


Abstract
The ribosomal stalk in bacteria is composed of four or six copies of L12 proteins arranged in dimers that bind to the adjacent sites on protein L10, spanning 10 amino acids each from the L10 C-terminus. To study why multiple L12 dimers are required on the ribosome, we created a chromosomally engineered Escherichia coli strain, JE105, in which the peripheral L12 dimer binding site was deleted. Thus JE105 harbors ribosomes with only a single L12 dimer. Compared to MG1655, the parental strain with two L12 dimers, JE105 showed significant growth defect suggesting suboptimal function of the ribosomes with one L12 dimer. When tested in a cell-free reconstituted transcription–translation assay the synthesis of a full-length protein, firefly luciferase, was notably slower with JE105 70S ribosomes and 50S subunits. Further, in vitro analysis by fast kinetics revealed that single L12 dimer ribosomes from JE105 are defective in two major steps of translation, namely initiation and elongation involving translational GTPases IF2 and EF-G. Varying number of L12 dimers on the ribosome can be a mechanism in bacteria for modulating the rate of translation in response to growth condition.
INTRODUCTION
The ribosomal ‘stalk’ is a finger-like protrusion on the large ribosomal subunit that constitutes one of the main interaction sites for the translation factors. The stalk is highly flexible and composed of four or six copies of L12 proteins arranged as dimers on the protein L10 (1,2). The L10 protein binds to ribosomal RNA (rRNA) (nucleotides 1030–1124 in Escherichia coli) next to the protein L11 at the base of the stalk (3,4). L12 is the only multicopy protein on the ribosome (5), its number varying from species to species (1). In E. coli, two L12 dimers and one L10 protein form a strong pentameric complex known as the L8 complex (6). L12 also exists in an N-terminally acetylated form called L7, thus often referred as L7/L12. Functional importance of this modification is not well understood, but thought to be linked with the stability of the L12 dimers on L10 (7). For convenience, we will refer to L7/L12 as only L12 in this article. In contrast to many ribosomal proteins L12 is highly acidic in nature, with a pI of 4.9 (8). In E. coli, an L12 monomer is composed of 120 amino acids with an approximate molecular mass of 12 kDa.
L12 proteins form strong dimers not only on the ribosome but also in solution. The dimerization involves the N-terminal domains (NTDs) composed of residues 1–37. In a L12 monomer, this domain is folded into two α-helices arranged in a hairpin conformation (9–12). The NTD continues to a flexible hinge region containing residues 38–49, which can change its conformation from a compact helix to an extended structure (12,13). The flexible nature of the hinge is important for the function of L12, especially for the mobility and varied localization of the globular C-terminal domain (CTD) during translation factor interaction (1,14). The CTD of L12 is highly conserved and consists of three α-helices and three β-sheets corresponding to residues 50–120 (12,14,15). It provides sites for interaction with the GTPase factors (16,17).
The mode of dimerization of the L12 proteins has been debated for a long time. The crystal structure of a truncated tetrameric L12 complex from Thermotoga maritima favored a ‘parallel dimerization’ mode where two adjacent L12 molecules both with compact helical hinges were proposed to form the core dimer (12). However, this model failed to justify the strong dimer interaction in L12 dimer, had no functional relevance and was contradicted by an NMR structure where both the hinges were seen in fully extended form (9,11). Thus it is now universally accepted that the L12 dimer is ‘antiparallel’ where NTDs of two L12 molecules form a four-helix bundle dimer occupying the same site on L10. This model is supported by the X-ray crystal structure of the L10–(L12 NTD)6 complex from T. maritima (1). It has been proposed (13) and later evidenced with FRET (18) that on the ribosome L12 dimer can exist with one hinge compact and the other extended. Although thought to be important for translation factor recruitment, physiological relevance of such conformation is not fully understood. Recent crystal structure of EF-G bound to the ribosome shows one interacting L12 molecule with an extended hinge segment (19). Thus flexibility and dynamics of the hinge is important for interaction of L12 CTD with the translation factors.
One unique feature of the L12 protein is that, unlike other ribosomal proteins, it makes no direct contact with the rRNA. L12 dimers link to the rRNA via the L10 protein. It was shown earlier with a plasmid-based construct that none of the L12 dimers could bind to the E. coli ribosome when 20 or more amino acids were deleted from the C terminus of L10. Furthermore, only one L12 dimer could bind if the last 10 amino acids were truncated from L10 (referred hereafter as L10Δ10) (20). Later, the high-resolution crystal structure of a complex containing L10 and 6 L12-NTDs from T. maritima identified the α8-helix at the C-terminus of protein L10 to be the site for the attachment of the L12 dimers (1). From this structure and the L10 truncation experiments, it was concluded that the L12 dimer binding sites at the CTD of L10 spanned 10 amino acids and although located adjacent to each other, they had different amino acid sequences.
Protein L12 plays a key role in protein synthesis. It has been suggested that the L12 dimers are required for optimal rates and low error frequency in protein synthesis (21,22). These proteins are known to be necessary for stimulation of GTPase activity of the translation factors, particularly EF-Tu and EF-G (23–25). In addition, a role of L12 CTDs in translation factor recruitment has recently been proposed (1). In the absence of L12, the binding of the elongation factors to the ribosome can be severely impaired (14,26). During initiation, L12 is important for the recognition of IF2·GTP on the 30S pre-initiation complex resulting in fast subunit association, although L12 does not play the role of GTPase activating protein (GAP) for IF2 (27).
One unanswered question is why multiple L12 dimers are present on the bacterial ribosome. Although it has been shown that the ribosome with a single L12 dimer bound to the protein L10Δ10 is active in protein synthesis in a poly-Phe system, the implications of such a genetic construct on the growth rate of the bacteria is not known. It is hard to imagine that a ribosome with multiple L12 dimers would have evolved if the ribosome with single L12 dimer was capable of carrying out all the steps of protein synthesis with equal efficiency and accuracy. In this report we describe the construction and characterization of an E. coli strain JE105 that lacks the peripheral L12 binding site on L10 and thereby produces homogeneous ribosomes with a L10Δ10-single L12 dimer. As expected, JE105 showed significant growth defect. Further, we investigated the reason for such compromised growth by testing the single L12 dimer ribosomes from JE105 in different assays in vitro. This study attempts to pinpoint in which translational steps multiple L12 dimers are required on the bacterial ribosome.
MATERIALS AND METHODS
Construction of the E. coli strain JE105
The last 33 nucleotides of the rplJ gene of E. coli MG1655 were replaced at its chromosomal locus with an ampicillin-resistance cassette (Amp) by means of lambda red recombineering (28,29) (Figure 1a). The Amp cassette was produced by PCR amplification from the plasmid PBR322 with primers 5′ACCATGAAAGAAGCTTCGGCTGGCAAACTGGTTCGTACTCTGtaatcgcagttatctttttaacgcattcgcttacgtataaactta3′ and 5′TAAGTTTATACGTAAGCGAATGCGTTAAAAAGATAACTGCGAttaagcagcttctttcgcatcacgaactgctgcagcagcttcttt3′ having 45 nucleotide homologies to the flanking sequences of the target region (shown in uppercase letters). Recombinants were selected on LB agar-amp (100 µg ml−1) plates and were further screened by PCR using forward primers upL10 – 5′tatccaggcctccgtcgaaga3′, and in Amp – 5 ′tgctgataaatctggagccg3′ in combination with the reverse primer downL12 – 5′tgtgaaacgctactggcgcct3′ (Figure 1b). One of the successful recombinants, named JE105 [MG1655 (rplJΔ30)-Amp], was used for this study. The replacement was confirmed by DNA sequencing.
Figure 1.

Construction and characterization of JE105 strain. (a) A scheme showing the steps for construction of JE105, where last 33 nucleotides of rplJ gene in the chromosome of E. coli MG1655 was replaced with an Amp cassette by means of lambda Red recombineering (left). While MG1655 ribosome has two L12 dimers bound to L10, JE105 ribosome lacked the peripheral L12 dimer due to deletion of the binding site (II) (right). (b) Agarose gel electrophoresis showing bands from colony PCR with MG1655 (lanes 3 and 5) and JE105 (lanes 2 and 4) using primers In Amp and Down L12 (lanes 2 and 3) and Up L10 and Down L12 (lanes 4 and 5). (c) The growth curves for MG1655 (black) and JE105 (red) in LB at 37°C. The generation times are listed in the box.
Growth-rate measurements
The growth rate of JE105 was compared with the parental strain MG1655 in LB medium at 37°C using a Bioscreen C analyzer (Figure 1c). Overnight cultures of JE105 and MG1655 were diluted ~1000 times in LB medium and measured in quadruplet during each experiment. The increase in absorbance at 595 nm as a function of time was used to calculate the generation times.
Purification and characterization of JE105 ribosomes, quantification of the L12 proteins
Tight coupled ribosomes (70S), and ribosomal subunits (50S and 30S) were prepared from the strains MG1655 (wild-type parental strain), MRE600 (commonly used lab strain for translation assays) and JE105 (L10Δ10-single L12 dimer) following protocols described in Johansson et al. (30). The composition of the ribosomes was checked by electrophoresis in a composite native gel at a low Mg2+ concentration (Figure 2a), in SDS–PAGE (only L8 complex) (Figure 2b) followed by quantitative Western blotting (Figure 2c), and also in a 2D-gel (Figure 2d).
Figure 2.

Characterization of single L12 dimer ribosomes from JE105. (a) Native composite gel electrophoresis with MG1655 and JE105 ribosomes. (b) SDS–PAGE analysis of the isolated L8 complex from MG1655 and JE105 ribosomes. (c) Quantitative Western blot with cell lysate and total ribosomal proteins from MG1655 and JE105, using antibodies against L12 and S1 proteins. Pure L12 protein was used as a control. (d) Comparison of the ribosomal proteins from JE105 and MG1655 (in the box) in 2D-gel electrophoresis. The L12 spots are labeled with the arrow.
The composite native gel containing 0.7% agarose and 2% polyacrylamide was prepared following the protocol of Nadano et al. (31), where 5 pmol of 70S ribosome from MG1655 and JE105 were subjected to electrophoresis for 2 hrs at 150 V. The gel was stained with Coomassie brilliant blue to detect the ribosomal subunits based on the ribosomal proteins (Figure 2a). L8 complex was isolated from the ribosomes by NH4Cl and ethanol wash. The isolated L8 complex together with 20 pmol of pure L12 protein were applied to a 14% SDS–PAGE and the migration of the L10 and L12 proteins was followed by Coomassie staining (Figure 2b). The cell lysate and the total ribosomal proteins from MG1655 and JE105 run on a similar SDS–PAGE were transferred onto a nitrocellulose membrane and blotted with antibodies against L12 (gift from Prof. J.P. Ballesta, Universidad Autonoma de Madrid, Spain) and S1 (as internal control). The relative quantity of the S1 and L12 proteins in each sample was determined by estimating the pixel ratio with Image Quant (GE Healthcare). JE105 ribosomes (10 pmol) were further analyzed in a 2D-gel and compared with those from MG1655 following the procedure of Geyl et al. (32). The stoichiometry of L12 proteins in these two ribosomes was quantified using L23 and L24 spots as internal standards.
Components for in vitro translation assays
All experiments were performed at 37°C in HEPES polymix buffer (pH 7.5) containing 5 mM NH4Cl, 5 mM Mg(OAc)2, 100 mM KCl, 0.5 mM CaCl2, 8 mM putrescine, 1 mM spermidine, 15 mM HEPES, 1 mM ATP and 1 mM DTT (33). All translation components were from E. coli. The XR7 MLL mRNA and fMet-tRNAfMet were prepared using standard lab protocols (27,34). Translation factors IF1, IF2, IF3, EF-Tu, EF-Ts and EF-G (in-house laboratory clones, all His-tagged) were overexpressed in E. coli and purified using affinity chromatography on a Ni-IMAC column (GE Healthcare).
Ribosome activity in full-length protein synthesis in vitro
The expression vector pET30-Ek/LIC-luc was constructed by PCR amplification of firefly luciferase gene (1650 bp) from the vector pBESTluc™ (Promega) with primers 5′-GACGACGACAAGATGGAAGACGCCAAAAACATAAAG-3′ and 5′-GAGGAGAAGCCCGGTTACAATTTGGACTTTCCGCCC followed by ligation into pET30 Ek/LIC (Novagen) according to the user manual. For comparison of the activity of the MG1655, MRE600 and JE105 ribosomes in their rate of synthesis of a full-length protein, 1 µM of 70S ribosomes or 1 µM of each of 50S and 30S subunit of the respective strains was added to a cell-free reconstituted transcription–translation system containing the pET30-Ek/LIC-luc plasmid (40 ng µl−1) and all other transcription and translation components such as T7-RNA polymerase, all translation factors (1–2 µM) and energy pump components (PEP, Phosphocreatine, MK and PK). Luciferase synthesis was followed over time in a GloMax® 20/20 luminometer (Promega) by analyzing 2 µl of the reaction mixture with luciferase assay mixture (Promega).
Subunit association
Two mixtures A and B were prepared in HEPES-polymix. Mixture A contained either 0.5 µM 30S alone or together with 2 µM each of mRNA (MLL), fMet-tRNAfMet, initiation factors IF1 and IF2 and 100 µM GTP. Mixture B contained 0.5–2.5 µM of 50S from MG1655, MRE600 or JE105. Both mixtures were incubated at 37°C for 5 min and rapidly mixed in a stopped-flow instrument (Applied Photophysics). The extent of 70S formation was monitored by the increase in Rayleigh light scattering at 425 nm (35). The rate constants were derived as in (27,34).
Single-round Pi release
The release of inorganic phosphate (Pi) due to GTP hydrolysis was monitored in a stopped-flow instrument by following instantaneous increase of MDCC fluorescence at 464 nm (λEx = 425 nm) upon Pi binding to MDCC-labeled phosphate binding protein (PBP-MDCC) (a kind gift from Prof. Martin Webb, MRC National Institute for Medical Research, London, UK) (Ka of Pi binding >500 µM−1 s−1) (36). To remove the pre-accumulated phosphates, a phosphate mop containing 0.1 U ml−1 of purine nucleoside phosphorylase, and 500 µM u7-methylguanosine were added to both mixtures.
During IF2-mediated subunit association, Pi release was measured in a setup similar to that for the subunit association, with 5 µM MDCC-PBP added to the 50S mix. For measuring GTP hydrolysis by EF-G, a mixture containing 0.5 µM 70S and 100 µM GTP was rapidly mixed with 2 µM EF-G and 5 µM MDCC-PBP in stopped flow. The curves were fitted with a single-exponential using ORIGIN 8.0 (Origin Lab Co.).
Dipeptide formation
Mixture A: 1 µM 70S (MRE600/MG1655/JE105) was pre-incubated at 37°C for 10 min with 2 µM IF1, IF2, IF3, mRNA MLL stop, [3H]fMet-tRNAfMet, 1 mM GTP, 50 µg ml−1 PK, 2 µg ml−1 MK and 10 mM PEP in HEPES-polymix buffer (37) to form proper initiation complex. Mixture B consisting of 10 µM EF-Tu, 5 µM EF-Ts, 10 µM tRNALeu, 0.2 mM of Leucine amino acid, 0.5 µM of LeuRS, 1 mM ATP, 1 mM GTP, 50 µg ml−1 pyruvate kinase (PK), 2 µg ml−1 myokinase (MK) and 10 mM phosphoenolpyruvate (PEP) was mixed and incubated at 37°C for 10 min to obtain proper EF-Tu-tRNA-amino acid-GTP ternary complexes (henceforth referred as EF-Tu-ternary complex). Equal volumes of both mixtures were rapidly mixed in a KinTek quench-flow instrument and quenched with 50% formic acid at different time points. The fraction of dipeptide formed in each time-point was estimated on an HPLC by comparing the dipeptide peak with the peak of unused [3H]fMet-tRNAfMet and plotted against time. The curves were fitted with single-exponential equation using ORIGIN 8.0.
Tripeptide formation
Tripeptide formation was monitored in a setup similar to the dipeptide assay, the difference being that in mixture B 5 µM EF-G was added in addition to all the components listed earlier. The data were treated similarly as for dipeptide experiments.
Binding of EF-G to the ribosome
A single cysteine variant of E. coli EF-G, EF-G 650-Cys (kind gift from Prof. Harry Noller, University of California, Santa Cruz, USA) was labeled uniformly with rhodamine (tetramethylrhodamine-5-maleimide) using the recommended protocol from Invitrogen. The resulting EF-G-rho (2 µM) was mixed rapidly with 70S ribosome (0.5 µM) and 100 µM GTP in a stopped-flow instrument. The increase of the fluorescence signal at 590 nm due to binding of the labeled EF-G to the ribosome was followed with time. The curves were fitted with single exponential equation using ORIGIN 8.0.
Average time analysis
For reactions involving two consecutive steps the ‘average time (t)’ for the second step was estimated as (1/kobs (second step) − 1/kobs (first step)), as shown earlier (27,38). Thus, the average time for GTP hydrolysis during initiation was (1/kobs (GTP hydrolysis) − 1/kobs (subunit association)). For reactions involving multiple steps, the average time for the middle step was estimated by subtracting the average times of steps one and three from the average time spent in the total reaction. Thus, the average time for EF-G driven steps during tripeptide formation was derived as [1/kobs (tripeptide) − 2 (1/kobs (dipeptide))]. The underlined assumption is that the average time for the second peptide bond formation including EF-Tu mediated steps would be essentially same as the first one measured by dipeptide formation.
RESULTS
Escherichia coli JE105—a strain with single L12 dimer ribosome
In E. coli, the rplJ gene codes for the L10 protein, the last 10 amino acids of L10 constitute the binding site for one L12 dimer (1,20). The nucleotides corresponding to these amino acids were substituted with an ampicillin resistance cassette (Amp) (~800 nt) on the chromosome of the wild-type E. coli strain MG1655 by means of lambda red recombineering (Figure 1a) (28,29). The resulting strain JE105 was expected to carry only one L12 dimer on the ribosome due to the loss of the peripheral L12 dimer binding site on L10 (Figure 1a). The substitution was confirmed by two PCR reactions. In the first PCR, with primers up L10 and down L12, JE105 produced a band ~800 bp larger than that from the parental strain MG1655 (~1200 bp), whereas in the second PCR, with primers in Amp (an internal primer for the amp cassette) and down L12 only JE105 produced a band of ~800 bp (Figure 1b).
JE105 showed significantly reduced fitness compared to MG1655
The fitness cost conferred by the loss of one L12 dimer-binding site from L10 (and hence presumably due to the loss of one L12 dimer from the ribosome) was evident by the smaller colony size of JE105 on LB-agar plate in comparison to the parental strain MG1655. Under standard growth condition (LB broth at 37°C), the generation time of JE105 was 46 min compared to around 24 min for the wild-type strain MG1655 (Figure 1c). Addition of ampicillin (100 µg ml−1) in the growth medium did not alter the growth pattern of JE105. Also, the overall expression of the L12 protein in JE105 remained the same as in MG1655 (Figure 2c) confirming that the insertion of Amp had no effect on the expression of the down-stream genes of the rplJ operon.
Composition of the JE105 ribosomes and quantification of L12 proteins
Purified 70S ribosomes from JE105 and MG1655 were characterized in the native composite gel at a low Mg2+ concentration so that the 50S and 30S subunits would migrate separately. Indeed, the ribosomal subunits produced two distinct bands; while the 30S subunits from both strains co-migrated to the same distance the 50S subunit from JE105 showed slightly faster migration than those from MG1655 (Figure 2a). This result, on the one hand, confirmed that the ribosomal subunits of the engineered strain JE105 were intact and homogeneous. On the other hand, it indicated that JE105 50S was somewhat lighter than that from MG1655. Since the resolution of the native composite gel was not very high, the deletion of only 10 amino acids from L10 would be insufficient to account for the difference in 50S migration. Instead, it could arise from the loss of some other ribosomal proteins, presumably one L12 dimer.
The JE105 and MG1655 ribosomal L8 protein complex, when separated by SDS–PAGE, migrated in a similar fashion with the exception of protein L10. In the JE105 sample, L10 migrated somewhat faster than that in MG1655 confirming that the chromosomal deletion of a portion of the rplJ gene indeed resulted in a truncated L10 protein (Figure 2b). This result also indicated that the C-terminally truncated L10 could assemble stably with the JE105 ribosome. No difference in migration was seen in the L12 bands although their amount was different in the two samples.
To quantify the relative amount of the L12 protein in the JE105 and MG1655 ribosomes and cell lysates, Western blotting was performed using antibodies against L12 and S1 protein, where S1 was used as an internal standard. The cell lysates from both strains produced L12 blots of almost equal intensity whereas the L12 blot in the MG1655 ribosome sample was visibly stronger than that in the JE105 ribosome (Figure 2c). When estimated from the pixel ratio of L12 and S1 blots, no significant variation in the L12 content in the cell lysate of MG1655 and JE105 was seen [(L12:S1)MG1655 lysate : (L12:S1)JE105 lysate ~ 1]. However, when ribosomes from the two strains were compared in a similar fashion, MG1655 showed approximately double the amount of L12 compared to JE105 [(L12:S1)MG1655 70S : (L12:S1)JE105 70S ~ 2]. Because MG1655 ribosome contained two L12 dimers, this result confirmed that only one L12 dimer was bound on the JE105 ribosome. However, the comparable amount of L12 in the cell-lysates in JE105 and MG1655 confirmed that the expression of the downstream genes (including rplL coding L12) was not affected by the genetic manipulation at the rplJ locus.
Purified 70S ribosomes from JE105 and MG1655 were further characterized and compared by 2D gel electrophoresis. The ribosomal protein spots were highly similar in JE105 and MG1655 ribosomes except for the L12 protein, which appeared smaller in JE105 (Figure 2d). When quantified from the pixel ratio of the L12 and L24 protein spots, the amount of L12 was found to be half in JE105 (L12:L24 = 0.78) compared to that in MG1655 (L12:L24 = 1.55). The ratio of L24 with another protein, L23, remained essentially the same (L23: L24 = 0.97 ± 2) in both samples ensuring that L24 served as a proper internal standard.
JE105 ribosomes are defective in synthesis of full-length proteins in vitro
JE105 ribosomes were compared with ribosomes from MG1655 and MRE600 (both of which contained two L12 dimers) for their activity in synthesis of a full-length protein firefly luciferase in vitro. For that, a cell-free transcription–translation system composed of highly active, purified components from E. coli was employed (27). JE105 ribosomes were notably slower in luciferase synthesis than the other two ribosomes (Figure 3a). Further, when ribosomal subunits from these strains were mixed in pair-wise combinations, the reactions with JE105 50S subunits showed slower increase in luminescence irrespective of the source of the 30S subunit (Figure 3b). Thus, it could be inferred that the defect in JE105 ribosomes did arise from its 50S subunit containing single L12-dimer. It is also noteworthy that the ribosomal subunits from MG1655 and MRE600 strains behaved very similarly in this assay and that the 30S subunits from JE105 were as active as those from MRE600.
Figure 3.

In vitro synthesis of firefly luciferase. Synthesis of full-length, active firefly luciferase followed by the increase in luminescence in a reconstituted transcription–translation system with 70S ribosomes (a) and ribosomal subunits (b) from MG1655, MRE600 (both containing two L12 dimers) and JE105 (single L12 dimer).
Single L12 dimer JE105 ribosomes are slow in IF2·GTP mediated initiation
Subunit association—The ribosomal subunits from JE105 were compared with those from MG1655 and MRE600 in the subunit association assay followed by Rayleigh light-scattering (34). When naked 50S and 30S subunits were mixed rapidly in a stopped-flow instrument, no significant difference in the rate of association was seen between the two strains (Figure 4a) suggesting that the presence of single or double L12 dimers on the 50S subunit was immaterial in the context of factor-free subunit association. This was expected from our previous work, where even a complete depletion of L12 proteins from the 50S subunits did not change the rate of association of the naked subunits (27). However, when subunit association was performed with a proper 30S pre-initiation complex programmed with mRNA, initiator tRNA and the initiation factors IF1 and IF2·GTP, the single L12 dimer containing JE105 50S subunits associated two times slower (Ka = 72 ± 5 µM−1s−1) than the MG1655 and MRE600 50S subunits containing two L12 dimers (Ka = 160 ± 5 µM−1s−1) (Figure 4b and Table 1). Further, 50S subunits from MRE600 and JE105 were titrated in the same assay and the linearly increasing observed rate of association was plotted against the 50S concentration. As shown in Figure 4c, at the higher concentration of 50S subunits, the incompetence of the single L12 dimer 50S in IF2-mediated subunit association was more pronounced in comparison with the double L12 dimer 50S. The role of L12 protein in IF2-mediated subunit association is a rather recent finding (27). Our present result strengthens that notion and suggests that multiple L12 dimers are required on the 50S subunit for proper interaction with IF2·GTP on the 30S pre-initiation complex, a key-step for rapid formation of the 70S initiation complex. It should be noted that both the titration curves with MRE600 and JE105 50S subunits passed through the origin, indicating that the samples were free from any kind of heterogeneity.
Figure 4.

Fast kinetics measurements of the steps of initiation. The time course of association of MRE600 (black trace), MG1655 (blue trace) (both with two L12 dimers) and JE105 (red trace) (with single L12 dimer) 50S subunits with naked 30S subunits (a), or with 30S preIC containing mRNA, fMet-tRNAfMet, IF1 and IF2·GTP (b) followed in stopped-flow by monitoring increase in light scattering at 430 nm. The insert in (b) shows the same reaction for prolonged time. (c) The plots showing linear dependence of the observed rates of subunit association on 50S concentration in reactions with 30S preIC. (d) Single round Pi release with MRE600 (black trace) and JE105 (red trace) ribosomes, measured in parallel to subunit association in the same reaction as in (b). The increase in MDCC fluorescence upon Pi binding to PBP-MDCC was monitored at 464 nm (λEx = 425 nm) (e) Schematic representation of translation initiation where average times for individual steps were estimated for two L12 dimer MRE600 (in black) and single L12 dimer JE105 (in red) ribosomes (see ‘Materials and Methods’ section for details).
Table 1.
Observed rates (kobs) and average times (t) for different steps of initiation and elongation with MG1655 (two L12 dimer) and JE105 (single L12 dimer) ribosomes
| MG1655 (double L12 dimer ribosome) |
JE105 (single L12 dimer ribosome) |
t′/t | |||||
|---|---|---|---|---|---|---|---|
| kobs (s−1) | 1/kobs (s) | t (ms) | 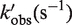 |
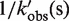 |
t′ (ms) | ||
| Initiation | |||||||
| Subunit association | 40 ± 5 | 0.025 | 25 | 20 ± 2 | 0.05 | 50 | 2.0 |
| GTP hydrolysis (Pi release) | 20 ± 1 | 0.05 | 25 | 13 ± 1 | 0.077 | 27 | 1.1 |
| Elongation | |||||||
| Dipeptide (ML) formation | 51 ± 7 | 0.02 | 20 | 50 ± 2 | 0.02 | 20 | 1.0 |
| Tripeptide (MLL) formation | 5 ± 1 | 0.2 | 200 | 3.3 ± 1 | 0.303 | 303 | 1.5 |
| EF-G driven steps | – | – | 160 | – | – | 263 | 1.6 |
The ‘average time (t)’ for GTP hydrolysis during initiation is estimated as (1/kobs (GTP hydrolysis) − 1/kobs (subunit association)) and the same for EF-G driven steps in elongation is estimated as [1/kobs (tripeptide) − 2 (1/kobs (dipeptide))].
GTP hydrolysis on IF2—to see whether the lack of one L12 dimer influenced GTP hydrolysis by IF2 after subunit association, we monitored the release of inorganic phosphate (Pi) over time in parallel to the measurement of subunit association using dual detectors in the stopped-flow instrument. In our previous work, no significant time delay (27,39) between GTP hydrolysis and Pi release was seen in the context of translation initiation. Thus, in the present study Pi release was considered to be reflecting GTP hydrolysis. The apparent rate of Pi release with single L12 dimer 50S subunits from JE105 (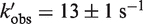 ) was slower than that with double L12 dimer 50S from MRE600 (kobs = 20 ± 1 s−1) (Figure 4d and Table 1). However, it should be noted that the observed rate of Pi release is a ‘compounded’ rate arising from at least two consecutive steps in initiation; subunit association and GTP hydrolysis. Thus, it was necessary to conduct ‘average time analysis’ (see ‘Materials and Methods’ section for details) in order to conclude which of these two steps was mainly responsible for the defect in JE105. The average time for subunit association with JE105 was ~50 ms (referred hereafter as ms) while for MRE600 (or MG1655) was ~25 ms. However, there was practically no difference in the average time for GTP hydrolysis (~25 ms) with these ribosomes (Figure 4e and Table 1). Thus, this result is consistent with our earlier result (27) that L12 does not influence GTPase activity of IF2.
) was slower than that with double L12 dimer 50S from MRE600 (kobs = 20 ± 1 s−1) (Figure 4d and Table 1). However, it should be noted that the observed rate of Pi release is a ‘compounded’ rate arising from at least two consecutive steps in initiation; subunit association and GTP hydrolysis. Thus, it was necessary to conduct ‘average time analysis’ (see ‘Materials and Methods’ section for details) in order to conclude which of these two steps was mainly responsible for the defect in JE105. The average time for subunit association with JE105 was ~50 ms (referred hereafter as ms) while for MRE600 (or MG1655) was ~25 ms. However, there was practically no difference in the average time for GTP hydrolysis (~25 ms) with these ribosomes (Figure 4e and Table 1). Thus, this result is consistent with our earlier result (27) that L12 does not influence GTPase activity of IF2.
Elongation of protein synthesis with single L12 dimer ribosomes
We have tested the single L12 dimer ribosomes from JE105 in the steps of elongation by monitoring the rate of formation of fMet-Leu dipeptide and fMet-Leu-Leu tripeptide starting from a complete 70S initiation complex (70S IC). The dipeptide experiment essentially integrated the steps starting from EF-Tu-Leu-tRNALeu·GTP binding, GTP hydrolysis and release of EF-Tu·GDP and Pi (referred as EF-Tu mediated steps) to the peptide bond formation. The rate of dipeptide formation with 70S IC from JE105 was about the same as with that from MG1655 and MRE600 (kobs = 51 ± 7 s−1) (Figure 5a). Although somewhat surprising, this result suggested no necessity of multiple L12 dimers in the steps involving EF-Tu. It also indicates that peptidyl transfer is independent of the number of L12 proteins on the ribosome.
Figure 5.

Dipeptide and tripeptide formation experiments; scheme of translation elongation. Comparison of the MRE600 (black trace), MG1655 (blue trace) (both contain two L12 dimers) and JE105 (red trace) (single L12 dimer) ribosomes in dipeptide (a) and tripeptide (b) formation assays. (c) Schematic representation of different steps of elongation showing the average time analysis for by MG1655/MRE600 (in black) and JE105 (in red) ribosomes. The average time for EF-G driven steps was estimated as [1/kobs (tripeptide) − 2 (1/kobs (dipeptide))].
When the single and double L12 dimer ribosomes were subjected to tripeptide assay starting from the respective 70S IC, MG1655 and MRE600 ribosomes were ~1.6 times faster than JE105 ribosomes (Figure 5b and Table 1). This assay included EF-G mediated steps, i.e. binding of EF-G·GTP, tRNA translocation, GTP hydrolysis and release of EF-G·GDP sandwiched between two rounds of EF-Tu action and peptidyl transfer. Because JE105 ribosomes did not show any defect in EF-Tu-mediated steps, we inferred that the defect in tripeptide formation originated from the steps involving EF-G. This inference was further supported by average time analysis. The average time spent in EF-G driven steps by JE105 ribosomes was ~260 ms in contrast to ~160 ms by MRE600 ribosomes while the average time in the EF-Tu mediated steps for both the ribosomes was ~20 ms (Figure 5c and Table 1).
Single L12 dimer ribosomes are defective in GTPase stimulation on EF-G
Early experiments suggested that L12 is involved in the stimulation of GTPase activity of EF-G (23,25). Later L12 has also been proposed to be involved in factor recruitment (1). To see whether the defect in tripeptide formation in JE105 ribosomes originated from either of these two steps or from both, we followed the binding of EF-G to the JE105, MG1655 and MRE600 70S ribosomes using a rhodamine-labeled single Cys 650 (EF-G-rho) variant of E. coli EF-G. The labeled protein showed 60% activity compared to the unlabeled EF-G in tripeptide assay. The binding of EF-G-rho to the ribosome was monitored by the increase in rhodamine fluorescence. Surprisingly, no difference in the rate EF-G-rho binding was seen with these ribosomes (Figure 6a). However, when GTP hydrolysis on EF-G was measured in the same experimental set up by following Pi release, JE105 showed a one and half to 2-fold slower rate compared to MG1655 and MRE600 (Figure 6b). Thus we conclude that single L12 dimer ribosomes are less competent in stimulation of GTPase activity on EF-G but not in EF-G binding as compared to double L12 dimer ribosomes.
Figure 6.

EF-G binding to the ribosome and GTP hydrolysis. Comparison of the naked 70S ribosomes from MRE600 (black trace), MG1655 (blue trace) (both contain two L12 dimers) and JE105 (red trace) (single L12 dimer) in (a) time course of binding of EF-G-rho followed by increase in rhodamine fluorescence at 590 nm and (b) in stimulation of GTP hydrolysis by EF-G measured by Pi release using MDCC-PBP.
DISCUSSION
Because of its unique features, the stalk protein L12 has drawn attention of the scientists since an early period of ribosome research. With the development of knowledge about the structure and function of the ribosome we have gained insight about these proteins too, especially about their functions in relation to the translational GTPase factors EF-Tu, EF-G and IF2. However, a complete picture of L12 action, especially linking its structure and dimer organization, has yet not been available. Thus it remained an open question why the bacterial ribosome has evolved to carry multiple copies of L12 dimers on the flexible ribosomal stalk. There have been some previous attempts to construct and characterize E. coli ribosomes carrying less than two L12 dimers (20,40). These studies took advantage of the fact that protein L12 (and also protein L10) could be removed from the ribosome by chemical treatment (41) and further re-associated to the ribosomal core lacking these protein.
In the study by Möller et al. (40), ribosomes containing different amount of L12 proteins were obtained by varying the amount of L12 protein during reconstitution on L12-depleted ribosomal core. These ribosomes were obviously heterogeneous in nature and the exact number of the L12 proteins on the individual ribosomes could not be determined. In a more recent work, by Griaznova and Traut (20), wild-type L10 was replaced by C-terminally truncated L10 protein in vivo by overexpression from a plasmid or in vitro by ribosomal reconstitution. One of their constructs, L10Δ10, which lacked the peripheral L12 dimer binding site, associated mostly a single L12 dimer on the ribosome. The in vitro reconstituted L10Δ10 ribosomes showed similar activity to the ribosomes reconstituted with full-length L10 in a multiple turnover EF-G dependent GTP hydrolysis assay, and also in a poly(U)-dependent poly-Phe synthesis assay. However, some heterogeneity in these ribosomes was unavoidable as their construction relied fully on the efficiency of L10 and L12 depletion and reconstitution. The in vivo reconstituted L10Δ10 ribosomes were not free from heterogeneity either, since the truncated L10 was expressed in the wild-type background. Surprisingly, a comparison of the reconstituted ribosomes with the untreated wild-type ribosomes was missing and the in vivo aspects of such reconstructions were ignored.
From a physiological point of view, we wondered what implications the loss of one L12 dimer would have on the fitness of the bacteria. This query led us to construct a chromosomally engineered, pure E. coli strain JE105, where the terminal L12 dimer binding site on the L10 protein was deleted, i.e. the JE105 ribosomes contained only single L12 dimer. JE105 showed highly compromised fitness compared to its parental strain MG1655 (wild-type) suggesting that one L12 dimer is not sufficient for full functionality of the ribosome and thus cannot support optimal growth of the bacteria. It has been shown recently with mass-spectroscopy that the number of L12 dimers varies in different growth stages of the archaebacteria Methanosarcina sp.; the ribosomes carry two L12 dimers in the lag phase but three in the exponential growth phase (42). Also, ribosomes extracted from E. coli, cultured under stressed condition, showed significantly increased N-acetylation of the L12 proteins, which is thought to increase the stability of the L12 dimers on L10 (7). Together these results indicate that there is a strong correlation between the number of L12 dimers on the ribosome and the bacterial growth rate. It also implies that ribosomes lacking one L12 dimer would be at least partially impaired in protein synthesis, resulting in slower growth.
To test this hypothesis, we compared single L12 dimer JE105 ribosomes with two L12 dimer ribosomes from MG1655 (wild-type) and MRE600 (K-12 strain) in the synthesis of firefly luciferase in vitro. Indeed, 70S ribosomes and 50S subunits from JE105 were slowest of all in luciferase synthesis suggesting that single L12 dimer ribosomes are defective in protein synthesis. However, it was not possible to pinpoint from this experiment exactly in which step(s) of translation the JE105 ribosomes were impaired. Therefore we conducted fast kinetics experiments to estimate the rates of the individual steps of translation under single-turnover conditions. Our results showed that single L12 dimer ribosomes are two times slower in IF2-mediated subunit association and about one and half times slower in EF-G mediated elongation than the double L12 dimer ribosomes. Although the relative contribution of the two defects towards the fitness of JE105 is difficult to judge, it can be assumed that the relatively small elongation defect would be amplified several times in the context of a long peptide and thus can be more effective. Furthermore, the defect in the EF-G driven steps is caused by decreased activity of the JE105 ribosomes in stimulation of GTP hydrolysis on EF-G. Presumably this would cause a slower release of the factor from the ribosome thereby delaying the entrance of the next EF-Tu-ternary complex.
Surprisingly, JE105 ribosomes did not show any impairment in dipeptide formation experiment that includes EF-Tu mediated steps other than peptidyl transfer. This seems apparently contradictory to the earlier reports that proposed L12's involvement in initial binding of EF-Tu-ternary complex (26) as well as in stimulation of EF-Tu GTPase (23). Our results show that a single L12 dimer is sufficient for interaction with EF-Tu-ternary complex. Even if JE105 ribosomes are defective in a sub-step during dipeptide formation, it must be insignificant in relation to the average time spent in the whole reaction. Further experiments will be needed to clarify the mode of interaction of L12 and EF-Tu. It is also notable that the two main reactions catalyzed by ribosomes, i.e. decoding of mRNA and peptide bond formation, are not dependent on the number of L12 dimers on the ribosome. This is not unexpected as these functions are mainly driven by rRNA in the ribosomal core and the participation of ribosomal stalk proteins is unlikely in this context.
This study brings forward a new insight regarding L12 interaction with translational GTPases on the ribosome. Since translational GTPases share common interaction sites on the conserved C-terminal domain of the L12, it was previously proposed that the L12 proteins interact with the translational GTPases in a generalized manner (16). However, our study with single L12 dimer ribosomes points towards two specialized roles of L12 proteins in relation to IF2 and EF-G. For IF2, multiple dimers of L12 proteins on the 50S subunit are needed for initial interaction with the factor on the 30S pre-initiation complex but not for its stimulation for GTP hydrolysis. In contrast, for EF-G, multiple L12 dimers are required to stimulate GTP hydrolysis on the factor rather than its recruitment. The role of the L12 proteins and importance of having multiple L12 dimers in translation termination, in relation to another G-protein RF3 remains to be seen.
In summary, we have created a pure strain of E. coli, JE105, carrying only a single L12 dimer on the ribosome and this strain grows significantly slower than the wild-type strain. We could further explain some of the causes of the fitness defect of JE105 by characterizing its ribosomes in vitro. JE105 ribosomes were defective in the initiation and elongation steps involving IF2 and EF-G. It should be noted that not necessarily all effects of varying L12 dimer copy number may have been detected in our experimental setup and further experiments will be needed to clarify all aspects. It is clear that the activity of the ribosome can be modulated by varying the number of loosely bound L12 dimers on it. This can be a regulatory mechanism in bacteria to tune the rate of protein synthesis in response to the growth condition.
FUNDING
Swedish Research Council (research grants 2009-5081, 2010-2619, 2006-267); Carl Tryggers Foundation (09:341, 10:330); Wenner Gren Foundation; Vinnova (P35533-1) and Knut and Alice Wallenberg Foundation (2009.0251 to S.S.). Funding for open access charge: Swedish Research Council (research grants 2009-5081, 2010-2619, 2006-267).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Prof. J.P. Ballesta, Prof. Harry Noller and Prof. Martin Webb for providing us plasmids or antibody used in this study. We are grateful to Prof. Knud H. Nierhaus (Max-Planck-Institut für Molekulare Genetik, Berlin) for helping with 2D-gel electrophoresis. We also thank Dr Terese Bergfors, Uppsala University, for helping in the manuscript preparation.
REFERENCES
- 1.Diaconu M, Kothe U, Schlunzen F, Fischer N, Harms JM, Tonevitsky AG, Stark H, Rodnina MV, Wahl MC. Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTPase activation. Cell. 2005;121:991–1004. doi: 10.1016/j.cell.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 2.Ilag LL, Videler H, McKay AR, Sobott F, Fucini P, Nierhaus KH, Robinson CV. Heptameric (L12)6/L10 rather than canonical pentameric complexes are found by tandem MS of intact ribosomes from thermophilic bacteria. Proc. Natl Acad. Sci. USA. 2005;102:8192–8197. doi: 10.1073/pnas.0502193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harms J, Schluenzen F, Zarivach R, Bashan A, Gat S, Agmon I, Bartels H, Franceschi F, Yonath A. High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell. 2001;107:679–688. doi: 10.1016/s0092-8674(01)00546-3. [DOI] [PubMed] [Google Scholar]
- 4.Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. Crystal structure of the ribosome at 5.5 A resolution. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 5.Hardy SJ. The stoichiometry of the ribosomal proteins of Escherichia coli. Mol. Gen. Genet. 1975;140:253–274. doi: 10.1007/BF00334270. [DOI] [PubMed] [Google Scholar]
- 6.Pettersson I, Hardy SJ, Liljas A. The ribosomal protein L8 is a complex L7/L12 and L10. FEBS Lett. 1976;64:135–138. doi: 10.1016/0014-5793(76)80267-0. [DOI] [PubMed] [Google Scholar]
- 7.Gordiyenko Y, Deroo S, Zhou M, Videler H, Robinson CV. Acetylation of L12 increases interactions in the Escherichia coli ribosomal stalk complex. J. Mol. Biol. 2008;380:404–414. doi: 10.1016/j.jmb.2008.04.067. [DOI] [PubMed] [Google Scholar]
- 8.Kaltschmidt E. Ribosomal proteins. XIV. Isoelectric points of ribosomal proteins of E. coli as determined by two-dimensional polyacrylamide gel electrophoresis. Anal. Biochem. 1971;43:25–31. doi: 10.1016/0003-2697(71)90103-5. [DOI] [PubMed] [Google Scholar]
- 9.Bocharov EV, Sobol AG, Pavlov KV, Korzhnev DM, Jaravine VA, Gudkov AT, Arseniev AS. From structure and dynamics of protein L7/L12 to molecular switching in ribosome. J. Biol. Chem. 2004;279:17697–17706. doi: 10.1074/jbc.M313384200. [DOI] [PubMed] [Google Scholar]
- 10.Christodoulou J, Larsson G, Fucini P, Connell SR, Pertinhez TA, Hanson CL, Redfield C, Nierhaus KH, Robinson CV, Schleucher J, et al. Heteronuclear NMR investigations of dynamic regions of intact Escherichia coli ribosomes. Proc. Natl Acad. Sci. USA. 2004;101:10949–10954. doi: 10.1073/pnas.0400928101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mulder FA, Bouakaz L, Lundell A, Venkataramana M, Liljas A, Akke M, Sanyal S. Conformation and dynamics of ribosomal stalk protein L12 in solution and on the ribosome. Biochemistry. 2004;43:5930–5936. doi: 10.1021/bi0495331. [DOI] [PubMed] [Google Scholar]
- 12.Wahl MC, Bourenkov GP, Bartunik HD, Huber R. Flexibility, conformational diversity and two dimerization modes in complexes of ribosomal protein L12. EMBO J. 2000;19:174–186. doi: 10.1093/emboj/19.2.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandra Sanyal S, Liljas A. The end of the beginning: structural studies of ribosomal proteins. Curr. Opin. Struct. Biol. 2000;10:633–636. doi: 10.1016/s0959-440x(00)00143-3. [DOI] [PubMed] [Google Scholar]
- 14.Leijonmarck M, Liljas A. Structure of the C-terminal domain of the ribosomal protein L7/L12 from Escherichia coli at 1.7 A. J. Mol. Biol. 1987;195:555–579. doi: 10.1016/0022-2836(87)90183-5. [DOI] [PubMed] [Google Scholar]
- 15.Liljas A, Gudkov AT. The structure and dynamics of ribosomal protein L12. Biochimie. 1987;69:1043–1047. doi: 10.1016/0300-9084(87)90004-6. [DOI] [PubMed] [Google Scholar]
- 16.Helgstrand M, Mandava CS, Mulder FA, Liljas A, Sanyal S, Akke M. The ribosomal stalk binds to translation factors IF2, EF-Tu, EF-G and RF3 via a conserved region of the L12 C-terminal domain. J. Mol. Biol. 2007;365:468–479. doi: 10.1016/j.jmb.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 17.Kothe U, Rodnina MV. Delayed release of inorganic phosphate from elongation factor Tu following GTP hydrolysis on the ribosome. Biochemistry. 2006;45:12767–12774. doi: 10.1021/bi061192z. [DOI] [PubMed] [Google Scholar]
- 18.Moens PD, Wahl MC, Jameson DM. Oligomeric state and mode of self-association of Thermotoga maritima ribosomal stalk protein L12 in solution. Biochemistry. 2005;44:3298–3305. doi: 10.1021/bi048015n. [DOI] [PubMed] [Google Scholar]
- 19.Gao YG, Selmer M, Dunham CM, Weixlbaumer A, Kelley AC, Ramakrishnan V. The structure of the ribosome with elongation factor G trapped in the posttranslocational state. Science. 2009;326:694–699. doi: 10.1126/science.1179709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griaznova O, Traut RR. Deletion of C-terminal residues of Escherichia coli ribosomal protein L10 causes the loss of binding of one L7/L12 dimer: ribosomes with one L7/L12 dimer are active. Biochemistry. 2000;39:4075–4081. doi: 10.1021/bi992621e. [DOI] [PubMed] [Google Scholar]
- 21.Kirsebom LA, Isaksson LA. Involvement of ribosomal protein L7/L12 in control of translational accuracy. Proc. Natl Acad. Sci. USA. 1985;82:717–721. doi: 10.1073/pnas.82.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettersson I, Kurland CG. Ribosomal protein L7/L12 is required for optimal translation. Proc. Natl Acad. Sci. USA. 1980;77:4007–4010. doi: 10.1073/pnas.77.7.4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mohr D, Wintermeyer W, Rodnina MV. GTPase activation of elongation factors Tu and G on the ribosome. Biochemistry. 2002;41:12520–12528. doi: 10.1021/bi026301y. [DOI] [PubMed] [Google Scholar]
- 24.Savelsbergh A, Mohr D, Kothe U, Wintermeyer W, Rodnina MV. Control of phosphate release from elongation factor G by ribosomal protein L7/12. EMBO J. 2005;24:4316–4323. doi: 10.1038/sj.emboj.7600884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savelsbergh A, Mohr D, Wilden B, Wintermeyer W, Rodnina MV. Stimulation of the GTPase activity of translation elongation factor G by ribosomal protein L7/12. J. Biol. Chem. 2000;275:890–894. doi: 10.1074/jbc.275.2.890. [DOI] [PubMed] [Google Scholar]
- 26.Kothe U, Wieden HJ, Mohr D, Rodnina MV. Interaction of helix D of elongation factor Tu with helices 4 and 5 of protein L7/12 on the ribosome. J. Mol. Biol. 2004;336:1011–1021. doi: 10.1016/j.jmb.2003.12.080. [DOI] [PubMed] [Google Scholar]
- 27.Huang C, Mandava CS, Sanyal S. The ribosomal stalk plays a key role in IF2-mediated association of the ribosomal subunits. J. Mol. Biol. 2010;399:145–153. doi: 10.1016/j.jmb.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 28.Costantino N, Court DL. Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants. Proc. Natl Acad. Sci. USA. 2003;100:15748–15753. doi: 10.1073/pnas.2434959100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ellis HM, Yu D, DiTizio T, Court DL. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl Acad. Sci. USA. 2001;98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johansson M, Bouakaz E, Lovmar M, Ehrenberg M. The kinetics of ribosomal peptidyl transfer revisited. Mol. Cell. 2008;30:589–598. doi: 10.1016/j.molcel.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 31.Nadano D, Aoki C, Yoshinaka T, Irie S, Sato TA. Electrophoretic characterization of ribosomal subunits and proteins in apoptosis: specific downregulation of S11 in staurosporine-treated human breast carcinoma cells. Biochemistry. 2001;40:15184–15193. doi: 10.1021/bi0108397. [DOI] [PubMed] [Google Scholar]
- 32.Geyl D, Bock A, Isono K. An improved method for two-dimensional gel-electrophoresis: analysis of mutationally altered ribosomal proteins of Escherichia coli. Mol. Gen. Genet. 1981;181:309–312. doi: 10.1007/BF00425603. [DOI] [PubMed] [Google Scholar]
- 33.Gao H, Zhou Z, Rawat U, Huang C, Bouakaz L, Wang C, Cheng Z, Liu Y, Zavialov A, Gursky R, et al. RF3 induces ribosomal conformational changes responsible for dissociation of class I release factors. Cell. 2007;129:929–941. doi: 10.1016/j.cell.2007.03.050. [DOI] [PubMed] [Google Scholar]
- 34.Antoun A, Pavlov MY, Tenson T, Ehrenberg MM. Ribosome formation from subunits studied by stopped-flow and Rayleigh light scattering. Biol. Proced. Online. 2004;6:35–54. doi: 10.1251/bpo71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Antoun A, Pavlov MY, Andersson K, Tenson T, Ehrenberg M. The roles of initiation factor 2 and guanosine triphosphate in initiation of protein synthesis. EMBO J. 2003;22:5593–5601. doi: 10.1093/emboj/cdg525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brune M, Hunter JL, Corrie JE, Webb MR. Direct, real-time measurement of rapid inorganic phosphate release using a novel fluorescent probe and its application to actomyosin subfragment 1 ATPase. Biochemistry. 1994;33:8262–8271. doi: 10.1021/bi00193a013. [DOI] [PubMed] [Google Scholar]
- 37.Tobin C, Mandava CS, Ehrenberg M, Andersson DI, Sanyal S. Ribosomes lacking protein S20 are defective in mRNA binding and subunit association. J. Mol. Biol. 2010;397:767–776. doi: 10.1016/j.jmb.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Johansson M, Ieong KW, Trobro S, Strazewski P, Aqvist J, Pavlov MY, Ehrenberg M. pH-sensitivity of the ribosomal peptidyl transfer reaction dependent on the identity of the A-site aminoacyl-tRNA. Proc. Natl Acad. Sci. USA. 2011;108:79–84. doi: 10.1073/pnas.1012612107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pavlov MY, Sanyal S, Ehrenberg M. Ribosomes:Structure, Function, and Dynamics. Wein: Springer Wien Newyork; 2011. pp. 129–141. [Google Scholar]
- 40.Moller W, Schrier PI, Maassen JA, Zantema A, Schop E, Reinalda H, Cremers AF, Mellema JE. Ribosomal proteins L7/L12 of Escherichia coli. Localization and possible molecular mechanism in translation. J. Mol. Biol. 1983;163:553–573. doi: 10.1016/0022-2836(83)90112-2. [DOI] [PubMed] [Google Scholar]
- 41.Hamel E, Koka M, Nakamoto T. Requirement of an Escherichia coli 50 S ribosomal protein component for effective interaction of the ribosome with T and G factors and with guanosine triphosphate. J. Biol. Chem. 1972;247:805–814. [PubMed] [Google Scholar]
- 42.Gordiyenko Y, Videler H, Zhou M, McKay AR, Fucini P, Biegel E, Muller V, Robinson CV. Mass spectrometry defines the stoichiometry of ribosomal stalk complexes across the phylogenetic tree. Mol. Cell. Proteomics. 2010;9:1774–1783. doi: 10.1074/mcp.M000072-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]