. Our results show that the absence of Pten leads to striking defects in embryonic morphogenesis because of its roles in collective cell migration, a role that could contribute to Pten-dependent tumor progression.
Pten is required for collective migration of the Anterior Visceral Endoderm (AVE)
Although the embryonic lethality of Pten mouse mutants was described previously (Di Cristofano et al., 1998; Podsypanina et al., 1999; Suzuki et al., 1998; Wang et al., 2010), the essential role of Pten in the specification of the anterior-posterior axis has not been recognized. This is due, at least in part, to the sensitivity of the Pten mutant phenotype to genetic background. As we show, an additional complication is that some of the null embryos with early defects in axis specification recover to form a single primitive streak at e8.5. Studies in chick have uncovered a negative feedback loop, in which the primitive streak is able to inhibit ectopic primitive streak formation (Bertocchini et al., 2004), which suggests a developmental mechanism that would allow embryos to recover from the specification of ectopic primitive streaks.
Although not all Pten−/− embryos show defects in the expression of primitive streak markers, all Pten null mutants show disruptions in the migration of the extraembryonic organizer, the AVE, as assayed by the distribution of Hex-positive cells. Some Pten−/− AVE cells fail to migrate at all, while others migrate in apparently randomized directions, so that AVE cells are dispersed to all sides of the embryo, rather than migrating as a coherent group along one side of the embryo.
The obligate relationship between AVE migration and axis specification has been documented in a number of different mutants in which the AVE does not move, including Otx2−/− (Perea-Gomez et al., 2001), Cripto−/− (Ding et al., 1998), β-catenin−/− (Huelsken et al., 2000) and Rac1−/− (Migeotte et al., 2010). In these mutants, AVE cells are specified but never move from the distal tip of the embryo; as a result cells with the identity of primitive streak cells are found in a ring around the proximal end of the embryo instead of along the presumptive posterior side of the embryo.
The variable defects in axis specification seen in Pten mutants are more similar to the phenotypes of mutants that only partially disrupt movement of the AVE. For example, in Nap1khlo/khlo mutant embryos, AVE cells do not migrate all the way to the presumptive extraembryonic border and some mutant embryos have more than one site of expression of primitive streak markers (Rakeman and Anderson, 2006). In Cer1−/−; Lefty1−/− double mutants the AVE has a broad irregular domain and the embryos can show ectopic primitive streaks (Perea-Gomez et al., 2002). Similarly, in Pten−/− mutants, AVE cells migrate some distance from the distal tip of the embryo, which is sufficient to allow specification of a single primitive streak in some embryos but leads to ectopic expression of streak markers in other mutants.
Our genetic experiments show that Pten is required in either the visceral endoderm or the extraembryonic ectoderm for AVE migration. Previous studies have shown that the extraembryonic ectoderm can influence the migration of the AVE: one group used embryological manipulations to show that signals from the extraembryonic ectoderm inhibit migration of the AVE (Rodriguez et al., 2005) and another group found that RNAi knockdown of Bmp4 blocked AVE cell migration (Soares et al., 2005). Because Bmp4 expression is normal in Pten mutants, it is possible that Pten might either influence Bmp4 signaling downstream of the ligand through a novel mechanism. It is also possible that Pten acts in another pathway in the extraembryonic ectoderm to control production of a factor that diffuses to visceral endoderm cells and influences their migration. We show that Pten is expressed and active in the visceral endoderm; given the nature of the cellular defects that we observe in the visceral endoderm, we think it is most likely that Pten acts cell autonomously in the visceral endoderm to promote AVE migration.
The defects in specification of the body axis seen in the Pten null mutants are similar to, but distinct from, the phenotypes we have seen previously in mutants that lack Rac1 or the WAVE complex regulator Nap1 (Migeotte et al., 2010; Rakeman and Anderson, 2006), and there are direct biochemical links among these proteins. Rac1 and Nap1 mutant AVE cells fail to move or do not migrate as far as wild-type AVE cells, whereas Pten mutant cells disperse in nearly random directions from their origin at the distal tip of the embryo. Rac1 and PIP3 act in concert at the leading edge of migrating cells to activate the WAVE complex and promote the formation of the branched actin structures that push the membrane forward (Oikawa et al., 2004; Padrick and Rosen, 2010). The plasma membranes of all faces of Pten mutant visceral endoderm cells have high level of PIP3, as shown by the high level of membrane-associated phospho-Akt. This high level of PIP3 is likely to lead to activity of the WAVE complex at all positions on the cell membrane, which would account for the ectopic F-actin puncta that we observe in the apical region of visceral endoderm cells. Although Pten mutant AVE cells do move some distance from their origin, these ectopic actin networks are likely to interfere with the oriented migration of AVE cells.
The defect in directional movement of Pten mutant AVE cells is remarkably similar to the phenotype of Dictyostelium amoebae that lack Pten (Iijima and Devreotes, 2002). Dictyostelium pten mutant cells are stimulated to move by chemoattractant, but they move in more random directions, with the net effect that they do not move as far as wild-type cells. Similarly, Pten mutant AVE cells can sense a developmental cue that stimulates their movement, but they do not move as far and they disperse instead of moving in a coordinated direction. The phenotype of mammalian hematopoietic cells that lack Pten appears to be more complex; it has been argued that SHIP1 is the principal PIP3 phosphatase that permits movement of neutrophils towards chemoattractants (Nishio et al., 2007) and that Pten is only important in neutrophil chemotaxis when cells need to prioritize multiple chemoattractants (Heit et al., 2008).
A general role for Pten in collective cell migration
Recent work has shown that the movement of the AVE is a collective migration, where epithelial organization is retained as cells migrate directionally while changing neighbors (Migeotte et al., 2010; Trichas et al., 2011). Pten has been shown to be important for directed migration of single cells, as in Dictyostelium amoebae and mammalian hematopoietic cells. Our findings demonstrate that Pten is also critical for polarized migration of non-hematopoietic mammalian cells, and, more surprisingly, that Pten is required for directional collective cell migration within an epithelial sheet. Thus in cells migrating collectively, as in cells migrating as individuals, migration is inefficient and uncoordinated in absence of Pten.
Our studies with the Pten epiblast deleted embryos show that Pten is also required after gastrulation for normal mesoderm migration. In contrast to previous results where overexpression of Pten disrupted the gastrulation EMT in the chick (Leslie et al., 2007), the gastrulation EMT appeared to be normal in Pten−/− and Pten epiblast deleted embryos. However, we observe incompletely penetrant defects in mesoderm migration, and completely penetrant defects in formation of a single heart tube, which depends on the migration of the definitive endoderm and cardiac mesoderm. These findings are consistent with experiments in the chick in which expression of truncated forms of Pten disrupted the directional migration of mesoderm away from the primitive streak (Leslie et al., 2007). We therefore conclude that Pten is important for the efficient migration of mesoderm away from the primitive streak. Although nascent mesoderm cells are mesenchymal, they retain junctions between cells and move in a type of collective migration (Winklbauer et al., 1992; Yang et al., 2008). Thus our findings suggest a general role for Pten in collective cell movement.
Because collective cell migration mediates the rearrangement of many tissues during development (Aman and Piotrowski, 2010), Pten is likely to have a broad role in tissue morphogenesis throughout development. In addition, it will be interesting to test whether Pten−/− cells in solid tumors show a similar tendency to disperse, a cellular behavior that could contribute to the invasiveness of Pten mutant tumors.
Supplementary Material
Expression of Brachyury (T) in mutants that showed aberrant axis specification. (A) 6% of PtenM1un/M1un embryos (1/17) expressed T in a ring at the embryonic/extraembryonic boundary; the remainder showed normal posterior T expression. (B, C) Two examples of e8.5 PtenM1un/M1un embryos with ectopic T expression (arrows).
Transverse sections of e8.5 wild-type (A, C, E) and Pten-epiblast deleted mutant (B, D, F) embryos stained for pHH3 (A,B), Anti-BrdU labels cells in S-phase (C, D), or anti-Active-caspase3 (E, F) labels cells undergoing apoptosis. No difference in the mitotic index or the proliferation index was detected at e8.5 in the neuroepithelium (see Table S1). The rate of apoptosis was low in both wild-type and mutant embryos. Scale bar = 100 μm.
The data from Supp. Table 2 are presented as the distance that the Hex-GFP expressing cells migrated along the proximal-distal axis, as a fraction of total embryo length (including the extraembryonic region) in wild-type (+/+), Pten−/+ heterozygotes and Pten−/− homozygotes at e5.75 and e6.5.
The distribution of F-actin at e5.75 in wild-type (A) and Pten−/− mutants (B). These images are the phalloidin (red)-only channels from Figures 6A and B. Most cells in the Pten−/− mutant visceral endoderm (A) show puncta of actin on the apical surface of the apical surface of the cell; arrows point to examples. These puncta are not apparent in wild-type embryos (B).
Highlights.
Pten mutant mouse embryos frequently show duplication of the anterior-posterior body axis
Pten is required in extraembryonic, rather than embryonic, tissues for axis specification
Pten is required for collective migration of the anterior visceral endoderm (AVE)
Pten mutant AVE cells disperse in nearly random directions
Pten mutants show ectopic apical foci of F-actin in cells of the visceral endoderm
Acknowledgments
We thank Heather Alcorn for technical assistance and P.-P. Pandolfi for Pten mutant mice. We thank the MSKCC Molecular Cytology and SEM Core facilities. We thank Nitya Ramkumar, Jamie Mahaffey and Hisham Bazzi for helpful comments on the manuscript. IM was supported by a Starr Foundation fellowship, and an FNRS post-doctoral fellowship. JGB was supported by a long-term EMBO post-doctoral fellowship. The work was supported by National Institutes of Health (NIH) grants HD035455 to KVA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aman A, Piotrowski T. Cell migration during morphogenesis. Dev Biol. 2010;341:20–33. doi: 10.1016/j.ydbio.2009.11.014. [DOI] [PubMed] [Google Scholar]
- Anzelon AN, Wu H, Rickert RC. Pten inactivation alters peripheral B lymphocyte fate and reconstitutes CD19 function. Nat Immunol. 2003;4:287–94. doi: 10.1038/ni892. [DOI] [PubMed] [Google Scholar]
- Bertocchini F, Skromne I, Wolpert L, Stern CD. Determination of embryonic polarity in a regulative system: evidence for endogenous inhibitors acting sequentially during primitive streak formation in the chick embryo. Development. 2004;131:3381–3390. doi: 10.1242/dev.01178. [DOI] [PubMed] [Google Scholar]
- Blando J, Portis M, Benavides F, Alexander A, Mills G, Dave B, Conti CJ, Kim J, Walker CL. PTEN deficiency is fully penetrant for prostate adenocarcinoma in C57BL/6 mice via mTOR-dependent growth. Am J Pathol. 2009;174:1869–79. doi: 10.2353/ajpath.2009.080055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–50. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constam DB, Robertson EJ. Tissue-specific requirements for the proprotein convertase furin/SPC1 during embryonic turning and heart looping. Development. 2000;127:245–54. doi: 10.1242/dev.127.2.245. [DOI] [PubMed] [Google Scholar]
- Cully M, Elia A, Ong SH, Stambolic V, Pawson T, Tsao MS, Mak TW. grb2 heterozygosity rescues embryonic lethality but not tumorigenesis in pten+/− mice. Proc Natl Acad Sci USA. 2004;101:15358–15363. doi: 10.1073/pnas.0406613101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Ding J, Yang L, Yan YT, Chen A, Desai N, Wynshaw-Boris A, Shen MM. Cripto is required for correct orientation of the anterior-posterior axis in the mouse embryo. Nature. 1998;395:702–707. doi: 10.1038/27215. [DOI] [PubMed] [Google Scholar]
- Eggenschwiler JT, Anderson KV. Dorsal and lateral fates in the mouse neural tube require the cell autonomous activity of the open brain gene. Dev Biol. 2000;227:648–660. doi: 10.1006/dbio.2000.9918. [DOI] [PubMed] [Google Scholar]
- Endersby R, Baker SJ. PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene. 2008;27:5416–30. doi: 10.1038/onc.2008.239. [DOI] [PubMed] [Google Scholar]
- Freeman D, Lesche R, Kertesz N, Wang S, Li G, Gao J, Groszer M, Martinez-Diaz H, Rozengurt N, Thomas G, Liu X, Wu H. Genetic background controls tumor development in PTEN-deficient mice. Cancer Res. 2006;66:6492–6496. doi: 10.1158/0008-5472.CAN-05-4143. [DOI] [PubMed] [Google Scholar]
- García-García MJ, Anderson KV. Essential role of glycosaminoglycans in Fgf signaling during mouse gastrulation. Cell. 2003;114:727–737. doi: 10.1016/s0092-8674(03)00715-3. [DOI] [PubMed] [Google Scholar]
- García-García MJ, Eggenschwiler JT, Caspary T, Alcorn HL, Wyler MR, Huangfu D, Rakeman AS, Lee JD, Feinberg EH, Timmer JR, Anderson KV. Analysis of mouse embryonic patterning and morphogenesis by forward genetics. Proc Natl Acad Sci USA. 2005;102:5913–5919. doi: 10.1073/pnas.0501071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, Lewis P, Pevny L, McMahon AP. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech Dev. 2002;119(Suppl 1):S97–S101. doi: 10.1016/s0925-4773(03)00099-6. [DOI] [PubMed] [Google Scholar]
- Heit B, Robbins SM, Downey CM, Guan Z, Colarusso P, Miller BJ, Jirik FR, Kubes P. PTEN functions to ‘prioritize’ chemotactic cues and prevent ‘distraction’ in migrating neutrophils. Nat Immunol. 2008;9:743–52. doi: 10.1038/ni.1623. [DOI] [PubMed] [Google Scholar]
- Huelsken J, Vogel R, Brinkmann V, Erdmann B, Birchmeier C, Birchmeier W. Requirement for β-catenin in anterior-posterior axis formation in mice. J Cell Biol. 2000;148:567–578. doi: 10.1083/jcb.148.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima M, Devreotes P. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 2002;109:599–610. doi: 10.1016/s0092-8674(02)00745-6. [DOI] [PubMed] [Google Scholar]
- Knobbe CB, Lapin V, Suzuki A, Mak TW. The roles of PTEN in development, physiology and tumorigenesis in mouse models: a tissue-by-tissue survey. Oncogene. 2008;27:5398–5415. doi: 10.1038/onc.2008.238. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29:404–411. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- Leslie NR, Yang X, Downes CP, Weijer CJ. PtdIns(3,4,5)P(3)-dependent and -independent roles for PTEN in the control of cell migration. Curr Biol. 2007;17:115–125. doi: 10.1016/j.cub.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, Tang L, Hla T, Zeng R, Li L, Wu D. Regulation of PTEN by Rho small GTPases. Nat Cell Biol. 2005;7:399–404. doi: 10.1038/ncb1236. [DOI] [PubMed] [Google Scholar]
- Liu P, Wakamiya M, Shea MJ, Albrecht U, Behringer RR, Bradley A. Requirement for Wnt3 in vertebrate axis formation. Nat Genet. 1999;22:361–365. doi: 10.1038/11932. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, Hassan AB, Volpin D, Bressan GM, Piccolo S. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci USA. 2003;100:3299–3304. doi: 10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meili R, Ellsworth C, Lee S, Reddy TB, Ma H, Firtel RA. Chemoattractant-mediated transient activation and membrane localization of Akt/PKB is required for efficient chemotaxis to cAMP in Dictyostelium. Embo J. 1999;18:2092–105. doi: 10.1093/emboj/18.8.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migeotte I, Omelchenko T, Hall A, Anderson KV. Rac1-dependent collective cell migration is required for specification of the anterior-posterior body axis of the mouse. PLoS Biol. 2010;8:e1000442. doi: 10.1371/journal.pbio.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migeotte I, Grego-Bessa J, Anderson KV. Rac1 mediates morphogenetic responses to intercellular signals in the gastrulating mouse embryo. Development. 2011;138:3011–3020. doi: 10.1242/dev.059766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio M, Watanabe K, Sasaki J, Taya C, Takasuga S, Iizuka R, Balla T, Yamazaki M, Watanabe H, Itoh R, Kuroda S, Horie Y, Förster I, Mak TW, Yonekawa H, Penninger JM, Kanaho Y, Suzuki A, Sasaki T. Control of cell polarity and motility by the PtdIns(3,4,5)P3 phosphatase SHIP1. Nat Cell Biol. 2007;9:36–44. doi: 10.1038/ncb1515. [DOI] [PubMed] [Google Scholar]
- Myers SA, Han JW, Lee Y, Firtel RA, Chung CY. A Dictyostelium homologue of WASP is required for polarized F-actin assembly during chemotaxis. Mol Biol Cell. 2005;16:2191–206. doi: 10.1091/mbc.E04-09-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg S, Ruvkun G. The C. elegans PTEN homolog, DAF-18, acts in the insulin receptor-like metabolic signaling pathway. Mol Cell. 1998;2:887–893. doi: 10.1016/s1097-2765(00)80303-2. [DOI] [PubMed] [Google Scholar]
- Oikawa T, Yamaguchi H, Itoh T, Kato M, Ijuin T, Yamazaki D, Suetsugu S, Takenawa T. PtdIns(3,4,5)P3 binding is necessary for WAVE2-induced formation of lamellipodia. Nat Cell Biol. 2004;6:420–426. doi: 10.1038/ncb1125. [DOI] [PubMed] [Google Scholar]
- Padrick SB, Rosen MK. Physical mechanisms of signal integration by WASP family proteins. Annu Rev Biochem. 2010;79:707–735. doi: 10.1146/annurev.biochem.77.060407.135452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons R. Human cancer, PTEN and the PI-3 kinase pathway. Semin Cell Dev Biol. 2004;15:171–176. doi: 10.1016/j.semcdb.2003.12.021. [DOI] [PubMed] [Google Scholar]
- Perea-Gomez A, Lawson KA, Rhinn M, Zakin L, Brulet P, Mazan S, Ang SL. Otx2 is required for visceral endoderm movement and for the restriction of posterior signals in the epiblast of the mouse embryo. Development. 2001;128:753–765. doi: 10.1242/dev.128.5.753. [DOI] [PubMed] [Google Scholar]
- Perea-Gomez A, Vella FD, Shawlot W, Oulad-Abdelghani M, Chazaud C, Meno C, Pfister V, Chen L, Robertson E, Hamada H, Behringer RR, Ang SL. Nodal antagonists in the anterior visceral endoderm prevent the formation of multiple primitive streaks. Dev Cell. 2002;3:745–756. doi: 10.1016/s1534-5807(02)00321-0. [DOI] [PubMed] [Google Scholar]
- Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci USA. 1999;96:1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakeman AS, Anderson KV. Axis specification and morphogenesis in the mouse embryo require Nap1, a regulator of WAVE-mediated actin branching. Development. 2006;133:3075–3083. doi: 10.1242/dev.02473. [DOI] [PubMed] [Google Scholar]
- Rivera-Perez JA, Mager J, Magnuson T. Dynamic morphogenetic events characterize the mouse visceral endoderm. Dev Biol. 2003;261:470–487. doi: 10.1016/s0012-1606(03)00302-6. [DOI] [PubMed] [Google Scholar]
- Rodriguez TA, Casey ES, Harland RM, Smith JC, Beddington RS. Distinct enhancer elements control Hex expression during gastrulation and early organogenesis. Dev Biol. 2001;234:304–16. doi: 10.1006/dbio.2001.0265. [DOI] [PubMed] [Google Scholar]
- Rodriguez TA, Srinivas S, Clements MP, Smith JC, Beddington RS. Induction and migration of the anterior visceral endoderm is regulated by the extra-embryonic ectoderm. Development. 2005;132:2513–2520. doi: 10.1242/dev.01847. [DOI] [PubMed] [Google Scholar]
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–34. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- Saga Y, Miyagawa-Tomita S, Takagi A, Kitajima S, Miyazaki J, Inoue T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development. 1999;126:3437–47. doi: 10.1242/dev.126.15.3437. [DOI] [PubMed] [Google Scholar]
- Skwarek LC, Boulianne GL. Great expectations for PIP: phosphoinositides as regulators of signaling during development and disease. Dev Cell. 2009;16:12–20. doi: 10.1016/j.devcel.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Soares ML, Torres-Padilla ME, Zernicka-Goetz M. Bone morphogenetic protein 4 signaling regulates development of the anterior visceral endoderm in the mouse embryo. Dev Growth Differ. 2008;50:615–621. doi: 10.1111/j.1440-169X.2008.01059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Rodriguez T, Clements M, Smith JC, Beddington RS. Active cell migration drives the unilateral movements of the anterior visceral endoderm. Development. 2004;131:1157–1164. doi: 10.1242/dev.01005. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Stocker H, Hafen E. Genetic control of cell size. Curr Opin Genet Dev. 2000;10:529–535. doi: 10.1016/s0959-437x(00)00123-4. [DOI] [PubMed] [Google Scholar]
- Sun X, Meyers EN, Lewandoski M, Martin GR. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev. 1999b;13:1834–1846. doi: 10.1101/gad.13.14.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Barco Barrantes I, Ho A, Wakeham A, Itie A, Khoo W, Fukumoto M, Mak TW. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- Takaoka K, Yamamoto M, Hamada H. Origin and role of distal visceral endoderm, a group of cells that determines anterior-posterior polarity of the mouse embryo. Nat Cell Biol. 2011;13:743–52. doi: 10.1038/ncb2251. [DOI] [PubMed] [Google Scholar]
- Trichas G, Joyce B, Crompton LA, Wilkins V, Clements M, Tada M, Rodriguez TA, Srinivas S. Nodal dependent differential localisation of dishevelled-2 demarcates regions of differing cell behaviour in the visceral endoderm. PLoS Biol. 2011;9:e1001019. doi: 10.1371/journal.pbio.1001019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van Dyke T, Cordon-Cardo C, Pandolfi PP. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite KA, Eng C. Protean PTEN: form and function. Am J Hum Genet. 2002;70:829–44. doi: 10.1086/340026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Karikomi M, Naidu S, Rajmohan R, Caserta E, Chen HZ, Rawahneh M, Moffitt J, Stephens JA, Fernandez SA, Weinstein M, Wang D, Sadee W, La Perle K, Stromberg P, Rosol TJ, Eng C, Ostrowski MC, Leone Gl. Allele-specific tumor spectrum in pten knockin mice. Proc Natl Acad Sci U S A. 2010;107:5142–7. doi: 10.1073/pnas.0912524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winklbauer R, Selchow A, Nagel M, Angres B. Cell interaction and its role in mesoderm cell migration during Xenopus gastrulation. Dev Dyn. 1992;195:290–302. doi: 10.1002/aja.1001950407. [DOI] [PubMed] [Google Scholar]
- Wu Y, Hannigan MO, Kotlyarov A, Gaestel M, Wu D, Huang CK. A requirement of MAPKAPK2 in the uropod localization of PTEN during FMLP-induced neutrophil chemotaxis. Biochem Biophys Res Commun. 2004;316:666–72. doi: 10.1016/j.bbrc.2004.02.107. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Saijoh Y, Perea-Gomez A, Shawlot W, Behringer RR, Ang SL, Hamada H, Meno C. Nodal antagonists regulate formation of the anteroposterior axis of the mouse embryo. Nature. 2004;428:387–392. doi: 10.1038/nature02418. [DOI] [PubMed] [Google Scholar]
- Yang X, Chrisman H, Weijer CJ. PDGF signalling controls the migration of mesoderm cells during chick gastrulation by regulating N-cadherin expression. Development. 2008;135:3521–30. doi: 10.1242/dev.023416. [DOI] [PubMed] [Google Scholar]
- Yeh CM, Liu YC, Chang CJ, Lai SL, Hsiao CD, Lee SJ. Ptenb mediates gastrulation cell movements via Cdc42/AKT1 in zebrafish. PLoS One. 2011;6:e18702. doi: 10.1371/journal.pone.0018702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27:5443–53. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- Yue Q, Groszer M, Gil JS, Berk AJ, Messing A, Wu H, Liu X. PTEN deletion in Bergmann glia leads to premature differentiation and affects laminar organization. Development. 2005;132:3281–91. doi: 10.1242/dev.01891. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression of Brachyury (T) in mutants that showed aberrant axis specification. (A) 6% of PtenM1un/M1un embryos (1/17) expressed T in a ring at the embryonic/extraembryonic boundary; the remainder showed normal posterior T expression. (B, C) Two examples of e8.5 PtenM1un/M1un embryos with ectopic T expression (arrows).
Transverse sections of e8.5 wild-type (A, C, E) and Pten-epiblast deleted mutant (B, D, F) embryos stained for pHH3 (A,B), Anti-BrdU labels cells in S-phase (C, D), or anti-Active-caspase3 (E, F) labels cells undergoing apoptosis. No difference in the mitotic index or the proliferation index was detected at e8.5 in the neuroepithelium (see Table S1). The rate of apoptosis was low in both wild-type and mutant embryos. Scale bar = 100 μm.
The data from Supp. Table 2 are presented as the distance that the Hex-GFP expressing cells migrated along the proximal-distal axis, as a fraction of total embryo length (including the extraembryonic region) in wild-type (+/+), Pten−/+ heterozygotes and Pten−/− homozygotes at e5.75 and e6.5.
The distribution of F-actin at e5.75 in wild-type (A) and Pten−/− mutants (B). These images are the phalloidin (red)-only channels from Figures 6A and B. Most cells in the Pten−/− mutant visceral endoderm (A) show puncta of actin on the apical surface of the apical surface of the cell; arrows point to examples. These puncta are not apparent in wild-type embryos (B).