

Abstract
Abstract
Neuromuscular acetylcholine receptors have long been a model system for understanding the mechanisms of operation of ligand-gated ion channels and fast chemical synapses. These five subunit membrane proteins have two allosteric (transmitter) binding sites and a distant ion channel domain. Occupation of the binding sites by agonist molecules transiently increases the probability that the channel is ion-permeable. Recent experiments show that the Monod, Wyman and Changeux formalism for allosteric proteins, originally developed for haemoglobin, is an excellent model for acetylcholine receptors. By using mutations and single-channel electrophysiology, the gating equilibrium constants for receptors with zero, one or two bound agonist molecules, and the agonist association and dissociation rate constants from both the closed- and open-channel conformations, have been estimated experimentally. The change in affinity for each transmitter molecule between closed and open conformations provides ∼–5.1 kcal mol−1 towards the global gating isomerization of the protein.
Anthony Auerbach (State University of New York at Buffalo) did undergraduate work in chemistry at Cornell University and postgraduate work with Jose del Castillo and Frederick Sachs. He has been at SUNY Buffalo working on acetylcholine receptor-channels since 1981. His focus is on understanding the energetics and internal machinery of ligand-gated ion channels.
 |
In 1965, Monod, Wyman and Changeux (Monod et al. 1965) (MWC) presented a beautifully simple model for allosteric proteins. With just two rules – (i) the protein reversibly adopts two global conformations and (ii) each conformation has a different affinity for the ‘allosteric’ ligand – the model could account for the sigmoidal nature of oxygen binding to haemoglobin. The relevance of MWC to acetylcholine receptor-channels (AChRs) was quickly recognized (Karlin, 1967), but in wild-type (wt) neuromuscular AChRs the less-than-fully-occupied open states of the scheme are rarely visited and, hence, are both difficult to quantify and irrelevant to describing cellular responses. As a consequence, the application of MWC to AChRs remained undeveloped. The model was widely believed to be correct, but with little supporting experimental proof.
The MWC mechanism (Fig. 1) is a cycle in which un-, mono- and di-liganded AChRs all undergo the same essential gating isomerization, with equilibrium constants E0, E1 and E2, respectively. In 1840, Germain Henri Hess discovered that in cyclic reaction schemes ‘…the quantity of heat evolved is always constant whether the combination is performed directly or whether it takes place indirectly and in different steps’ (Leicester & Klickstein, 1968). For the outer cycle in Fig. 1 this means that the product of the equilibrium constants for the clockwise path is equal to that for the anti-clockwise path. We use the symbols Kd and Jd for the equilibrium dissociation constants for binding to closed and open AChRs. Therefore, according to Hess's law, E2/E0=λ2 where λ=Kd/Jd. This affinity ratio, sometimes called the coupling constant, quantifies the ‘bang’ provided by the ligand to promote gating. λ is the factor by which the gating equilibrium constant increases with each added agonist molecule, and the natural logarithm of λ is proportional to the energy derived from the change in affinity for each agonist molecule used to promote gating. If we can measure both E2 and E0 then we can compute λ and estimate this energy.
Figure 1. The MWC model for AChRs.
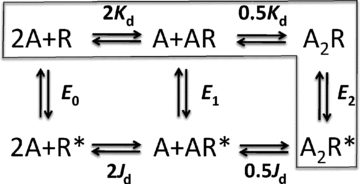
R is the low-affinity, closed-channel conformation, R* is the high-affinity, open-channel conformation and A is the agonist. Kd and Jd are the agonist equilibrium dissociation constants to R and R*. En is the gating equilibrium constant with n bound agonist molecules. The two binding sites of the adult mouse AChR are equivalent. Desensitized states (not shown) would be attached to both R and R* states, although this process mainly proceeds from R*. The boxed states are the ‘main’ states of the AChR that generate cellular responses. The unboxed states are rarely visited but knowledge of the corresponding equilibrium constants is essential because they complete the cycle and, therefore, allow the estimation of λ=Kd/Jd. The logarithm of λ is the energy derived from the change in agonist affinity that powers R↔R* gating.
It is relatively easy to measure E2 (Fig. 2). When agonists are applied at high concentrations, for example to a cell expressing neuromuscular AChRs, the single-channel currents occur as clusters of openings that arise from multiple cycles of binding and gating from an individual AChR. The silent periods between clusters are epochs when all of the AChRs are desensitized. By measuring the durations of intervals only within clusters, events associated with desensitization are removed from the analysis. Making intra-cluster interval duration measurements over a range of agonist concentrations allows the estimation of E2 (and Kd). In adult mouse wild-type (wt) AChRs at –70 mV and 23°C, E2wt,ACh≍ 25. When the two AChR transmitter sites are both occupied by ACh, the channel adopts the open conformation, transiently, with a ∼96% probability.
Figure 2. Estimating E2.
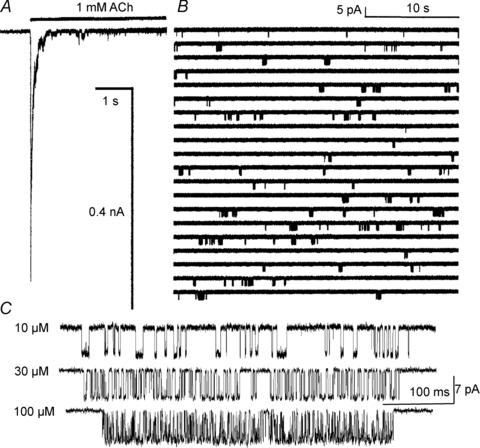
A, a step to a high ACh concentration in an outside-out patch. The rapid activation (downward) reflects binding to R and A2R↔A2R* gating (boxed states, Fig. 1) and the slower decay phase reflects entry into desensitized states (mainly from A2R*). Note the openings even after prolonged application. B, steady-state currents from a cell-attached patch (1 mm ACh). Channel openings (down) occur in clusters that represent the binding and gating activity of a single AChR. In the silent periods between clusters all AChRs in the patch are desensitized. C, the probability of being open within a cluster decreases with lower agonist concentrations because the un- and mono-liganded R states are increasingly occupied. Desensitization is removed from the analysis by fitting only intra-cluster interval durations by the ‘main’ part of the MWC scheme. E2≍ 25 and Kd≍ 150 μm for ACh and wt adult mouse AChRs (23°C, −70 mV).
Measuring E0, the ‘allosteric’ constant, is more difficult. In wt AChRs unliganded openings are both rare and brief and accurate estimates of E0 (and, hence, λ) cannot be readily obtained. Early measures of spontaneous activity of wt AChRs, from either ion flux (Neubig et al. 1982) or electrophysiology (Jackson, 1986) experiments, showed that E0 was small, ∼10−7. Later, mutations were used to increase the frequency of unliganded openings. It was found that adding even more mutations of amino acids that were far from the pore and that were known to decrease E2 also decreased the frequency of spontaneous openings (Grosman & Auerbach, 2000). This suggested that the spontaneous openings did not arise from a local fluctuation in the structure of the pore but rather from a global isomerization of the entire protein, as predicted by MWC.
In order to gain an accurate estimate of E0wt we made use of a database of >1000 different AChR mutants, all of which had been quantified (at the single-channel level) with regard to their effects on E2. Most of the mutations increased E2 and were far from the transmitter binding site, so we assumed that the increase was caused only by a parallel increase in E0 and that they had no effect on λ. As shown in Fig. 3, in these mutant AChRs the frequency of spontaneous openings was greater than in wt AChRs. Moreover, combining mutations further increased E0 (Purohit & Auerbach, 2009). If the effect of each separate mutation on E0 was energetically independent of the others, then the net effect of a combination should be the product of the fold-changes in E0 for each mutation alone. These high gain-of-function combinations could result in spontaneous openings occurring in discrete clusters, each of which reflected the unliganded gating of a single AChR. This clustering indicates that AChRs can enter and recover from states associated with desensitization even if there are no ligands present at the transmitter binding sites.
Figure 3. Estimating E0.
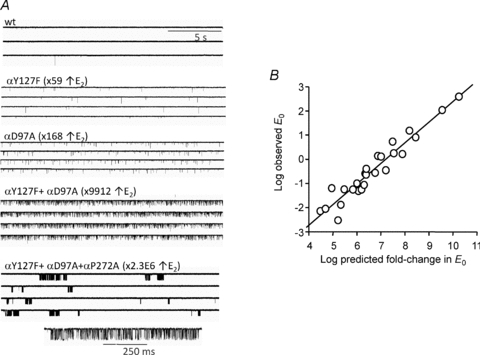
A, unliganded openings are rare in wt AChRs, but increase in frequency in AChRs with mutations that increase E2. Combining mutations further increases unliganded openings, and with very high gain-of-function combinations leads to clusters that reflect the gating behaviour of a single channel in the absence of agonists. B, the predicted fold change in E2 (assuming mutations only alter E0 and have energetically independent effects) and the observed single-channel E0 for the combinations are correlated. The extrapolated ‘observed’ value when the predicted fold change is unity is E0wt≍ 7 × 10−7.
Figure 3B shows a plot of the observed E0 values for a large number of different mutant combinations plotted against the fold-change in E0 predicted from the two assumptions, namely that the mutations only changed E0 and had independent effects. The correlation between the observed and predicted values was linear (on a log scale) over about 6 orders of magnitude, so the assumptions were approximately valid. In order to estimate E0 for wt AChRs we extrapolated the correlation to the condition where the predicted fold change in E0 was equal to 1. The result was E0wt≍ 7 × 10−7.
Mono-liganded openings, too, are infrequent in wt AChRs and represent only a small fraction of the current, even at low agonist concentrations. Early attempts at gaining an estimate of E1 were made by assuming that a brief open component arose from AChRs with only one binding site occupied (Lingle et al. 1992; Hallermann et al. 2005). We took a different approach (Fig. 4) (Jha & Auerbach, 2010). The mutation αW149M nearly eliminates the ability of the transmitter binding site to bind ACh but has little effect on E0 (Purohit & Auerbach, 2010). There are two transmitter binding sites per AChR, so when both mutant and wt α-subunit cDNAs are added to the transfection cocktail (along with wt non-α-subunits) four different types of AChR are expressed, some with two wt binding sites, some with two mutant (non-operational) binding sites and some ‘hybrids’ with one wt and one mutant binding site.
Figure 4. Estimating E1.
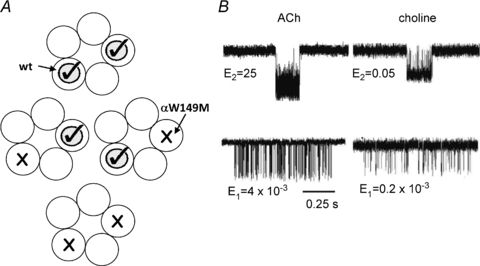
A, expressing both wt and αW149M mutant subunits results in AChRs with either two, one or zero operational transmitter binding sites. B, currents cluster from AChRs with two (top traces) or one (bottom traces) functional binding site(s). The current amplitudes are smaller for choline because of channel block by this agonist. Both ACh and choline, at concentrations that lead to full occupation of the wt site and low occupation of the mutant site, generate mono-liganded current clusters. To assist the analysis, the AChRs had an independent gain-of-function background mutation, either αS269I (ACh) or αP272A (choline). For these agonists the two wt binding sites are equivalent and independent so only one population of mono-liganded clusters is apparent. After correcting for the background, E2ACh≍ 25 and E2choline≍ 0.046, and E1ACh≍ 0.0048 and E1choline≍ 1.6 × 10−4. Using the relationship E0=E12/E2 we estimate from both the ACh and choline results that E0≍ 7 × 10−7.
Figure 4B shows example currents from the wt and hybrid AChRs. Although we might have seen two different hybrid species (with one or the other binding site functional), only one type was apparent. Both hybrid species were indeed expressed because the currents split into two distinct populations upon the addition of a reporter mutation. By analysing the intervals from the hybrid clusters we estimate that the gating equilibrium of monoliganded AChRs is E1wt,ACh≍ 4.3 × 10−3.
From these experiments we conclude that the two AChR binding sites have the same affinities for ACh in both the closed- and open-channel conformations (the same λ). Therefore, E2/E1=E1/E0=λ. Because we had experimental estimates for both E2 and E1, it was a simple matter to calculate E0. We did this using either ACh or choline as the agonist and got the same result: E0wt≍ 7 × 10−7.
It is significant that two completely different experimental approaches led to the same quantitative estimate of E0. Without any bound ligands (in the presence of just water; inorganic ions do not alter E0) each AChR opens briefly (for ∼85 μs) and rarely (approximately once every 2 min). With just one bound ACh molecule it opens for ∼140 μs once every ∼33 ms, and with two bound agonists it opens for ∼400 μs after a delay of only ∼15 μs. Clearly, the main effect of ACh at the binding sites is to speed the rate constant for channel opening.
From the relationship λ=√(E2/E0) we estimate that in wt AChRs, λACh= 6000. Each bound ACh molecule provides a ‘bang’ of −5.1 kcal mol−1 (=−0.59lnλ) when the protein gates from the closed-channel to the open-channel conformation. The fuzzy term ‘partial agonist’ can now be replaced with a number (λ) or, better yet, an energy value that can be applied to both mutant and wt AChRs of varying subunit composition. An agonist is a molecule that provides energy for gating (λ > 1; a negative energy), and the more negative the energy, the less ‘partial’ the agonist. For example, λcarbachol= 2760 and λcholine= 275, which translate to energies of −4.7 and −3.3 kcal mol−1, respectively (Jadey et al. 2011). Since there are two transmitter binding sites, this energy is like a Richter scale for earthquakes. In the AChR, two ACh molecules trigger approximately a magnitude 10 earthquake, but choline only a magnitude 6.6. An inverse agonist is a ligand that takes away energy from the intrinsic isomerization (λ < 1; a positive energy).
These experiments show without a doubt that un-, mono- and di-liganded AChRs undergo the same essential global closed↔open gating isomerization. To further solidify the applicability of the MWC model to AChRs it was also necessary to demonstrate that agonists could associate to and dissociate from the higher-affinity, open conformation. We again turned to mutations that increased E0 and, hence, E2 (Grosman & Auerbach, 2001). When these constructs are activated by a low concentration of ACh, openings occur in ‘bursts’ that reflect only closed and open di-liganded AChRs (Colquhoun & Sakmann, 1998). In wt receptors such bursts are almost always terminated by channel closing followed by the dissociation of an agonist molecule, but in the mutants this exit route from AR* was reduced to an extent that bursts usually ended either by desensitization or by agonist dissociation from the open conformation. Analyses of desensitization rates and burst durations with different mutants led to an estimate of the rate constant for the dissociation of ACh from the open conformation. These experiments are evidence that agonists come and go from both the closed and open conformations of the AChR. Channel gating greatly changes the ability of the agonist to leave the binding site, but has only a small effect on the ability of the agonist to enter the binding site. All of the rates and states of the MWC model for AChRs are shown in Fig. 5.
Figure 5. The parameters of the MWC model for adult mouse neuromuscular AChRs.
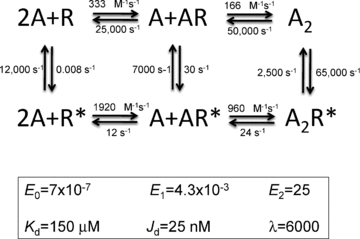
The rate constants are approximate (−70 mV, 23°C, ACh). The corresponding equilibrium constants are shown below. The coupling constant, λ= (E2/E0), is 6000 for ACh, which corresponds to an energy of −5.1 kcal mol −1 per ACh molecule.
Some conclusions can be drawn from the fact that most AChR laboratory mutations only change E0. It is reasonable to conclude that most natural mutations also have the same effect. Therefore in this protein the process of natural selection appears to be mostly about adjusting the intrinsic tendency of the protein to open and is not much directed to changing the ‘bang’ from ligands. It appears that λ is determined by only a few amino acids, all immediately at the transmitter binding site. Because most AChR mutations increase E0, we conclude that this receptor can readily open on its own accord. With time, random mutations that decrease spontaneous activity have been selected as being advantageous, as long as they still allow for a sufficiently high rate and probability of channel opening at the synapse. We also conclude that most natural AChR mutations that cause human disease (slow channel congenital myasthenic syndromes) do so because they increase E0 (Zhou et al. 1999). As a consequence, the decays of synaptic currents are prolonged and constitutive activity produces an inward current leak at the neuromuscular junction. In addition, the efficacy of choline, present at high concentrations at the synapse, increases in parallel with E0 to produce physiologically-inappropriate receptor activation.
The independence of action of mutations on E0 provides a simple path towards engineering AChR function (Jadey et al. 2011). By combining mutations having different effects on the allosteric constant and by using agonists with different λ values we can design and control the rate constants for channel opening and closing, at least over a 50 million-fold range in equilibrium constant. This ability opens the door to studying a wider range of mutations and agonists than had previously been possible. That the effects of mutations are largely independent further suggests that there is little long-range transfer of the energy arising from side-chain gating motions. The energy changes resulting from large-scale, closed↔open changes in conformation, as revealed by structural studies of related receptors (Bocquet et al. 2009; Hilf & Dutzler, 2009), apparently are mostly independent of the side-chain energy changes (Gupta & Auerbach, 2011).
Having an accurate estimate of E0 in AChRs is a beginning and not an end. We can now define agonist action quantitatively, according to the energy it provides to promote gating. We may be able to pinpoint the specific interactions at the binding site that give rise to these energies, atom by atom. It is conceivable that such a map of ligand-protein energies will help us in developing structural models of AChRs and new ligands with properties that can be forecast in advance. In addition, measuring and mapping energy will help us understand the mechanism by which the ‘bang’ from the ligand propagates to the distant ion channel domain to regulate ionic conductance. Combining knowledge of AChR structures with energy measurements (derived by using the MWC formalism) will lead us to a deeper understanding of how this protein works.
It is an open question whether or not other receptors adhere to the MWC model. Certainly, mutations of GABA, NMDA and P2X receptor-channels can increase the probability of being open in the absence of agonists. MWC is also a good approximation for the BK channel, a relative of tetrameric receptors. Given the success of the cyclic reaction scheme with AChRs, it is likely that its application to other ligand-gated ion channels and allosteric proteins will be as informative.
Acknowledgments
I thank all of my students for comments on the manuscript. This work is supported by NIH (NS-23513 and NS-64969).
References
- Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Sakmann B. From muscle endplate to brain synapses: a short history of synapses and agonist-activated ion channels. Neuron. 1998;20:381–387. doi: 10.1016/s0896-6273(00)80982-4. [DOI] [PubMed] [Google Scholar]
- Grosman C, Auerbach A. Kinetic, mechanistic, and structural aspects of unliganded gating of acetylcholine receptor channels: a single-channel study of second transmembrane segment 12′ mutants. J Gen Physiol. 2000;115:621–635. doi: 10.1085/jgp.115.5.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosman C, Auerbach A. The dissociation of acetylcholine from open nicotinic receptor channels. Proc Natl Acad Sci U S A. 2001;98:14102–14107. doi: 10.1073/pnas.251402498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Auerbach A. Mapping heat exchange in an allosteric protein. Biophys J. 2011;100:904–911. doi: 10.1016/j.bpj.2010.12.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallermann S, Heckmann S, Dudel J, Heckmann M. Short openings in high resolution single channel recordings of mouse nicotinic receptors. J Physiol. 2005;563:645–662. doi: 10.1113/jphysiol.2004.080606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilf RJ, Dutzler R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature. 2009;457:115–118. doi: 10.1038/nature07461. [DOI] [PubMed] [Google Scholar]
- Jackson MB. Kinetics of unliganded acetylcholine receptor channel gating. Biophys J. 1986;49:663–672. doi: 10.1016/S0006-3495(86)83693-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadey SV, Purohit P, Bruhova I, Gregg TM, Auerbach A. Design and control of acetylcholine receptor conformational change. Proc Natl Acad Sci U S A. 2011;108:4328–4333. doi: 10.1073/pnas.1016617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A, Auerbach A. Acetylcholine receptor channels activated by a single agonist molecule. Biophys J. 2010;98:1840–1846. doi: 10.1016/j.bpj.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin A. On the application of ‘a plausible model’ of allosteric proteins to the receptor for acetylcholine. J Theor Biol. 1967;16:306–320. doi: 10.1016/0022-5193(67)90011-2. [DOI] [PubMed] [Google Scholar]
- Leicester HM, Klickstein HS. A Source Book in Chemistry, 1400–1900. Harvard University Press; 1968. p. 331. [Google Scholar]
- Lingle CJ, Maconochie D, Steinbach JH. Activation of skeletal muscle nicotinic acetylcholine receptors. J Membr Biol. 1992;126:195–217. doi: 10.1007/BF00232318. [DOI] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions : a plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- Neubig RR, Boyd ND, Cohen JB. Conformations of Torpedo acetylcholine receptor associated with ion transport and desensitization. Biochemistry. 1982;21:3460–3467. doi: 10.1021/bi00257a032. [DOI] [PubMed] [Google Scholar]
- Purohit P, Auerbach A. Unliganded gating of acetylcholine receptor channels. Proc Natl Acad Sci U S A. 2009;106:115–120. doi: 10.1073/pnas.0809272106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P, Auerbach A. Energetics of gating at the apo-acetylcholine receptor transmitter binding site. J Gen Physiol. 2010;135:321–331. doi: 10.1085/jgp.200910384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Engel AG, Auerbach A. Serum choline activates mutant acetylcholine receptors that cause slow channel congenital myasthenic syndromes. Proc Natl Acad Sci U S A. 1999;96:10466–10471. doi: 10.1073/pnas.96.18.10466. [DOI] [PMC free article] [PubMed] [Google Scholar]