


Abstract
The Gram-negative plant-pathogenic bacterium Xanthomonas campestris pv. vesicatoria (Xcv) is an important model to elucidate the mechanisms involved in the interaction with the host. To gain insight into the transcriptome of the Xcv strain 85–10, we took a differential RNA sequencing (dRNA-seq) approach. Using a novel method to automatically generate comprehensive transcription start site (TSS) maps we report 1421 putative TSSs in the Xcv genome. Genes in Xcv exhibit a poorly conserved −10 promoter element and no consensus Shine-Dalgarno sequence. Moreover, 14% of all mRNAs are leaderless and 13% of them have unusually long 5′-UTRs. Northern blot analyses confirmed 16 intergenic small RNAs and seven cis-encoded antisense RNAs in Xcv. Expression of eight intergenic transcripts was controlled by HrpG and HrpX, key regulators of the Xcv type III secretion system. More detailed characterization identified sX12 as a small RNA that controls virulence of Xcv by affecting the interaction of the pathogen and its host plants. The transcriptional landscape of Xcv is unexpectedly complex, featuring abundant antisense transcripts, alternative TSSs and clade-specific small RNAs.
INTRODUCTION
At a staggering pace new high-throughput sequencing technologies have helped to unveil the transcriptional complexity of many organisms in all kingdoms of life (1–3). The recently developed differential RNA sequencing approach (dRNA-seq) has yet added a new perspective. dRNA-seq, based on a selective enrichment of native 5′-ends, has been shown to accurately and cost-effectively identify transcription start sites (TSSs) and RNA processing sites for whole genomes (4). In addition to the obvious advantages for the analysis of 5′-UTR or promoter elements, dRNA-seq allows distinguishing independently transcribed short non-coding and coding RNAs from post-transcriptional processes such as maturation (4). However, a fully-automated method to annotate and statistically evaluate TSSs in large dRNA-seq data sets has been missing so far. Here, we sketch a procedure to automatically identify TSSs.
Transcriptome analyses in plant pathogenic bacteria so far mainly focused on coding regions and the regulon controlling type III secretion [e.g. (5,6)]. A recent deep sequencing analysis of Pseudomonas syringae identified many small RNA (sRNA) candidates, most of which, however, await validation by independent methods (7).
The Gram-negative plant pathogenic γ-proteobacterium Xanthomonas campestris pv. vesicatoria (Xcv) is the causal agent of bacterial spot disease on pepper and tomato and is of great economic importance in regions with a warm and humid climate (8). Xcv serves as a model system to elucidate the molecular communication between plant pathogens and their hosts and to characterize bacterial virulence strategies. Genome analysis predicted 4726 open reading frames (ORFs) in the Xcv strain 85–10 (9), yet the overall gene structure and non-coding RNA output of this model pathogen are still poorly understood.
Essential for pathogenicity of Xcv on susceptible host plants is the type III secretion (T3S) system, encoded by the hrp [hypersensitive response (HR) and pathogenicity] gene cluster (10). In Xcv, as in most Gram-negative bacterial pathogens, the T3S nanomachine translocates a suite of effector proteins into the plant cell where they manipulate host cellular processes to the benefit of the pathogen, e.g. by suppression of basal plant defense responses (9,11–13). hrp mutants do not grow in plant tissue, and they no longer cause disease in susceptible plants and the HR in resistant plants (10). The HR is a local, rapid programmed cell death at the site of infection, which coincides with arrest of bacterial multiplication in the plant (14,15).
The T3S system is transcriptionally induced in certain minimal media and in the plant (16,17). Key regulatory proteins are the OmpR-type response regulator HrpG, which is activated by unknown plant signals and controls the expression of a genome-wide regulon including hrp, type III effector and putative virulence genes (16–19). HrpG-mediated activation of gene expression depends in most cases on the AraC-type transcriptional activator HrpX (18), which binds to a conserved motif (plant-inducible promoter; PIP box) in the promoters of target genes (20). The identification of a point mutation in HrpG (termed HrpG*), which renders the protein constitutively active, was key for the analysis of T3S and the identification of putative virulence factors that are cotranscribed with the T3S system (19,21). An open question was whether virulence gene expression in Xcv is post-transcriptionally regulated, for instance by sRNAs. Here, we provide for the first time an insight into the transcriptional landscape of a plant pathogenic bacterium and the involvement of sRNAs in its virulence.
MATERIALS AND METHODS
RNA isolation for 454 pyrosequencing, RACE analysis and northern blot
RNA was isolated from NYG-grown Xcv strains 85–10 and 85* (exponential growth phase) by phenol extraction and treated with DNase I (Roche). For RACE and northern blot analyses, RNA was isolated from NYG-grown Xcv strains in exponential and stationary growth phases, as described (22). RACE analyses were carried out as described (23) with modifications [for detailed information see Supporting Information (SI)]. Northern blots were performed as described (24) using 10 µg RNA, 5–10 pmol [γ-32P]-ATP end-labeled oligodeoxynucleotides (Supplementary Table S1). Hybridization signals were visualized with a phosphoimager (FLA-3000 Series, Fuji). Northern blot hybridizations were performed at least twice with independently isolated RNA.
Construction of cDNA libraries for dRNA-seq and 454 pyrosequencing
Prior to RNA treatment and cDNA synthesis, equal amounts of RNA from the two Xcv strains 85–10 and 85* were mixed. dRNA-seq libraries were prepared according to Sharma et al. (2010) and sequenced with a Roche 454 sequencer using FLX and Titanium chemistry (see SI).
Annotation of transcription start sites
We aimed at the automated identification of TSSs based on the discrimination between narrow clusters of dRNA-seq reads that might represent a TSS and the distribution of individual read starts. The density of read starts varies across the genome and can be modeled locally by a Poisson distribution with a parameter λ. We used fixed-length intervals of size l to determine λr = sr/l from the number of read starts sr in the region r. The parameter λave models the average genome wide arrival rate of read starts. λ is defined as λr/λave. The corresponding Poisson distribution F(k,λ) describes the probability that at most k read starts are observed at a given genomic position. We used library 1 to determine λm for the background distribution of read starts. Similarly, library 2 was used to obtain λp to model the distribution biased towards the TSS.
A TSS is defined as the genomic position at which the observed number of read starts in library 2 significantly exceeds the background distribution of read starts in library 1. The significance of a putative TSS was determined as follows: for each genomic position, the difference of the number of read starts P in library 2 and M in library 1, D = P–M, was calculated. The difference of two Poisson distributed variables, D, follows a Skellam distribution (25) whose cumulative distribution function is given by
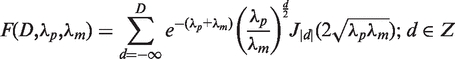 |
where J|d| is the modified Bessel function of the first kind and integer order |d|. Furthermore, 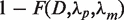 represents the probability that a difference of at least D read starts is observed given the normalized rates of read starts λp and λm. To reduce the influence of window sizes and local variation of transcriptional activity a sliding window of size x was shifted by y nucleotides along the genome and each site was tested t = x/y times for being a TSS. The p-value was obtained using the geometric mean
represents the probability that a difference of at least D read starts is observed given the normalized rates of read starts λp and λm. To reduce the influence of window sizes and local variation of transcriptional activity a sliding window of size x was shifted by y nucleotides along the genome and each site was tested t = x/y times for being a TSS. The p-value was obtained using the geometric mean
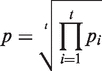 |
where pi denotes the P-value obtained in the i-th test. Note that only sites with a minimum expression of three read starts within a distance of ≤5 nt were tested. Furthermore, we excluded sites in the vicinity of perfectly aligned hit blocks, i.e. stacks of hits that all share a common 5′- and 3′-end. To determine λr, we selected a region size of 500 nt. For the sliding window approach an offset of 50 nt was used. All potential TSSs significant to the p = 0.05 level are listed in Supplementary Table S2. In order to achieve a high positive predictive value for data sets of similar size, these parameters have been fixed globally in our study and may have to be adjusted for the application of the method to other data sets.
Evaluation of the automated TSS annotation method
To evaluate the predictive power of the automated TSS annotation method we used Helicobacter pylori and its manually curated TSS map (4) as reference. A data set of comparable size to the Xcv data set was generated. Reads overlapping with annotated tRNA or rRNA genes were excluded. From the H. pylori data set 40 385 mapped reads of the treated library and 49 845 reads of the untreated library were randomly selected and contained 392 manually annotated TSSs which were used as reference class. TSSs were predicted using the same parameter settings (500 nt window size, 50 nt offset; 0.05 p-value cutoff) as for the Xcv data set. 566 genomic positions met the criteria for being TSS candidates, i.e. the clustering of at least three read starts. These positions represent putative TSSs and were statistically evaluated with the automatic TSS annotation approach, according to (26). The results are summarized in an extended confusion matrix (Supplementary Table S9).
Estimation of expression level
To estimate the expression level of CDSs in Xcv likely to exhibit a proximal promoter, we selected 1276 annotated CDSs in a head-to-head arrangement. The set comprised 549 CDSs with and 727 without annotated TSS. Due to the limited sequencing depth of our data set we combined reads of both libraries and evaluated the coverage of the first 100 nt of CDSs (Supplementary Figure S2).
Detailed information about additional methods is provided in SI.
Further supporting information and the raw sequencing data are available at the official institutional website of the University of Leipzig (http://www.bioinf.uni-leipzig.de/publications/supplements/10-035).
RESULTS
Mapping of sequencing reads
To analyze the primary transcriptome of Xcv, total RNA of strain 85–10 and its derivative 85* were mixed (SI and Supplementary Table S1). Xcv strain 85* carries a chromosomal point mutation in hrpG (hrpG*) leading to expression of the Hrp-regulon. cDNAs were synthesized from total RNA (untreated library; hereafter library 1) and RNA enriched for primary transcripts (treated library; hereafter library 2), respectively (4). dRNA-seq analysis resulted in 160 349 reads for library 1 and 149 596 reads for library 2. A total of 84% of the reads were mapped to the Xcv genome using the program segemehl (27). As previously described, Xcv contains two identical copies of the 5S, 23S and 16S rRNA clusters, respectively, and 56 tRNA loci (9). A total of 63% of the reads of library 1 and 68% of library 2 reads mapped to these genes although the processed rRNAs and tRNAs were expected to be depleted in library 2. Closer examination revealed that the majority of tRNA-read starts in library 2 correspond to the presumed RNase P processing sites rather than TSSs (Supplementary Figure S1). To verify our observations we analyzed all reads overlapping tRNAs in the Helicobacter pylori dRNA-seq data set (4), which supports our findings (Supplementary Figure S1). The abundance of library 2 tRNA reads mapping to putative RNase P processing sites might be due to stable secondary structures formed after RNase P cleavage thus protecting mature tRNAs from exonuclease degradation. We, therefore, discarded the reads mapping to rRNA and tRNA loci and analyzed the remaining 49 845 and 40 385 reads in more detail. While reads of library 1 cover entire genes, the read starts of library 2 are shifted towards the 5′-end of primary transcripts, which permits precise mapping of the TSS of a given gene (Figure 1A, e.g. XCV0520), as described (4).
Figure 1.

Identification of TSSs, promoter elements and analysis of 5′-UTRs. (A) Distribution of dRNA-seq reads in the chromosomal locus of Xcv 85–10 spanning genes XCV0519 to XCV0524. Annotated CDSs and RNAcode high-scoring segments are highlighted in green and blue, respectively. Sequencing reads of library 1 (black) and library 2 (red) are shown on top for the (+)-strand and below for the (−)-strand. Predicted TSSs and corresponding classes are indicated in red. (B) Venn diagram illustrating the TSS classes. TSSs found maximal 300-bp upstream of coding sequences are classified as primary. Internal TSSs are found within and antisense TSSs on the opposite strand of genes (±100 bp). Orphan TSSs do not belong to other classes. (C) Sequence analysis identified a T/A-rich promoter element for 1205 of 1421 putative TSSs. The histogram depicts the position of the conserved sequence pattern relative to the annotated TSSs at position +1. (D) 5′-UTR length distribution. The x-axis is split into linear (0–50) and logarithmic (51–300) scales. The top of the histogram gives the percentage of leaderless (≤10 bp), short (≤50 bp) and longer UTRs (>50 bp).
A statistical model to annotate TSSs
Most of the TSS maps published to date are derived from tedious manual inspection of sequencing data (4,24,28) or using ad hoc heuristics complemented by manual inspection (29–31). Here, we aimed at the automated identification of TSSs based on well-defined criteria, i.e. to discriminate between potential TSSs and the background distribution of read starts. This background, however, is not uniform across the genome but varies depending on gene expression levels. We therefore modeled read starts by Poisson distributions depending on the expression level in a well-defined genomic neighbourhood. Comparing the two libraries, a TSS is defined as a position where the observed difference of read starts in both libraries significantly exceeds the expected differences of read starts modeled by a Skellam distribution from which p-values are readily derived (see ‘Materials and Methods’ section).
Annotation of TSSs
In total, 1372 chromosomal and 49 TSSs on the large plasmid pXCV183 of Xcv (Figure 1B and Supplementary Table S2) were identified. The data confirm TSSs determined previously for selected pathogenicity genes, e.g. hrcU and hrpB1 (20,32). Nevertheless, the majority of TSSs annotated in our study should be considered as putative. TSSs were classified into four categories, i.e. (i) primary TSSs located up to 300 bp 5′ of an annotated translation start, (ii) internal TSSs within an annotated coding sequence (CDS), (iii) antisense TSSs that map to the opposite strand of CDSs ± 100 bp and (iv) orphan TSSs that do not belong to the other three categories. Most of the annotated TSSs are primary TSSs (831) and probably correspond to the 5′-end of mRNAs. Overall, CDSs that lack an assigned TSS exhibit much lower expression levels than CDSs with an annotated TSS (see ‘Materials and Methods’ section and Supplementary Figure S2).
As illustrated in Figure 1B, TSSs can belong to more than one category, e.g. the assumed primary TSS of XCV0523 is also antisense to XCV0522 (Figure 1A). Interestingly, 10% (86/831) of primary TSSs are also classified as internal. Thus, some neighboring CDSs previously supposed to be cotranscribed as part of a polycistronic mRNA can also be transcribed from alternative promoters. As illustrated for XCV0522 (Figure 1A), we identified 71 putative TSSs which are located within the first 50 bp of annotated CDSs suggesting that previously annotated translation starts have to be revisited (Supplementary Table S3). Furthermore, 345 TSSs are located antisense to annotated genes. Interestingly, 41% of these TSSs are also classified as primary TSSs, including 16 TSSs that correspond to overlapping mRNAs in an antisense orientation (Supplementary Table S2). 49 antisense TSSs are positioned in the 3′-region (±100 bp) of annotated sense genes (Supplementary Table S4). In total, antisense reads map to 22% of all nucleotides that belong to annotated CDSs irrespective of read numbers, the presence of a TSS and the expression of the corresponding CDSs. The majority of these antisense reads lack automatically assigned TSSs and do not accumulate in clusters and thus, might not be originated from defined antisense genes. We also compared the sense- and antisense-read coverage of all annotated CDSs in Xcv and did not observe a correlation (data not shown).
Most bacterial δ70-dependent promoters contain conserved sequence elements, i.e. −35 (TTGACA) and −10 (TATAAT) elements present in Escherichia coli (33). In Xcv, there is a weakly conserved T/A-rich motif in the proximity of −10 regions, however, other conserved promoter elements and a Shine-Dalgarno (SD) motif are missing (Figure 1C). This might be due to the high G + C content (65%) of the Xcv genome (9) and is discussed below.
Analysis of 5′-UTRs revealed unexpected size diversity
The lengths of 5′-UTRs deduced from 831 putative primary TSSs range from 0 to >300 bp, with the majority being between 10 and 50 bp (Figure 1D). Surprisingly, 14% of the mRNAs (118 of 831) are leaderless, i.e. their 5′-UTR consists of <10 bp with respect to the annotated genome sequence of Xcv (9). Many of the corresponding genes presumably have housekeeping functions (Supplementary Table S5). In addition, the 5′-UTRs of type III effectors were manually inspected. TSSs of 11 described type III effectors from Xcv strain 85–10 (9,13) were mapped in this study (Supplementary Table S5). The promoter regions of nine effector genes contain a PIP box (consensus TTCG-N16-TTCG) (20). The assumed lengths of the 5′-UTRs of avrBs2, xopE2, xopJ1 and xopO are average. Curiously, the avrRxv mRNA is leaderless, and six mRNAs (avrBs1, xopAA, xopB, xopC, xopD and xopN) (9,13) contain unusually long 5′-UTRs, ranging from 173 to 678 bp. Consequently, the CDSs of some effector genes might be considerably larger than previously described (9). Overall, 13% of the Xcv 5′-UTRs are unusually long (150–300 bp; Supplementary Table S5).
Northern blot analysis confirmed 23 sRNAs in Xcv
A computational scan for known RNA elements in Xcv identified already annotated tRNAs, rRNAs and the recently described ptaRNA1 (34). In addition, we identified eight putative riboswitches and widely conserved RNAs, i.e. RNase P-, RtT-, SRP-, tmRNA and 6S-RNA (Figure 2 and Supplementary Table S6). Based on our dRNA-seq data, most of these transcripts were strongly expressed and TSSs were annotated for four of the housekeeping RNAs and five of the predicted riboswitches (Supplementary Table S6). The genes located downstream of the riboswitch candidates are either known to be involved in the respective riboswitch-controlled pathways in other bacteria or, as in case of yybP/ykoY candidates, presumably encode membrane proteins (35–37) (Supplementary Table S6).
Figure 2.

Expression of selected Xcv sRNAs and antisense RNAs depends on HrpG and HrpX. Total RNA isolated from exponential (exp) and stationary phase cultures (stat) of (a) Xcv strain 85–10, (b) 85–10 expressing hrpG* from pFG72-1 and (c) 85–10ΔhrpX carrying pFG72-1 was analyzed by northern blot. Arrows and filled squares indicate signals corresponding to the expected full-length RNA and processing products obtained by transcriptome sequencing, respectively. The open square indicates the expected size of full-length asX4 determined by RACE analysis. The expected size of sX4 according to the sequencing data is marked by an asterisk. 5S rRNA (lower panel) was probed as loading control.
Prior to the automated TSS prediction, we selected 89 sRNA candidates by manual inspection of the sequencing data with a focus on intergenic regions. We used northern blot analysis to experimentally validate sRNA candidates and analyzed their potential coregulation with the T3S system. To this end, RNA was isolated from exponential and stationary phase cultures of NYG-grown Xcv strains 85–10, 85–10 expressing HrpG* and a derivative lacking hrpX (85–10ΔhrpXphrpG*), respectively. Northern hybridizations confirmed 23 new sRNAs, whereas remaining candidates either appeared to correspond to longer transcripts, i.e. UTRs of mRNAs, or were poorly detectable. The latter can be explained by their low abundance in the dRNA-seq data (data not shown).
After completion of bioinformatic analyses, seven verified sRNAs turned out to correspond to cis-encoded antisense RNAs, termed asX1-7 (Table 1, Figure 2 and Supplementary Figure S3). We detected dRNA-seq reads mapping to both antisense RNA and mRNA for six of these transcripts and a few reads mapping to the CDS complementary to asX4, respectively (data not shown). The remaining 16 sRNAs mapped to intergenic regions and were termed sX1-15 and 6S (Table 1, Figures 2, 3A, 4A and Supplementary Figure S3). Intriguingly, three sRNAs (sX15, asX6, asX7) are encoded on the large plasmid, two of which (asX7 and sX15) are in antisense orientation to each other (Table 1 and Supplementary Figure S3). Most sRNA genes were constitutively expressed under the conditions tested, and appeared to accumulate in stationary growth phase either due to higher transcription rates or increased stability, e.g. sX14 and 6S (Figure 2). Interestingly, expression/accumulation of five intergenic sRNAs and three antisense RNAs was affected by the key regulators of hrp gene expression, HrpG and HrpX, suggesting a role of these sRNAs or their targets in the interaction of Xcv with the plant. HrpX-dependent induction of sRNA expression was observed for asX4, sX5, sX8 (Figure 2) and sX12 (see below), whereas sX11 appeared to be HrpG/HrpX-dependently repressed (Supplementary Figure S3). In case of sX4 (Figure 2) and the antisense RNAs asX1 and asX5 (Supplementary Figure S3) the sRNA stability appeared to depend on HrpG and HrpX as well as on the growth phase.
Table 1.
Verified sRNAs (sX) and antisense RNAs (asX) in Xcv
| RNA (Strand) | TSS categorya | Start-Stopb | Library 2c | Library 1c | Expected length (nt)d | Detected length (nt)e | HrpG/HrpX dependencyf | Conservationg |
|---|---|---|---|---|---|---|---|---|
| sX1 (−) | primary: XCV0067 | 78978–78799 | 8 | 5 | 180 | 190 | – | A (5); B (4); C1 (4); C2 (5); C3 (6); D1–3 (4); E (3) |
| 78978–78816 | 8 | 5 | 163 | 170 | ||||
| 78978–78937 | 8 | 5 | 42 | 50 | ||||
| sX2 (+) | orphan | 85087–85196 | 11 | 0 | 110 | 110 | – | A; B; C1–3; D1–3 |
| 85109–85196 | 0 | 5 | 88 | 85 | ||||
| sX3 (+) | orphan | 1233578–1233669 | 28 | 5 | 92 | 85 | – | A |
| 1233611–1233669 | 0 | 14 | 59 | 60 | ||||
| sX4 (+) | orphan | 1235373–1235528 | 5 | 1 | 156 | 280–300 | stability | A; B; C1–3 |
| sX5 (−) | – | 1899107–1899037 | 2 | 0 | 71 | 70 | HrpX–induced | A; B; C1–3; D1–3; G1–2 |
| 1899085–1899037 | 35 | 41 | 49 | 50 | ||||
| sX6 (+) | antisense: XCV1748 | 1971505–1971845 | 15 | 1 | 341 | 350 | – | A; B; C1–3; D1–3 |
| sX7 (+) | – | 1995660–1995754 | 1 | 57 | 95 | 85 | – | A; B; C1–3; D1–3; E |
| sX8 (+) | orphan | 2740875–2740992 | 7 | 3 | 118 | 110 | HrpX–induced | A; F1; H; I; J |
| 2740875–2740956 | 7 | 3 | 82 | 85 | ||||
| sX9 (+) | – | 2929816–2929890 | 2 | 1 | 75 | 75 | – | A; B |
| sX10 (+) | – | 3850318–3850496 | 25 | 17 | 179 | 180 | – | A (5); B (4); C1 (4); C2 (5); C3 (6); D1–3 (4); E (3) |
| 3850415–3850496 | 14 | 6 | 82 | 80 | ||||
| sX11 (+) | orphan | 4069950–4070087 | 35 | 10 | 138 | 130 | HrpG/HrpX–repressed | A; B; C1–3; D1–3 |
| 4070000–4070087 | 0 | 5 | 88 | 100 | ||||
| sX12 (+) | – | 4358796–4358873 | 4 | 2 | 78 | 67 | HrpX–induced | A; B; C1–3; D1–3 |
| sX13 (−) | – | 4810196–4810082 | 2 | 73 | 115 | 105 | – | A; B; C1–3; D1–3; E; G1–2 |
| sX14 (−) | – | 5040690–5040599 | 2 | 16 | 92 | 85 | – | A; B; C–3; D1–3 |
| sX15 (+) | antisense: XCVd0106 and primary: XCVd0107 | #116378–116536 | 80 | 8 | 159 | 150 | – | Ap; G1 (10); G2 |
| #116438–116536 | 2 | 4 | 99 | 100 | ||||
| 6S (+) | orphan | 4037865–4038084 | 2627 | 442 | 220 | 220 | – | A; B; C1–3; D1–3; E; F1–4; G1–2 |
| 4037865–4037952 | 2627 | 442 | 88 | 90 | ||||
| asX1 (+) | – | 447108–447223 | 2 | 0 | 116 | 110 | stability | A; B; C1–3; D1–3;E; F1–4; G1–2 |
| asX2 (−) | orphan | 3290997–3290913 | 12 | 2 | 85 | 75 | – | A; B |
| asX3 (+) | orphan | 4498825–4499001 | 16 | 0 | 177 | 70–600 | – | A; D1–3 |
| asX4 (−) | antisense: XCV4106 | 4701994–4701856 | 23 | 7 | 139 | 140 | HrpX–induced | A; B; C1–3; D1–3 |
| 4701994–4701686 | 23 | 7 | 309 | – | ||||
| asX5 (+) | orphan | 4757260–4757445 | 21 | 2 | 186 | 190 | stability | A |
| 4757360–4757445 | 1 | 7 | 86 | 85 | ||||
| asX6 (+) | antisense: XCVd0099 | #109851–109943 | 35 | 6 | 93 | 90 | – | Ap; Bp; Kp; Lp |
| #109851–109875 | 35 | 6 | 25 | 30 | ||||
| asX7 (−) | – | #116528–116459 | 4 | 4 | 70 | 60 | – | Ap; G1 (10); G2 |
aClassification of the automatically annotated TSS (Figure 1B). bThe 5′- and 3′-positions of the respective dRNA-seq-read clusters on the Xcv chromosome and plasmid pXCV183 (indicated by #). Positions highlighted in bold indicate an automatically annotated TSS (see SI; Supplementary Table S2). Underlined numbers correspond to transcript ends which were verified by 5′- and 3′-RACE, respectively. The underlined 3′-end of asX4 was identified only by RACE. cNumber of read starts at the respective start position given in column ‘Start-Stop’. dTranscript length deduced from dRNA-seq. etranscript size and fHrpG/HrpX dependency of sRNA/antisense RNA accumulation determined by northern blot (Figures 2, 3, 4 and Supplementary Figure S3); ‘stability’ indicates altered amounts of sRNA processing products in dependency of HrpG and/or HrpX. Constitutive expression is indicated by ‘-’. gsequence conservation among other bacteria (see SI). Strains containing homologous sequences and the respective accession numbers are given below. Numbers in brackets indicate the number of homologous sequences in the respective strains if more than one homolog was identified.
A: X. campestris pv. vesicatoria 85–10 (NC_007508)
Ap: X. campestris pv. vesicatoria 85–10 plasmid pXCV183 (NC_007507)
B: X. axonopodis pv. citri 306 (NC_003919)
Bp: X. axonopodis pv. citri 306 plasmid pXAC64 (NC_003922)
C1: X. campestris pv. campestris ATCC 33913 (NC_003902)
C2: X. campestris pv. campestris 8004 (NC_007086)
C3: X. campestris pv. campestris B100 (NC_010688)
D1: X. oryzae pv. oryzae MAFF 311018 (NC_007705)
D2: X. oryzae pv. oryzae KACC 10331 (NC_006834)
D3: X. oryzae pv. oryzae PXO99A (NC_010717)
E: X. albilineans GPE PC73 (NC_013722)
F1: Xylella fastidiosa 9a5c (NC_002488)
F2: Xylella fastidiosa Temecula1 (NC_004556)
F3: Xylella fastidiosa M12 (NC_010513)
F4: Xylella fastidiosa M23 (NC_010577)
G1: Stenotrophomonas maltophilia K279a (NC_010943)
G2: S. maltophilia R551-3 (NC_011071)
H: Burkholderia xenovorans LB400 (NC_007951)
I: Acidovorax sp. JS42 (NC_008782)
J: Bordetella petrii DSM 12804 (NC_010170)
Kp: Ralstonia solanacearum CMR15 plasmid pRSC35 (FP885893)
Lp: X. citri plasmid pXcB (AY228335)
Figure 3.

sX6 encodes a small protein. (A) Expression analysis of the sX6 transcript. Total RNA isolated from exponential (exp) and stationary phase cultures (stat) of (a) Xcv strain 85–10, (b) 85–10 expressing hrpG* from pFG72-1 and (c) 85–10ΔhrpX carrying pFG72-1 was analyzed by northern blot. The expected signal according to sequencing data is indicated by an arrow. 5S rRNA (lower panel) was probed as loading control. (B) Expression of the sX6 protein. Derivatives of Xcv strain 85–10 (wt) carrying promoterless empty vector pBRM-P (−) and sX6-c-Myc expression construct, respectively, were grown to OD600 = 0.7. Protein extracts were analyzed by immunoblotting using c-Myc epitope-specific and GroEL-specific antibodies.
Figure 4.

sX12 is involved in virulence of Xcv. (A) sX12 is HrpX-dependently expressed. Total RNA isolated from exponential (exp) and stationary phase cultures (stat) of (a) Xcv strain 85–10, (b) Xcv expressing hrpG* from pFG72-1 and (c) a derivative deleted in hrpX and carrying pFG72-1 was analyzed by northern blot. The right panel shows a northern blot with RNA from (d) Xcv strain 85–10 and (e) an sX12 deletion mutant carrying empty vector pLAFR6, respectively, and (f) an sX12 deletion mutant ectopically expressing sX12 from psX12. The expected RNA size is indicated by an arrow. The asterisk denotes an unspecific signal. 5S rRNA (lower panel) was probed as loading control. (B) sX12 contributes to virulence and the HR. Strains used in (A) (right panel) were inoculated at a density of 1.25 × 108 CFU ml−1 into leaves of susceptible ECW and resistant ECW-10R pepper plants. Disease symptoms were photographed at 7 days post-inoculation (dpi). The HR was visualized by ethanol bleaching of the leaves at 2 days post-inoculation. Dashed lines indicate the inoculation site.
Processing of sRNAs
In general, the dRNA-seq data and northern blots suggest that Xcv sRNAs do not accumulate as primary transcripts but undergo growth-phase dependent processing. However, in most cases the apparent sizes of full-length and processed sRNAs in northern blots were in agreement with the dRNA-seq data, e.g. sX8 and 6S RNA (Figure 2 and Table 1). In addition to full-length and processing products, northern blots detected unexpectedly long signals, up to 900 nt, for the antisense RNAs asX1, asX2, asX3, asX6 and asX7 (Supplementary Figure S3). These signals may be caused by alternative termination of transcription. The sequencing data also suggest that sX7, sX13 and sX14 represent processing products of longer transcripts since reads mapping to these loci are predominantly found in library 1, and no TSS was identified in library 2 (Table 1). For selected RNAs the 5′- and 3′-ends were determined by RACE (Table 1). While the 5′-end of the antisense RNA asX4 is identical to the TSS identified by dRNA-seq, the 3′-region is 170 nt longer suggesting the presence of a processing site.
Phylogenetic distribution of sRNAs from Xcv
While sX3 and asX5 are unique for Xcv, homology searches revealed that 10 sRNA genes are exclusively found in sequenced Xanthomonas species that encode a hrp-T3S system (Table 1). Four of the latter sRNAs, including sX12 described in more detail below, and asX5 were coregulated with the T3S system.
Two intergenic sRNAs, sX1 and sX10 (Table 1; Supplementary Figure S3) are highly similar in sequence and structure. Three additional homologous genes are predicted and expressed in Xcv and might therefore be considered as an sRNA family. As three to six copies of members of this gene family are found in other Xanthomonas species (Table 1), we propose a functional redundancy of the respective sRNAs.
Interestingly, 10 homologs of the plasmid-encoded and complementary Xcv sX15 and asX7 genes are present in the chromosome of Stenotrophomonas maltophilia strain K279a (38) (Table 1). Moreover, asX6, which is also located on pXCV183 of Xcv, is conserved in plasmids of X. axonopodis pv. citri strain 306 (39), Ralstonia solanacearum strain CMR15 (40) and X. citri (41) (Table 1). A rather erratic phylogenetic distribution was observed for sX8 since homologs are predicted in a small subset of the known genomes of both beta- and gamma-proteobacteria (Table 1). Interestingly, this holds true also for the gene cluster upstream of sX8 which suggests a common evolutionary origin of this region. This type of phylogenetic pattern has in particular been observed for toxin/anti-toxin systems and suggests frequent horizontal transmissions (42).
sX6 encodes a small protein
Using RNAcode, a program, which was applied for the detection of novel protein coding genes in E. coli (43), 24 potential short ORFs were predicted in the Xcv genome (see SI and Supplementary Table S7). dRNA-seq reads mapped to 12 of these loci. One example is sX6 (341 nt), which is constitutively expressed (Figure 3A) and has a predicted coding capacity of 80 amino acids including a signal peptide in the N-terminal region. We generated a translational fusion of sX6 with a C-terminal c-Myc epitope tag, under control of the native sX6 promoter (44), and introduced the expression construct into Xcv strain 85–10. As shown in Figure 3B, a fusion protein of the predicted molecular mass (∼12 kDa) was detectable in protein extracts of Xcv.
Besides sX6, TSSs for two of the predicted ORFs with a coding capacity of 36 and 67 amino acids, respectively, were predicted (Supplementary Table S7). Interestingly, homologs of genes for the three small proteins are exclusively found in xanthomonads encoding a hrp-T3S system.
sX12 contributes to virulence
The fact that several sRNAs are expressed under control of the T3S system regulators suggested a possible role in virulence. Here, we focused on sX12 whose size of 78 nt was confirmed by 5′- and 3′-RACE (Table 1). As mentioned above, expression of sX12 is HrpX-dependently induced and accumulates in stationary growth phase (Figure 4A). To assess the contribution of sX12 to virulence we generated a deletion mutant derivative of strain 85–10 (ΔsX12). While growth of strain ΔsX12 in planta was as wild-type (Supplementary Figure S4), plant reactions were altered. Disease symptoms in leaves of infected susceptible (ECW) and the HR in resistant (ECW-10R) pepper plants were delayed with strain ΔsX12 when compared to the wild-type (Figure 4B). The ΔsX12 mutant phenotype was complemented by ectopic expression of sX12 under control of its own promoter (Figure 4). We also performed T3S assays to analyze whether the delay in plant reactions by strain ΔsX12 might be due to reduced protein levels of T3S system components, e.g. the conserved apparatus component HrcJ, or the secretion of T3S substrates, i.e. the translocon protein HrpF. However, the detected protein amounts and the secretion of HrpF were comparable for the wild-type and the ΔsX12 mutant (Supplementary Figure S4).
DISCUSSION
The dRNA-seq-based analysis of the Xcv transcriptome led to remarkable insights into the transcriptional landscape of this important model plant pathogen and identified an sRNA with a role in virulence. In this study, we have devised a new method to automatically generate maps of TSSs for dRNA-seq data sets alleviating the need for manual inspection and allowing application of dRNA-seq also for larger genomes than Xcv. In contrast to earlier dRNA-seq approaches, mostly based on laborious manual inspection of sequencing data (4,24,28), the presented computational approach provides a measure of statistical confidence and ensures that predictions are comparable between different studies as demonstrated by our comparative analysis between manual and automated annotation of the previously published H. pylori transcriptome (4). While the sensitivity of 82% demonstrates the method's capability of recovering manually annotated TSS at exactly the same position, a positive predictive value of 72% indicates its reliability (Supplementary Table S9). However, to dynamically adjust parameters such as significance levels the method remains subject to further research. We used only exact matches of the manual and automated TSS map for this analysis. The number of false positives and negatives might therefore be overestimated and suffers from biases introduced by manual inspection. Several parameters including window sizes to determine local expression levels, minimum coverage and significance thresholds to control for sensitivity and specificity have been fixed globally for this study.
We annotated 1421 putative TSSs in Xcv (Supplementary Table S2) including riboswitches and genes for conserved housekeeping and novel sRNAs. Interestingly, 178 TSSs correspond to antisense transcripts including six that map to type III effector genes and to hrcC, which is transcriptionally induced by HrpX and essential for T3S and pathogenicity (Supplementary Tables S2 and S4) (10,45). The potential role of post-transcriptional regulation in Xcv is further supported by the finding that 22% of all nucleotides that belong to annotated CDSs are covered by antisense reads. Nevertheless, the majority of these reads might be derived from promiscuous transcription initiation as it was also suggested for E. coli (46). It remains to be clarified whether the identified antisense transcripts in Xcv represent functional gene products or the transcription itself has a regulatory function.
We identified 831 putative primary TSSs, which were assigned to 17.35% of the 4726 annotated CDSs (Figure 1B and Supplementary Table S2) (9). Similarly, in the archeon Methanosarcina mazei TSSs for ∼20% of the CDSs were assigned (24). A considerably larger number of TSSs corresponding to 60% of the CDSs were recently mapped in the plant symbiont Sinorhizobium meliloti (31) and the human pathogen H. pylori (>50%) (4). This might be explained by the plethora of conditions analyzed and/or the higher number of sequencing reads and is supported by our finding that TSSs in Xcv are predominantly assigned to CDSs with high expression level whereas CDSs without assigned TSS are generally weakly or not expressed (Supplementary Figure S2).
In Xcv, the majority of 5′-UTRs appears to be <50 bp (Figure 1D), which is characteristic for bacteria (3). Surprisingly, there is no clear consensus sequence for ribosome binding. A recent study (47) analyzed the evolutionary process of translation initiation in prokaryotes and found that a SD-initiated translation in xanthomonads is unlikely. In good agreement with this, we identified an unexpected high number of leaderless mRNAs in Xcv (Supplementary Table S5) suggesting an alternative mechanism of ribosome guidance. In Xcv, transcription of 82% of the leaderless mRNAs starts with AUG which was shown to be essential for stable ribosome binding to these transcripts in E. coli (48).
Unusually long 5′-UTRs as identified here for Xcv might be indicative of extensive post-transcriptional regulation, e.g. by sRNA-mediated modulation of mRNA translation or transcript stability. Since also 5′-UTRs of genes that encode type III effector proteins are unusually long (Supplementary Table S5), this might indicate a role of these 5′-UTRs in virulence. For instance, the genes for the type III effector proteins XopN and XopAA, shown to be important virulence factors of Xcv (49,50), comprise 5′-UTRs of 173 and 477 bp, respectively (Supplementary Table S5). In H. pylori, mRNAs of genes involved in pathogenesis also carry long 5′-UTRs (4).
Another potential implication of the high number of unusually long 5′-UTRs in Xcv is that the respective CDSs might be longer than predicted by the genome annotation as shown recently for the type III effector protein XopD (51). On the other hand, a number of CDSs are presumably shorter than annotated, because 71 internal TSSs are located within the first 50 bp of annotated CDSs (Supplementary Table S3). We also identified 12 expressed new loci with potential coding capacity (Supplementary Table S7) exemplified by sX6 that encodes an 80 amino acid protein (Figure 3). Hence, this study contributes to a first refinement of CDS annotation in Xcv.
sRNAs represent important post-transcriptional regulators involved in a variety of processes such as quorum sensing (52) and virulence (53). In this study, the combination of manual and automatic inspection of the cDNA sequencing data and northern blots verified 23 sRNAs in Xcv, seven of which represent antisense RNAs (Table 1). For six of the antisense RNAs we also detected expression of the complementary mRNAs. It should be noted, however, that our data do not allow distinguishing between cells that express both transcripts at the same time and cells that either express the mRNA or the antisense RNA.
Notably, expression of five intergenic sRNAs and three antisense RNAs verified in this study was affected by the master regulators of Xcv virulence, HrpG and/or HrpX (Table 1) (16,18,19). Coregulation of sRNA expression with the T3S system clearly suggests a role of these transcripts in the interaction of Xcv with its host plant. As a proof-of-principle, we have demonstrated that sX12 contributes to virulence of Xcv (Figure 4B). Lack of sX12 does not affect bacterial growth inside the host and T3S, i.e. bacterial fitness is not impaired (Supplementary Figure S4). What might be the targets of sX12? Preliminary experiments did not reveal an effect of the absence of sX12 on selected hrp (T3S) genes, i.e. transcript and protein accumulation was unaltered. Instead of regulating mRNA targets, sX12 might control gene expression in a different manner, e.g. by binding to proteins, DNA or metabolites. Furthermore, sX12 might impinge on the efficiency of the T3S system, similar to the Salmonella typhimurium sRNA IsrJ which accumulates under infection conditions. IsrJ positively contributes to invasion and effector translocation (54).
After our analysis was complete, the identification of eight sRNAs in Xanthomonas oryzae pv. oryzae (Xoo) strain PXO99A was reported (55). In agreement with our data, the Xoo sRNAs, Xoo3, Xoo4 and Xoo6, represent orthologs of the Xcv RNAs sX14, asX4 and sX1, respectively (Table 1). Contrary to Xoo4 (55), which is 145 nt, our analyses revealed that asX4 in Xcv is 309-nt long and encoded antisense to an annotated CDS. We also identified potential TSSs for the Xcv homologs of Xoo1 and Xoo5, whereas Xcv lacks homologs of described bacterial sRNA genes except for housekeeping RNAs. Vice versa, the majority of sRNAs identified in Xcv is restricted to the genera Xanthomonas, Xylella and Stenotrophomonas (Table 1) and thus, reflects the current taxonomy (56). An estimation of the total number of sRNAs in Xcv is hampered by the relatively small number of sequence reads and the fact that, for example, TSSs of sRNA genes in the proximity (≤300 bp) of downstream CDSs are classified as primary TSSs (see sX1; Table 1).
A remarkable finding of this study is the indication of frequent processing of Xcv sRNAs, which appears to be growth-phase dependent. In several studies, sRNA processing was shown to affect sRNA activity, e.g. GlmZ from E. coli which is cleaved and thus inactivated (57,58). The E. coli sRNA IstR-1 is rendered inactive by RNase III-dependent cleavage upon sRNA-mRNA interaction (59). In contrast, MicX from Vibrio cholerae is stabilized by RNaseE-mediated cleavage which does not impair its interaction with target-mRNAs (60). Which ribonucleases are involved in processing of the Xcv sRNAs is not known.
The analysis of additional knock-out mutants is needed to assess sRNA functions in Xcv. In case of virulence phenotypes, a challenge will be the identification of the targets. Besides possible effects of sRNAs on mRNAs the target can also be an RNA-binding protein. To the best of our knowledge, the only reported sRNAs involved in the regulation of virulence gene expression in plant pathogenic bacteria are members of the RsmB family which was studied in Erwinia carotovora ssp. carotovora. RsmB antagonizes the RNA-binding protein RsmA that acts as translational repressor (61–63). Although a major virulence function was reported for RsmA from X. campestris pv. campestris the interacting sRNAs are not known yet (5). The latter is complicated by the lack of CsrB/RsmB sequence homologs in xanthomonads.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supporting Information (SI), Supplementary Figures S1–4, Supplementary Tables S1–9 and Supplementary References [64–76].
FUNDING
Deutsche Forschungsgemeinschaft as part of the priority program ‘Sensory and Regulatory RNAs in Prokaryotes’ (SPP 1258, to U.B., P.F.S. and J.V.); ‘Graduiertenkolleg’ (GRK 1591, to U.B.); Bundesministerium für Bildung und Forschung (‘GenoMik-Plus’ network, to U.B.); LIFE Leipzig Research Center for Civilization Diseases, Universität Leipzig; European Social Fund and the Free State of Saxony. Funding for open access charge: Deutsche Forschungsgemeinschaft as part of the priority program ‘Sensory and Regulatory RNAs in Prokaryotes’ (SPP 1258, to U.B., P.F.S. and J.V.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to B. Rosinsky and C. Kretschmer for technical assistance. The authors thank Richard Reinhardt (MPI for Molecular Genetics, Berlin, Germany) for 454 sequencing and Daniela Büttner for helpful comments on the manuscript.
REFERENCES
- 1.van Vliet AH. Next generation sequencing of microbial transcriptomes: challenges and opportunities. FEMS Microbiol. Lett. 2010;302:1–7. doi: 10.1111/j.1574-6968.2009.01767.x. [DOI] [PubMed] [Google Scholar]
- 2.Croucher NJ, Thomson NR. Studying bacterial transcriptomes using RNA-seq. Curr. Opin. Microbiol. 2010;13:619–624. doi: 10.1016/j.mib.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sorek R, Cossart P. Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat. Rev. Genet. 2010;11:9–16. doi: 10.1038/nrg2695. [DOI] [PubMed] [Google Scholar]
- 4.Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermüller J, Reinhardt R, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464:250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 5.Chao NX, Wei K, Chen Q, Meng QL, Tang DJ, He YQ, Lu GT, Jiang BL, Liang XX, Feng JX, et al. The rsmA-like gene rsmAXcc of Xanthomonas campestris pv. campestris is involved in the control of various cellular processes, including pathogenesis. Mol. Plant-Microbe Interact. 2008;21:411–423. doi: 10.1094/MPMI-21-4-0411. [DOI] [PubMed] [Google Scholar]
- 6.Plener L, Manfredi P, Valls M, Genin S. PrhG, a transcriptional regulator responding to growth conditions, is involved in the control of the type III secretion system regulon in Ralstonia solanacearum. J. Bacteriol. 2010;192:1011–1019. doi: 10.1128/JB.01189-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Filiatrault MJ, Stodghill PV, Bronstein PA, Moll S, Lindeberg M, Grills G, Schweitzer P, Wang W, Schroth GP, Luo S, et al. Transcriptome analysis of Pseudomonas syringae identifies new genes, noncoding RNAs, and antisense activity. J. Bacteriol. 2010;192:2359–2372. doi: 10.1128/JB.01445-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones JB, Stall RE, Bouzar H. Diversity among xanthomonads pathogenic on pepper and tomato. Annu. Rev. Phytopathol. 1998;36:41–58. doi: 10.1146/annurev.phyto.36.1.41. [DOI] [PubMed] [Google Scholar]
- 9.Thieme F, Koebnik R, Bekel T, Berger C, Boch J, Büttner D, Caldana C, Gaigalat L, Goesmann A, Kay S, et al. Insights into genome plasticity and pathogenicity of the plant pathogenic bacterium Xanthomonas campestris pv. vesicatoria revealed by the complete genome sequence. J. Bacteriol. 2005;187:7254–7266. doi: 10.1128/JB.187.21.7254-7266.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonas U, Schulte R, Fenselau S, Minsavage GV, Staskawicz BJ, Stall RE. Isolation of a gene-cluster from Xanthomonas-campestris pv. vesicatoria that determines pathogenicity and the hypersensitive response on pepper and tomato. Mol. Plant-Microbe Interact. 1991;4:81–88. [Google Scholar]
- 11.Szczesny R, Büttner D, Escolar L, Schulze S, Seiferth A, Bonas U. Suppression of the AvrBs1-specific hypersensitive response by the YopJ effector homolog AvrBsT from Xanthomonas depends on a SNF1-related kinase. New Phytol. 2010;187:1058–1074. doi: 10.1111/j.1469-8137.2010.03346.x. [DOI] [PubMed] [Google Scholar]
- 12.Büttner D, Bonas U. Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol. Rev. 2010;34:107–133. doi: 10.1111/j.1574-6976.2009.00192.x. [DOI] [PubMed] [Google Scholar]
- 13.White FF, Potnis N, Jones JB, Koebnik R. The type III effectors of Xanthomonas. Mol. Plant Pathol. 2009;10:749–766. doi: 10.1111/j.1364-3703.2009.00590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White FF, Yang B, Johnson LB. Prospects for understanding avirulence gene function. Curr. Opin. Plant Biol. 2000;3:291–298. doi: 10.1016/s1369-5266(00)00082-0. [DOI] [PubMed] [Google Scholar]
- 15.Klement Z. In: Phytopathogenic Prokaryotes. Mount MS, Lacy GH, editors. Vol. 2. New York: Academic Press; 1982. pp. 149–177. [Google Scholar]
- 16.Wengelnik K, Bonas U. HrpXv, an AraC-type regulator, activates expression of five of the six loci in the hrp cluster of Xanthomonas campestris pv. vesicatoria. J. Bacteriol. 1996;178:3462–3469. doi: 10.1128/jb.178.12.3462-3469.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulte R, Bonas U. Expression of the Xanthomonas campestris pv. vesicatoria hrp gene cluster, which determines pathogenicity and hypersensitivity on pepper and tomato, is plant inducible. J. Bacteriol. 1992;174:815–823. doi: 10.1128/jb.174.3.815-823.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wengelnik K, Van den Ackerveken G, Bonas U. HrpG, a key hrp regulatory protein of Xanthomonas campestris pv. vesicatoria is homologous to two-component response regulators. Mol. Plant-Microbe Interact. 1996;9:704–712. doi: 10.1094/mpmi-9-0704. [DOI] [PubMed] [Google Scholar]
- 19.Noel L, Thieme F, Nennstiel D, Bonas U. cDNA-AFLP analysis unravels a genome-wide hrpG-regulon in the plant pathogen Xanthomonas campestris pv. vesicatoria. Mol. Microbiol. 2001;41:1271–1281. doi: 10.1046/j.1365-2958.2001.02567.x. [DOI] [PubMed] [Google Scholar]
- 20.Koebnik R, Krüger A, Thieme F, Urban A, Bonas U. Specific binding of the Xanthomonas campestris pv. vesicatoria AraC-type transcriptional activator HrpX to plant-inducible promoter boxes. J. Bacteriol. 2006;188:7652–7660. doi: 10.1128/JB.00795-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wengelnik K, Rossier O, Bonas U. Mutations in the regulatory gene hrpG of Xanthomonas campestris pv. vesicatoria result in constitutive expression of all hrp genes. J. Bacteriol. 1999;181:6828–6831. doi: 10.1128/jb.181.21.6828-6831.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hartmann RK, Bindereif A, Schön A, Westhof E. Handbook of RNA biochemistry. Vol. 2. Germany: Wiley-VCH, Weinheim; 2005. pp. 636–637. [Google Scholar]
- 23.Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EG, Margalit H, Altuvia S. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr. Biol. 2001;11:941–950. doi: 10.1016/s0960-9822(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 24.Jäger D, Sharma CM, Thomsen J, Ehlers C, Vogel J, Schmitz RA. Deep sequencing analysis of the Methanosarcina mazei Gö1 transcriptome in response to nitrogen availability. Proc. Natl Acad. Sci. USA. 2009;106:21878–21882. doi: 10.1073/pnas.0909051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skellam JG. The frequency distribution of the difference between two Poisson variates belonging to different populations. J. R. Stat. Soc. Ser. A. 1946;109:296. [PubMed] [Google Scholar]
- 26.Fawcett T. An introduction to ROC analysis. Pattern Recogn. Lett. 2006;27:861–874. [Google Scholar]
- 27.Hoffmann S, Otto C, Kurtz S, Sharma CM, Khaitovich P, Vogel J, Stadler PF, Hackermüller J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009;5:10. doi: 10.1371/journal.pcbi.1000502. 1371/journal.pcbi.1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albrecht M, Sharma CM, Reinhardt R, Vogel J, Rudel T. Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res. 2010;38:868–877. doi: 10.1093/nar/gkp1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wurtzel O, Sapra R, Chen F, Zhu Y, Simmons BA, Sorek R. A single-base resolution map of an archaeal transcriptome. Genome Res. 2010;20:133–141. doi: 10.1101/gr.100396.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitschke J, Georg J, Scholz I, Sharma CM, Dienst D, Bantscheff J, Voss B, Steglich C, Wilde A, Vogel J, et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc. Natl Acad. Sci. USA. 2011;108:2124–2129. doi: 10.1073/pnas.1015154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schlüter JP, Reinkensmeier J, Daschkey S, Evguenieva-Hackenberg E, Janssen S, Jänicke S, Becker JD, Giegerich R, Becker A. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genomics. 2010;11:1111–1245. doi: 10.1186/1471-2164-11-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fenselau S, Bonas U. Sequence and expression analysis of the hrpB pathogenicity operon of Xanthomonas campestris pv. vesicatoria which encodes eight proteins with similarity to components of the Hrp, Ysc, Spa, and Fli secretion systems. Mol. Plant-Microbe Interact. 1995;8:845–854. doi: 10.1094/mpmi-8-0845. [DOI] [PubMed] [Google Scholar]
- 33.Burgess RR, Anthony L. How sigma docks to RNA polymerase and what sigma does. Curr. Opin. Microbiol. 2001;4:126–131. doi: 10.1016/s1369-5274(00)00177-6. [DOI] [PubMed] [Google Scholar]
- 34.Findeiß S, Schmidtke C, Stadler PF, Bonas U. A novel family of plasmid-transferred anti-sense ncRNAs. RNA Biol. 2010;7:120–124. doi: 10.4161/rna.7.2.11184. [DOI] [PubMed] [Google Scholar]
- 35.Barrick JE, Corbino KA, Winkler WC, Nahvi A, Mandal M, Collins J, Lee M, Roth A, Sudarsan N, Jona I, et al. New RNA motifs suggest an expanded scope for riboswitches in bacterial genetic control. Proc. Natl Acad. Sci. USA. 2004;101:6421–6426. doi: 10.1073/pnas.0308014101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang JX, Breaker RR. Riboswitches that sense S-adenosylmethionine and S-adenosylhomocysteine. Biochem. Cell Biol. 2008;86:157–168. doi: 10.1139/O08-008. [DOI] [PubMed] [Google Scholar]
- 37.Winkler WC, Breaker RR. Regulation of bacterial gene expression by riboswitches. Annu. Rev. Microbiol. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- 38.Crossman LC, Gould VC, Dow JM, Vernikos GS, Okazaki A, Sebaihia M, Saunders D, Arrowsmith C, Carver T, Peters N, et al. The complete genome, comparative and functional analysis of Stenotrophomonas maltophilia reveals an organism heavily shielded by drug resistance determinants. Genome Biol. 2008;9:R74. doi: 10.1186/gb-2008-9-4-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.da Silva AC, Ferro JA, Reinach FC, Farah CS, Furlan LR, Quaggio RB, Monteiro-Vitorello CB, Van Sluys MA, Almeida NF, Alves LM, et al. Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature. 2002;417:459–463. doi: 10.1038/417459a. [DOI] [PubMed] [Google Scholar]
- 40.Remenant B, Coupat-Goutaland B, Guidot A, Cellier G, Wicker E, Allen C, Fegan M, Pruvost O, Elbaz M, Calteau A, et al. Genomes of three tomato pathogens within the Ralstonia solanacearum species complex reveal significant evolutionary divergence. BMC Genomics. 2010;11:379. doi: 10.1186/1471-2164-11-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El Yacoubi B, Brunings AM, Yuan Q, Shankar S, Gabriel DW. In planta horizontal transfer of a major pathogenicity effector gene. Appl. Environ. Microbiol. 2007;73:1612–1621. doi: 10.1128/AEM.00261-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makarova KS, Wolf YI, Koonin EV. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct. 2009;4:10. doi: 10.1186/1745-6150-4-19. 1186/1745-6150-1184-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Washietl S, Findeiß S, Müller SA, Kalkhof S, von Bergen M, Hofacker IL, Stadler PF, Goldman N. RNAcode: robust discrimination of coding and noncoding regions in comparative sequence data. RNA. 2011;17:578–594. doi: 10.1261/rna.2536111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szczesny R, Jordan M, Schramm C, Schulz S, Cogez V, Bonas U, Büttner D. Functional characterization of the Xcs and Xps type II secretion systems from the plant pathogenic bacterium Xanthomonas campestris pv. vesicatoria. New Phytol. 2010;187:983–1002. doi: 10.1111/j.1469-8137.2010.03312.x. [DOI] [PubMed] [Google Scholar]
- 45.Wengelnik K, Marie C, Russel M, Bonas U. Expression and localization of HrpA1, a protein of Xanthomonas campestris pv. vesicatoria essential for pathogenicity and induction ofthe hypersensitive reaction. J. Bacteriol. 1996;178:1061–1069. doi: 10.1128/jb.178.4.1061-1069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dornenburg JE, Devita AM, Palumbo MJ, Wade JT. Widespread antisense transcription in Escherichia coli. MBio. 2010;46:e00024–10. doi: 10.1128/mBio.00024-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakagawa S, Niimura Y, Miura K, Gojobori T. Dynamic evolution of translation initiation mechanisms in prokaryotes. Proc. Natl Acad. Sci. USA. 2010;107:6382–6387. doi: 10.1073/pnas.1002036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brock JE, Pourshahian S, Giliberti J, Limbach PA, Janssen GR. Ribosomes bind leaderless mRNA in Escherichia coli through recognition of their 5′-terminal AUG. RNA. 2008;14:2159–2169. doi: 10.1261/rna.1089208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim JG, Li X, Roden JA, Taylor KW, Aakre CD, Su B, Lalonde S, Kirik A, Chen Y, Baranage G, et al. Xanthomonas T3S Effector XopN Suppresses PAMP-Triggered Immunity and Interacts with a Tomato Atypical Receptor-Like Kinase and TFT1. Plant Cell. 2009;21:1305–1323. doi: 10.1105/tpc.108.063123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morales CQ, Posada J, Macneale E, Franklin D, Rivas I, Bravo M, Minsavage J, Stall RE, Whalen MC. Functional analysis of the early chlorosis factor gene. Mol. Plant-Microbe Interact. 2005;18:477–486. doi: 10.1094/MPMI-18-0477. [DOI] [PubMed] [Google Scholar]
- 51.Canonne J, Marino D, Noel LD, Arechaga I, Pichereaux C, Rossignol M, Roby D, Rivas S. Detection and functional characterization of a 215 amino acid N-terminal extension in the xanthomonas type III effector XopD. PLoS One. 2010;5:e15773. doi: 10.1371/journal.pone.0015773. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Bejerano-Sagie M, Xavier KB. The role of small RNAs in quorum sensing. Curr. Opin. Microbiol. 2007;10:189–198. doi: 10.1016/j.mib.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 53.Papenfort K, Vogel J. Regulatory RNA in bacterial pathogens. Cell Host Microbe. 2010;8:116–127. doi: 10.1016/j.chom.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 54.Padalon-Brauch G, Hershberg R, Elgrably-Weiss M, Baruch K, Rosenshine I, Margalit H, Altuvia S. Small RNAs encoded within genetic islands of Salmonella typhimurium show host-induced expression and role in virulence. Nucleic Acids Res. 2008;36:1913–1927. doi: 10.1093/nar/gkn050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liang H, Zhao YT, Zhang JQ, Wang XJ, Fang RX, Jia YT. Identification and functional characterization of small non-coding RNAs in Xanthomonas oryzae pathovar oryzae. BMC Genomics. 2011;12:10. doi: 10.1186/1471-2164-12-87. 1186/1471-2164-1112-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cutino-Jimenez AM, Martins-Pinheiro M, Lima WC, Martin-Tornet A, Morales OG, Menck CF. Evolutionary placement of Xanthomonadales based on conserved protein signature sequences. Mol. Phylogenet. Evol. 2010;54:524–534. doi: 10.1016/j.ympev.2009.09.026. [DOI] [PubMed] [Google Scholar]
- 57.Kalamorz F, Reichenbach B, Marz W, Rak B, Görke B. Feedback control of glucosamine-6-phosphate synthase GlmS expression depends on the small RNA GlmZ and involves the novel protein YhbJ in Escherichia coli. Mol. Microbiol. 2007;65:1518–1533. doi: 10.1111/j.1365-2958.2007.05888.x. [DOI] [PubMed] [Google Scholar]
- 58.Urban JH, Vogel J. Two seemingly homologous noncoding RNAs act hierarchically to activate glmS mRNA translation. PLoS Biol. 2008;6:e64. doi: 10.1371/journal.pbio.0060064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vogel J, Argaman L, Wagner EG, Altuvia S. The small RNA IstR inhibits synthesis of an SOS-induced toxic peptide. Curr. Biol. 2004;14:2271–2276. doi: 10.1016/j.cub.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 60.Davis BM, Waldor MK. RNase E-dependent processing stabilizes MicX, a Vibrio cholerae sRNA. Mol. Microbiol. 2007;65:373–385. doi: 10.1111/j.1365-2958.2007.05796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cui Y, Chatterjee A, Liu Y, Dumenyo CK, Chatterjee AK. Identification of a global repressor gene, rsmA, of Erwinia carotovora subsp. carotovora that controls extracellular enzymes, N-(3-oxohexanoyl)-L-homoserine lactone, and pathogenicity in soft-rotting Erwinia spp. J. Bacteriol. 1995;177:5108–5115. doi: 10.1128/jb.177.17.5108-5115.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu Y, Cui Y, Mukherjee A, Chatterjee AK. Characterization of a novel RNA regulator of Erwinia carotovora ssp. carotovora that controls production of extracellular enzymes and secondary metabolites. Mol. Microbiol. 1998;29:219–234. doi: 10.1046/j.1365-2958.1998.00924.x. [DOI] [PubMed] [Google Scholar]
- 63.Cui Y, Chatterjee A, Yang H, Chatterjee AK. Regulatory network controlling extracellular proteins in Erwinia carotovora subsp. carotovora: FlhDC, the master regulator of flagellar genes, activates rsmB regulatory RNA production by affecting gacA and hexA (lrhA) expression. J. Bacteriol. 2008;190:4610–4623. doi: 10.1128/JB.01828-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berezikov E, Thuemmler F, van Laake LW, Kondova I, Bontrop R, Cuppen E, Plasterk RH. Diversity of microRNAs in human and chimpanzee brain. Nat. Genet. 2006;38:1375–1377. doi: 10.1038/ng1914. [DOI] [PubMed] [Google Scholar]
- 65.Bailey TL, Elkan C. The value of prior knowledge in discovering motifs with MEME. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1995;3:21–29. [PubMed] [Google Scholar]
- 66.Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hertel J, de Jong D, Marz M, Rose D, Tafer H, Tanzer A, Schierwater B, Stadler PF. Non-coding RNA annotation of the genome of Trichoplax adhaerens. Nucleic Acids Res. 2009;37:1602–1615. doi: 10.1093/nar/gkn1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Will S, Reiche K, Hofacker IL, Stadler PF, Backofen R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007;3:e65. doi: 10.1371/journal.pcbi.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Büttner D, Nennstiel D, Klüsener B, Bonas U. Functional analysis of HrpF, a putative type III translocon protein from Xanthomonas campestris pv. vesicatoria. J. Bacteriol. 2002;184:2389–2398. doi: 10.1128/JB.184.9.2389-2398.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rossier O, Van den Ackerveken G, Bonas U. HrpB2 and HrpF from Xanthomonas are type III-secreted proteins and essential for pathogenicity and recognition by the host plant. Mol. Microbiol. 2000;38:828–838. doi: 10.1046/j.1365-2958.2000.02173.x. [DOI] [PubMed] [Google Scholar]
- 71.Canteros BI. 1990. Ph.D. thesis. University of Florida, Gainesville, FL. [Google Scholar]
- 72.Menard R, Sansonetti PJ, Parsot C. Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexneri entry into epithelial cells. J. Bacteriol. 1993;175:5899–5906. doi: 10.1128/jb.175.18.5899-5906.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bonas U, Stall RE, Staskawicz B. Genetic and structural characterization of the avirulence gene avrBs3 from Xanthomonas campestris pv. vesicatoria. Mol. Gen. Genet. 1989;218:127–136. doi: 10.1007/BF00330575. [DOI] [PubMed] [Google Scholar]
- 74.Huguet E, Hahn K, Wengelnik K, Bonas U. hpaA mutants of Xanthomonas campestris pv. vesicatoria are affected in pathogenicity but retain the ability to induce host-specific hypersensitive reaction. Mol. Microbiol. 1998;29:1379–1390. doi: 10.1046/j.1365-2958.1998.01019.x. [DOI] [PubMed] [Google Scholar]
- 75.Figurski DH, Helinski DR. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl Acad. Sci. USA. 1979;76:1648–1652. doi: 10.1073/pnas.76.4.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gardner PP, Daub J, Tate J, Moore BL, Osuch IH, Griffiths-Jones S, Finn RD, Nawrocki EP, Kolbe DL, Eddy SR, et al. Rfam: Wikipedia, clans and the ‘decimal’ release. Nucleic Acids Res. 2010;39:D141–145. doi: 10.1093/nar/gkq1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.