

Abstract
Non-technical summary
Spontaneous bursting, in which neurons spontaneously fire clusters of action potentials, underlies a variety of neuronal functions, including breathing and sleep rhythms. The cellular mechanisms that underlie spontaneous burst generation are poorly understood. Here, we show that calcium-permeable ion channels, recently shown to be localized to the site of action potential initiation in the initial segment of axons, are crucial for the generation of spontaneous bursts in auditory brainstem neurons. Block of calcium influx at this site was sufficient to convert spontaneous bursting neurons into neurons which fired in a regular pattern. Block could also be mediated by the neurotransmitter dopamine, which alters calcium channel activity through a kinase dependent mechanism. These results are the first to show that spontaneous firing mode can be controlled by neuromodulators acting on a specific cellular compartment, and highlight the importance of these calcium channels in the generation of spontaneous neuronal rhythm.
Abstract
Spontaneously active neurons typically fire either in a regular pattern or in bursts. While much is known about the subcellular location and biophysical properties of conductances that underlie regular spontaneous activity, less is known about those that underlie bursts. Here, we show that T-type Ca2+ channels localized to the site of action potential initiation in the axon initial segment play a pivotal role in spontaneous burst generation. In auditory brainstem interneurons, axon initial segment Ca2+ influx is selectively downregulated by dopaminergic signalling. This regulation has marked effects on spontaneous activity, converting the predominant mode of spontaneous activity from bursts to regular spiking. Thus, the axon initial segment is a key site, and dopamine a key regulator, of spontaneous bursting activity.
Introduction
Intrinsic action potential (AP) generation in the absence of synaptic input is common to many neuronal circuits, and contributes to diverse functions including volitional motor control, reflexes, hormonal secretion and central pattern generation (Hausser et al. 2004; Ramirez et al. 2004; Feldman & Del Negro, 2006; Walter et al. 2006; Stojilkovic et al. 2009). Spontaneously active neurons exhibit two general states: ‘tonic’ activity characterized by single, simple spikes at a relatively constant rate, and ‘bursting’ activity that cycles from periods of quiescence to periods of high-frequency spiking. The pattern of firing is determined by specific complements of ion channels. In tonically active cells, subthreshold membrane potential (Vm) oscillations are established by an interplay between depolarizing currents (often Na+, but also Ca2+ or non-selective cation channels) and hyperpolarizing K+ currents (Do & Bean, 2003; Puopolo et al. 2007; de Oliveira et al. 2010; Khaliq & Bean, 2010). Interestingly, subthreshold Na+ currents that drive a neuron to threshold arise from the same channels that mediate the rising phase of the AP (Taddese & Bean, 2002; Astman et al. 2006; Fleidervish et al. 2010). These channels are restricted to the proximal section of the axon, termed the axon initial segment (AIS), suggesting that the AIS is important not only as the site of AP initiation, but also for establishing tonic rhythm.
In bursting neurons, additional contributions, often made by T-type Ca2+ channels, sustain long-lasting depolarizations (Huguenard & McCormick, 1992; Swensen & Bean, 2003; Womack & Khodakhah, 2004; Liu & Shipley, 2008; Wang et al. 2009; Cain & Snutch, 2010). In contrast to subthreshold Na+ conductances known to be expressed in the AIS, it remains unclear whether T-type conductances that support spontaneous bursting are restricted to a specific neuronal compartment, or are instead distributed amongst multiple structures. Cartwheel cells of the dorsal cochlear nucleus (DCN) offer a unique opportunity to probe this question. These glycinergic interneurons express T-type Ca2+ channels in both their dendrites and AIS (Bender & Trussell, 2009). Recently, we found that dopamine downregulates Ca2+ influx through AIS, but not dendritic, T-type channels. Moreover, this regulation is independent of Na+ or K+ channel modulation, and therefore allows one selective control over a single, compartmentally restricted population of ion channels (Bender et al. 2010).
Here, we took advantage of this neuromodulatory pathway to test the role of AIS T-type channels in the generation of spontaneous bursts. Even though dendritic T-type channels support depolarizations and Ca2+ transients during spike bursts in these and other cells (Roberts et al. 2008; Errington et al. 2010), we found that AIS T-type conductances, and their regulation via dopamine, play a predominant role in shaping spontaneous activity. While dopamine produces relatively modest reductions in AP-evoked AIS Ca2+ influx (Bender et al. 2010), it was sufficient to switch spontaneously active neurons from burst firing to largely tonic firing. This suggests that AIS ion channels determine ongoing firing modes, and furthermore identify a new role for dopamine in regulating spontaneous neuronal activity.
Methods
Electrophysiology
All procedures were in accordance with OHSU IACUC guidelines. Following anaesthesia, coronal brainstem slices (210 μm) were made from P17–26 CBA or C57BL/6J mice. D3−/− mice were genotyped by PCR and GlyT2-GFP mice were genotyped by visualizing fluorescence through the skull of <P2 animals (Roberts et al. 2008; Bender et al. 2010). Cutting solution contained (in mm): 87 NaCl, 25 NaHCO3, 25 glucose, 75 sucrose, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2 and 7 MgCl2; bubbled with 5% CO2–95% O2; 4°C. Following cutting, slices were incubated in the same solution for 30 min at 33°C, then at room temperature until recording. Extracellular recording solution contained (in mm): 130 NaCl, 3 KCl, 2.4 CaCl2, 1.3 MgSO4, 1.2 KH2PO4, 20 NaHCO3, 3 Na-Hepes, 10 glucose; bubbled with 5% CO2–95% O2; 32–34°C. To avoid dye saturation, CaCl2 was reduced to 1 mm and MgSO4 was raised to 2.7 mm for all imaging experiments in which Ca2+ transients were evoked by AP trains in whole-cell current clamp. For all other experiments, 2.4 mm CaCl2 was used. All recordings were performed in 0.5 μm strychnine, 10 μm SR95531, 10 μm 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide (NBQX), and 50 μm d-AP5 or 5 μm 3-(R)-2-carboxypiperazin-4-propyl-1-phosphonic acid ((R)-CPP) to block synaptic activity.
For loose-seal voltage-clamp recordings, electrodes (Rseal= 12–15 MΩ) were filled with (in mm): 141 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, pH 7.25–7.3. For current clamp recordings (Rseries < 10 MΩ), electrodes contained (in mm): 113 potassium gluconate, 9 Hepes, 4.5 MgCl2, 0.1 EGTA, 14 Tris2-phosphocreatine, 4 Na2-ATP, 0.3 Tris-GTP; 0.25 Fluo-5F; 0.02 Alexa 594; ∼290 mosmol l−1, pH: 7.2–7.25. Voltages were corrected for a 12 mV junction potential. Electrophysiological data were recorded at 20–50 kHz and filtered at 10 kHz using a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA), and acquired with an ITC-18 (Instrutech, Port Washingon, NY, USA) interface and Igor Pro (Wavemetrics, Lake Oswego, OR, USA) software. Cell-attached action currents were detected using a variable amplitude template matching algorithm (AxoGraph) and confirmed manually. Data were analysed from 180 s epochs immediately before and 8 min after drug administration. Control data were analysed over the same time course, with no drug added. Average firing rates were calculated as total spikes-per-epoch/180; instantaneous rates were the average of all instantaneous frequencies within that epoch. Ca2+ transients were acquired with a two-photon microscope and analysed as described previously (Bender et al. 2010), using a laser tuned to 810 nm. Ca2+ transients were presented as averages of 20 events per site and expressed as ΔG/Gsat× 100, where Gsat was the maximal fluorescence in saturating Ca2+ (2 mm).
Perforated-patch recordings
Recording electrodes were tip-filled with standard intracellular solution (see above), backfilled by pipette solution to which amphotericin B was added (300 μg ml−1, made fresh from 0.6 mg ml−1 stock in DMSO; final [DMSO]: 0.05%). Pipette capacitance was compensated 90–100%. Gigaohm seals were established on cartwheel cells and perforation was assessed by monitoring response to current pulses (–5 to –50 pA, 100 ms). Perforation was evident within 5 min, reaching steady state within 30–45 min (Baseline Rseries: 37 ± 3 MΩ, quinpirole Rseries: 35 ± 4 MΩ). Vm was held to <–75 mV with constant bias current if resting Vm > –70 mV. If resting Vm was <–70 mV, no bias current was added. Membrane integrity was assessed by monitoring evoked AP waveforms, since fast afterhyperpolarizations are clearly reduced when whole-cell configuration is established (Kim & Trussell, 2007).
Targeted iontophoresis of Ni2+ or Na+
Cartwheel cells expressing GFP were visualized in GlyT2-GFP mice. Only spontaneously bursting neurons with spatially segregated axons and dendrites were chosen for analysis. A second pipette (20 MΩ tip resistance) containing 100 mm NiCl2 or 200 mm NaCl was guided near a neurite by simultaneously visualizing GFP fluorescence and a differential interference contrast (DIC)-like image generated by projecting laser light transmitted through the preparation onto a diode. Ni2+ was iontophoresed with 9.1 ± 2.8 nA for 4 s (range: 1.5–15 nA net, compensating for –20 nA constant retention current). Appropriate current amplitude was determined empirically at AIS locations and was not altered when the pipette was relocated to the dendrite. The pipette was subsequently repositioned to the AIS to ensure continued function. AIS and dendritic Ca2+ imaging experiments used 10 nA ejection currents. AP-evoked Ca2+ transients were imaged –3.6 s (baseline), 3.4 s (Ni2+), and 9.4 s (wash) from iontophoresis onset. Currents of 15 nA were used with Na+ to mimic the maximum Ni2+ current. Phosphates were excluded from the recording solution to prevent precipitation.
Chemicals
Fluo-5F pentapotassium salt and Alexa Fluor 594 hydrazide Na+ salt were from Invitrogen. SR95531, d-AP5, R-CPP and NBQX were from Ascent Bristol, UK. (–)-Quinpirole hydrochloride and GF-109203X were from Tocris Bioscience (Ellisville, MO, USA). All others were from Sigma-Aldrich.
Statistics
All data are shown as means ± standard error of the mean (SEM). An ANOVA followed by a Mann–Whitney U test was used unless otherwise noted (significance: P < 0.05). For Kolmogorov–Smirnov (KS) tests, data were converted to cumulative probability distributions.
Results
Loose-seal recordings were made from cartwheel cells identified by laminar position, morphology, and, when spontaneously active, their ability to fire characteristic AP bursts. Of 261 putative cartwheel cells sampled, 172 were quiescent. Of the remainder, some (42) largely fired high-frequency bursts, characterized by instantaneous AP frequencies >100 Hz (Fig. 1A and B), while others (47) predominantly fired simple spikes. Cells were classified as ‘bursting’ or ‘simple spiking’ if >40% or <20% of all instantaneous AP frequencies were >100 Hz, respectively. Note that all cells classified as simple spiking fired at least one burst, thereby identifying them as cartwheel cells (Golding & Oertel, 1997).
Figure 1. Dopamine alters cartwheel cell spontaneous bursting activity.
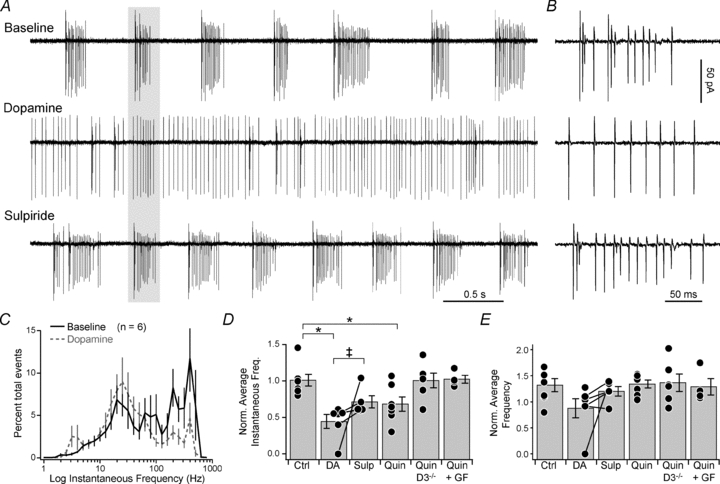
A, cell-attached recording of spontaneously bursting cell in the presence of dopamine and dopamine+sulpiride. Data from a single cell. B, expanded time base, highlighting spikes in grey bar in A. C, log distribution of instantaneous AP frequency, normalized to total events per cell. Bars are SEM. D, instantaneous spike rate, normalized to baseline. Dots are single cells, bars are SEM. Ctrl, control; DA, dopamine; Sulp, sulpiride; Quin, quinpirole; GF, GF-109203X. Lines connecting dots indicate results were from same cell. *P < 0.05, Mann–Whitney U test; ‡P < 0.05, Wilcoxon's signed rank test. E, average spike rate, normalized to baseline. Format as in D.
Dopamine downregulates Ca2+ influx through T-type channels localized to the AIS (Bender et al. 2010). To determine if this modulation has an impact on spontaneous activity, we applied 50 μm dopamine to spontaneously bursting cartwheel cells. Dopamine dramatically altered spontaneous spiking, reducing the percentage of high-frequency events (>100 Hz) from 48 ± 4 to 17 ± 4% (Fig. 1A–C, P < 0.005, paired t test). Consistent with an effective loss of bursting events, the overall distribution of instantaneous frequencies was shifted leftward (Fig. 1C, P < 0.005, KS test), and the average instantaneous frequency, normalized per cell to baseline rates, was lower (0.45 ± 0.09 of baseline, n = 6, P < 0.005 vs. control). Interestingly, the average spike frequency (see Methods) was not altered (Fig. 1E; 0.88 ± 0.18 of baseline, P > 0.05 vs. control), and long periods of quiescence were less frequent (Fig. 1A), indicating that dopamine reorganizes spike pattern without altering overall spike rate.
Dopamine's effects were partially reversed by the type 3 dopamine receptor (D3R) antagonist sulpiride (Fig. 1A, B, D and E; 200 nm, 0.72 ± 0.08 of baseline, n = 5 (3 cells in the continued presence of dopamine, ruling out D1-family contributions), P < 0.05, Wilcoxon's signed rank test vs. dopamine), and mimicked by the D3R agonist quinpirole (1 μm, 0.68 ± 0.10 of baseline, n = 7, P < 0.05 vs. control). Further, quinpirole did not alter spiking activity in D3R−/− animals (1.01 ± 0.10 of baseline, n = 6, P = 0.6) or when protein kinase C (PKC) activity was inhibited with GF-109203X (1 μm, 1.03 ± 0.05 of baseline, n = 4, P = 0.7). These results are consistent with the known molecular basis for AIS Ca2+ channel modulation in cartwheel cells, which requires both D3R and PKC activity (Bender et al. 2010).
To determine if downregulation of AIS Ca2+ influx is sufficient to mediate these changes in spike output, we mimicked the effects of dopamine by selective block of AIS Ca2+ channels via local iontophoresis of the T- and R-type antagonist Ni2+. Experiments were performed in mice expressing GFP in cartwheel cells, allowing us to target Ni2+-containing pipettes to specific sites along the axon or dendrite. Cells that had clear spatial separation between primary dendrites and the AIS were selected, and iontophoretic pipettes were placed near the axon, ∼15 μm from the axon hillock (Fig. 2A). In spontaneously bursting neurons, AIS Ni2+ iontophoresis produced a marked change in spiking, comparable to dopamine (Fig. 2B–D, % events >100 Hz, baseline: 55 ± 7%, Ni2+: 19 ± 9%, n = 4, P < 0.02, paired t test; Ni2+ average instantaneous frequency: 0.49 ± 0.07 of baseline, P < 0.01, repeated measures ANOVA). Burst firing resumed rapidly after current offset (Fig. 2B–D, wash instantaneous frequency: 0.96 ± 0.09 of baseline), indicating that leak of Ni2+ from the iontophoretic pipette was minimal.
Figure 2. Partial block of AIS Ca2+ prevents spontaneous burst generation.

A, overlay of GFP fluorescence (green) and DIC-like image (greyscale) imaged with 2-photon microscopy. Axon exits cell at bottom left and extends from soma at 45 deg. Inset, outline of neuron and pipettes, highlighting relative positions. B, single trials in which either Ni2+ or Na+ was iontophoresed onto the AIS. Ni2+ and Na+ data were collected from different cells. Scale applies to both. C, log distribution of instantaneous frequencies, normalized to total events per cell. Black, baseline; red, AIS Ni2+; cyan, wash 3–7s following Ni2+ application offset. Bars are SEM. D, average instantaneous spike rate, normalized to baseline. Dots are single cells, bars are SEM. Lines indicate results from same cell. Wa, wash; Dend, iontophoresis onto dendritic branch. *P < 0.001, repeated measures ANOVA (baseline, Ni2+, wash). E, top, simultaneous whole-cell recording (top) with evoked AP train (resting Vm: –81 mV). Bottom, concomitant Ca2+ imaging in AIS and primary dendrite before, during and after AIS Ni2+ iontophoresis. F, AIS and dendritic Ca2+ transients during and after Ni2+ iontophoresis, normalized to baseline. Format and statistics as in D.
Iontophoresis produced local field currents, which varied in amplitude based on the relative locations of iontophoretic and recording pipettes, recording seal resistance, and applied iontophoretic current. To ensure that Ni2+, and not the local current, was driving changes in spike output, we substituted Na+ for Ni2+ and applied the maximum iontophoretic current used in Ni2+ experiments, which should evoke comparable currents but have little impact on the local ionic solution. Though Na+ current artifacts were of comparable size to those in Ni2+ (Ni2+: –29 ± 10 pA, n = 4, Na+: –76 ± 23 pA, n = 3; P = 0.09, unpaired t test), Na+ iontophoresis had no effect on spike patterns (Fig. 2B and D; instantaneous frequency: 0.95 ± 0.04 of baseline, P = 0.8, repeated measures ANOVA).
To ensure that Ni2+ iontophoresis was restricted to the AIS, and that block of AIS Ca2+ was necessary for changes in spike output, we performed two experiments. First, the iontophoretic pipette was relocated to a proximal dendritic branch on the opposite side of the soma, ∼15 μm from its somatic origin. Dendritic Ni2+ application had no effect on spike output (Fig. 2D; instantaneous frequency: 1.02 ± 0.05 of baseline, n = 4, P = 0.3, repeated measures ANOVA). Second, the spread of Ni2+ from the AIS was assessed by imaging simultaneously AP-evoked Ca2+ influx in the AIS and dendrite. Using the same iontophoresis protocol, we found that Ni2+ reduced AP-evoked Ca2+ transients to 0.63 ± 0.07% of baseline in the AIS (Fig. 2E and F, n = 4, P < 0.001, repeated measures ANOVA), comparable to previous reports with dopamine and quinpirole (Bender et al. 2010). In contrast, dendritic Ca2+ transients were not altered by Ni2+ application to the AIS (1.05 ± 0.05 of baseline, n = 4, P = 0.4, repeated measures ANOVA). Thus, effects of AIS Ni2+ iontophoresis closely match those of dopamine on AIS Ca2+ influx, both in spatial restriction to the AIS and in magnitude of AIS Ca2+ block.
To test more specifically the function of T-type Ca2+ in burst generation we used the novel antagonist TTA-P2 (Shipe et al. 2008; Dreyfus et al. 2010) at a concentration (1 μm), which specifically blocks T-type channels, but not R-type channels also found in the AIS (Choe et al. 2011). TTA-P2 reduced AP-evoked AIS Ca2+ influx by 47 ± 3% (Fig. 3A and B; n = 6, P < 0.0001, one-sample t test), confirming our previous results with mibefradil and Ni2+ (Bender & Trussell, 2009). In contrast to dopamine's partial block of AIS T-type channels (Bender et al. 2010), removal of all T-type current by TTA-P2 (Dreyfus et al. 2010) completely blocked spontaneous bursting (Fig. 3C–E). The percentage of >100 Hz events was reduced from 48 ± 5% to 1.8 ± 0.7% (P < 0.001, paired t test; P < 0.0001, KS test), and the average instantaneous frequency was reduced to 0.11 ± 0.04 of baseline (P < 0.01 vs. control). Similar to dopamine, the average frequency was not altered (1.20 ± 0.24 of baseline, P = 0.7 vs. control).
Figure 3. Selective T-type Ca2+ channel antagonist mimics dopamine.

A, cartwheel cell filled with Alexa 594 via whole-cell recording (pipette masked), and imaged with a 2-photon microscope. Image is a maximum intensity z-stack from 75 images collected at 1 μm increments. Axon, highlighted by arrows, exits soma at the far right. B, APs (middle) were evoked via somatic current injection (top) and concomitant Ca2+ transients were imaged in the AIS (bottom). The T-type antagonist TTA-P2 reduced Ca2+ transients (grey). C, cell-attached recording of spontaneously bursting cell in the presence of TTA-P2. Data from a single cell. D, expanded time base, highlighting spikes within grey bar in C. E, log distribution of instantaneous frequencies, normalized to total events per cell. Bars are SEM.
Local Ni2+ iontophoresis, combined with pharmacological block of dopaminergic modulation and T-type channels, strongly suggests that perturbations in AIS T-type channel function are sufficient to regulate bursting activity; however, they do not rule out other mechanisms that could contribute to these changes. For example, D2-family receptors, which includes D3R, are coupled to inward rectifier K+ currents (GIRK) that can alter Vm and Rin, thereby altering spontaneous spiking (Kuzhikandathil & Oxford, 2000). Previous experiments did not suggest that cartwheel cells expressed dopamine-mediated GIRK currents, but these results were obtained from whole-cell recordings where the endogenous cytosol was dialysed (Bender et al. 2010). We therefore repeated these experiments using perforated-patch techniques, which leave the intracellular milieu largely intact (Rae et al. 1991). Again, 1 μm quinpirole had no effect on Vm (baseline: –80.4 ± 0.8 mV, quinpirole: –80.4 ± 0.9 mV, n = 5, P = 0.9, paired t test) or Rin (baseline: 96 ± 19 MΩ, quinpirole: 96 ± 20 MΩ, P = 0.8, paired t test), suggesting that D2 family-activated GIRK currents do not contribute to changes in spike output.
If dopamine acts exclusively on AIS T-type channels, then this modulatory pathway should have little impact on spontaneously simple spiking cartwheel neurons, which possibly never hyperpolarize to levels that relieve T-type channel inactivation (Cain & Snutch, 2010). To test this, we activated D3 receptors in simple spiking neurons with quinpirole (1 μm). Quinpirole had no effect on simple spiking cells (Fig. 4A and B; quinpirole instantaneous rate: 1.14 ± 0.07 of baseline, n = 6, control: 1.19 ± 0.09, n = 5, P = 0.6; quinpirole average rate: 1.24 ± 0.18, control: 1.23 ± 0.08, P = 0.7; KS test: P = 0.06). Moreover, simple spiking activity was not affected by the antagonist sulpiride (Fig. 4C; sulpiride instantaneous rate: 1.05 ± 0.05 of baseline, n = 4, P = 0.2 vs. control; sulpiride average rate: 1.26 ± 0.08, P = 0.6; KS test: P = 0.9), suggesting that slices of DCN lack endogenous dopamine tone.
Figure 4. Dopamine does not alter simple spiking cartwheel cell activity.

A, cell-attached recording of spontaneously simple spiking cell in the presence of quinpirole. Data are from a single cell. B and C, log distribution of instantaneous frequencies in quinpirole or sulpiride, normalized to total events per cell. Bars are SEM.
Discussion
Here, we described a novel mechanism by which neurons regulate the mode of spontaneous electrical activity. T-type Ca2+ channels underlie bursting in a variety of neurons, and subtle changes in their biophysical characteristics have a strong influence on AP output (Tscherter et al. 2011). While dendritic Ca2+ influx indeed contributes to spontaneous bursting activity (Fig. 3) (Womack & Khodakhah, 2004; Del Negro et al. 2011), it is regulated further by conductances localized to the site of spike initiation.
Selective modulation of AIS Ca2+ current was sufficient to markedly alter cartwheel cell activity, reversibly transforming bursting cells into tonically active cells (Figs 1 and 2). Previous data indicated that dopamine, delivered via exogenous or endogenous sources, weakens AP-evoked AIS Ca2+ influx by 30–40% (Bender et al. 2010). Further, this modulatory pathway is highly specific for AIS T-type channels; neither Na+ nor K+ nor dendritic T-type channels are affected by D3R signalling (Bender et al. 2010). Indirect effects on Ca2+-activated K+ conductances can also be excluded, since decreases in the activity of these channels should facilitate, not block, bursting (Kim & Trussell, 2007), and previous data suggest that these channels are not found in the cartwheel AIS (Bender & Trussell, 2009). Thus, these results demonstrate that small alterations in ion channel function can have profound effects on neuronal output, provided those changes are optimally targeted to proper neuronal compartments.
Ca2+ channels, most commonly T-type isoforms, appear to be localized to the AIS in a variety of neurons (Bender & Trussell, 2009; Yu et al. 2010). Dopamine dampens evoked burst firing in multiple cell types (Stanzione et al. 1984; Gulledge & Jaffe, 1998; Ding & Perkel, 2002; Maurice et al. 2004; Tseng & O'Donnell, 2004), possibly by alterations in AIS Ca2+ influx. Further, mechanisms that directly affect AIS Vm, including ionotropic receptor signalling onto the AIS (Szabadics et al. 2006), could affect T-type channel availability, raising the potential that similar switches in spontaneous activity exist even in cells that lack AIS dopamine signalling.
Dopaminergic modulation at the AIS was shown previously to require type 3 dopamine receptors and PKC (Bender et al. 2010), and here, dopamine-based changes in spontaneous activity were not observed in D3−/− mice or when PKC activity was inhibited. These results, combined with those demonstrating that dopaminergic effects can be mimicked by selective block of AIS Ca2+ influx (Fig. 2), strongly suggest that changes in AIS Ca2+ channel function mediate the observed effects. Based on whole-cell T-type current kinetics and sensitivity to subunit selective antagonists, we have hypothesized that CaV3.2 isoforms are localized to the AIS (Bender et al. 2010). This modulatory pathway is distinct from other known molecular mechanisms by which dopamine alters CaV3.2 channel activity. In cultured H295R cells, the concerted action of type 1 and type 2 dopamine receptor activation, through a Gβ-protein kinase A signalling cascade, are required to inhibit CaV3.2 channels (Hu et al. 2009). In contrast, AIS regulation more closely resembles neurokinin 1 receptor-mediated modulation of CaV3.2, which requires Gαq11 signalling and PKC (Rangel et al. 2010). Importantly, D3 receptors can couple to Gαq11 (Newman-Tancredi et al. 1999), though future studies will be required to determine whether these mechanisms are involved in AIS modulation.
A variety of neuromodulators alter cartwheel cell activity, either at synaptic inputs (Zhao & Tzounopoulos, 2011), or by altering AP output (Bender et al. 2010; Kuo & Trussell, 2011). Dopaminergic signalling in DCN acts on AIS Ca2+ channels via volume transmission (Bender et al. 2010), suggesting that it lacks specificity for particular cartwheel cells; however, the availability of T-type conductances may impose an activity filter, conferring specificity to only those cartwheel cells sufficiently hyperpolarized to relieve T-type channel inactivation (Cain & Snutch, 2010). Dopamine released across the DCN may therefore bias the cartwheel cell network towards non-bursting behaviour.
What effect would this have on DCN function? One current hypothesis is that cartwheel cell bursts contribute to the suppression of background noise by inhibiting DCN efferent neurons, in turn increasing the salience of external stimuli (Oertel & Young, 2004; Shore, 2005; Roberts & Portfors, 2008). The character of background noise changes as one enters a novel environment, and as such, the synaptic inputs that evoke cartwheel cell bursts should also change with environment. Excitatory synapses onto cartwheel cells exhibit robust plasticity (Fujino & Oertel, 2003; Tzounopoulos et al. 2004), enabling the acquisition of new associations that suppress now-relevant background noise. It is possible that dopamine makes additional contributions by suppressing bursts that no longer correspond to background sound. Indeed, novelty is a major cue for increased spiking activity in midbrain dopamine neurons (Schultz, 2007). An understanding of the dynamics of catecholamine signalling in the DCN will therefore not only provide clues into how these neuromodulators shape DCN function, but may also provide insight into the role the DCN plays in auditory processing.
Acknowledgments
We are grateful to members of the Trussell and Williams labs for comments on this work, and to S. Kuo and M. Roberts for critically reading this manuscript. We thank D. Grandy for knockout mice, and A. Truitt and C. Borges-Merjane for genotyping expertise. This research was supported by NIH grants DC011080 (K.J.B.), and NS028901 (L.O.T.).
Glossary
- AP
action potential
- AIS
axon initial segment
- Vm
membrane potential
- DIC
differential interference contrast
- DCN
dorsal cochlear nucleus
- PKC
protein kinase C
Author contributions
K.J.B. and L.O.T. designed experiments and wrote the paper. K.J.B. performed experiments and analyses. V.N.U. and J.J.R. developed and provided TTA-P2.
Disclosures
J.J.R. and V.N.U. are employees of Merck and potentially own stock and/or stock options in the company.
Author's present address
K. J. Bender: Gallo Research Center, University of California, San Francisco, Emeryville, CA 94608, USA.
References
- Astman N, Gutnick MJ, Fleidervish IA. Persistent sodium current in layer 5 neocortical neurons is primarily generated in the proximal axon. J Neurosci. 2006;26:3465–3473. doi: 10.1523/JNEUROSCI.4907-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, Ford CP, Trussell LO. Dopaminergic modulation of axon initial segment calcium channels regulates action potential initiation. Neuron. 2010;68:500–511. doi: 10.1016/j.neuron.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, Trussell LO. Axon initial segment Ca2+ channels influence action potential generation and timing. Neuron. 2009;61:259–271. doi: 10.1016/j.neuron.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain SM, Snutch TP. Contributions of T-type calcium channel isoforms to neuronal firing. Channels (Austin) 2010;4:475–482. doi: 10.4161/chan.4.6.14106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe W, Messinger RB, Leach E, Eckle VS, Obradovic A, Salajegheh R, Jevtovic-Todorovic V, Todorovic SM. TTA-P2 is a potent and selective blocker of T-type calcium channels in rat sensory neurons and a novel antinociceptive agent. Mol Pharmacol. 2011;80:900–910. doi: 10.1124/mol.111.073205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira RB, Howlett MC, Gravina FS, Imtiaz MS, Callister RJ, Brichta AM, van Helden DF. Pacemaker currents in mouse locus coeruleus neurons. Neuroscience. 2010;170:166–177. doi: 10.1016/j.neuroscience.2010.06.028. [DOI] [PubMed] [Google Scholar]
- Del Negro CA, Hayes JA, Rekling JC. Dendritic calcium activity precedes inspiratory bursts in preBotzinger complex neurons. J Neurosci. 2011;31:1017–1022. doi: 10.1523/JNEUROSCI.4731-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Perkel DJ. Dopamine modulates excitability of spiny neurons in the avian basal ganglia. J Neurosci. 2002;22:5210–5218. doi: 10.1523/JNEUROSCI.22-12-05210.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do MT, Bean BP. Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron. 2003;39:109–120. doi: 10.1016/s0896-6273(03)00360-x. [DOI] [PubMed] [Google Scholar]
- Dreyfus FM, Tscherter A, Errington AC, Renger JJ, Shin HS, Uebele VN, Crunelli V, Lambert RC, Leresche N. Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of ITwindow. J Neurosci. 2010;30:99–109. doi: 10.1523/JNEUROSCI.4305-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington AC, Renger JJ, Uebele VN, Crunelli V. State-dependent firing determines intrinsic dendritic Ca2+ signaling in thalamocortical neurons. J Neurosci. 2010;30:14843–14853. doi: 10.1523/JNEUROSCI.2968-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Del Negro CA. Looking for inspiration: new perspectives on respiratory rhythm. Nat Rev Neurosci. 2006;7:232–242. doi: 10.1038/nrn1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleidervish IA, Lasser-Ross N, Gutnick MJ, Ross WN. Na+ imaging reveals little difference in action potential-evoked Na+ influx between axon and soma. Nat Neurosci. 2010;13:852–860. doi: 10.1038/nn.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino K, Oertel D. Bidirectional synaptic plasticity in the cerebellum-like mammalian dorsal cochlear nucleus. Proc Natl Acad Sci U S A. 2003;100:265–270. doi: 10.1073/pnas.0135345100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding NL, Oertel D. Physiological identification of the targets of cartwheel cells in the dorsal cochlear nucleus. J Neurophysiol. 1997;78:248–260. doi: 10.1152/jn.1997.78.1.248. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Jaffe DB. Dopamine decreases the excitability of layer V pyramidal cells in the rat prefrontal cortex. J Neurosci. 1998;18:9139–9151. doi: 10.1523/JNEUROSCI.18-21-09139.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausser M, Raman IM, Otis T, Smith SL, Nelson A, du Lac S, Loewenstein Y, Mahon S, Pennartz C, Cohen I, Yarom Y. The beat goes on: spontaneous firing in mammalian neuronal microcircuits. J Neurosci. 2004;24:9215–9219. doi: 10.1523/JNEUROSCI.3375-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Depuy SD, Yao J, McIntire WE, Barrett PQ. Protein kinase A activity controls the regulation of T-type CaV3.2 channels by Gβγ dimers. J Biol Chem. 2009;284:7465–7473. doi: 10.1074/jbc.M808049200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. Simulation of the currents involved in rhythmic oscillations in thalamic relay neurons. J Neurophysiol. 1992;68:1373–1383. doi: 10.1152/jn.1992.68.4.1373. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Bean BP. Pacemaking in dopaminergic ventral tegmental area neurons: depolarizing drive from background and voltage-dependent sodium conductances. J Neurosci. 2010;30:7401–7413. doi: 10.1523/JNEUROSCI.0143-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Trussell LO. Ion channels generating complex spikes in cartwheel cells of the dorsal cochlear nucleus. J Neurophysiol. 2007;97:1705–1725. doi: 10.1152/jn.00536.2006. [DOI] [PubMed] [Google Scholar]
- Kuo SP, Trussell LO. Spontaneous spiking and synaptic depression underlie noradrenergic control of feed-forward inhibition. Neuron. 2011;71:306–318. doi: 10.1016/j.neuron.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzhikandathil EV, Oxford GS. Dominant-negative mutants identify a role for GIRK channels in D3 dopamine receptor-mediated regulation of spontaneous secretory activity. J Gen Physiol. 2000;115:697–706. doi: 10.1085/jgp.115.6.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Shipley MT. Multiple conductances cooperatively regulate spontaneous bursting in mouse olfactory bulb external tufted cells. J Neurosci. 2008;28:1625–1639. doi: 10.1523/JNEUROSCI.3906-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Mercer J, Chan CS, Hernandez-Lopez S, Held J, Tkatch T, Surmeier DJ. D2 dopamine receptor-mediated modulation of voltage-dependent Na+ channels reduces autonomous activity in striatal cholinergic interneurons. J Neurosci. 2004;24:10289–10301. doi: 10.1523/JNEUROSCI.2155-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman-Tancredi A, Cussac D, Audinot V, Pasteau V, Gavaudan S, Millan MJ. G protein activation by human dopamine D3 receptors in high-expressing Chinese hamster ovary cells: A guanosine-5′-O-(3-[35S]thio)- triphosphate binding and antibody study. Mol Pharmacol. 1999;55:564–574. [PubMed] [Google Scholar]
- Oertel D, Young ED. What's a cerebellar circuit doing in the auditory system? Trends Neurosci. 2004;27:104–110. doi: 10.1016/j.tins.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Puopolo M, Raviola E, Bean BP. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci. 2007;27:645–656. doi: 10.1523/JNEUROSCI.4341-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Ramirez JM, Tryba AK, Pena F. Pacemaker neurons and neuronal networks: an integrative view. Curr Opin Neurobiol. 2004;14:665–674. doi: 10.1016/j.conb.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Rangel A, Sanchez-Armass S, Meza U. Protein kinase C-mediated inhibition of recombinant T-type Cav3.2 channels by neurokinin 1 receptors. Mol Pharmacol. 2010;77:202–210. doi: 10.1124/mol.109.058727. [DOI] [PubMed] [Google Scholar]
- Roberts MT, Bender KJ, Trussell LO. Fidelity of complex spike-mediated synaptic transmission between inhibitory interneurons. J Neurosci. 2008;28:9440–9450. doi: 10.1523/JNEUROSCI.2226-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts PD, Portfors CV. Design principles of sensory processing in cerebellum-like structures. Early stage processing of electrosensory and auditory objects. Biol Cybern. 2008;98:491–507. doi: 10.1007/s00422-008-0217-1. [DOI] [PubMed] [Google Scholar]
- Schultz W. Multiple dopamine functions at different time courses. Annu Rev Neurosci. 2007;30:259–288. doi: 10.1146/annurev.neuro.28.061604.135722. [DOI] [PubMed] [Google Scholar]
- Shipe WD, Barrow JC, Yang ZQ, Lindsley CW, Yang FV, Schlegel KA, Shu Y, Rittle KE, Bock MG, Hartman GD, Tang C, Ballard JE, Kuo Y, Adarayan ED, Prueksaritanont T, Zrada MM, Uebele VN, Nuss CE, Connolly TM, Doran SM, Fox SV, Kraus RL, Marino MJ, Graufelds VK, Vargas HM, Bunting PB, Hasbun-Manning M, Evans RM, Koblan KS, Renger JJ. Design, synthesis, and evaluation of a novel 4-aminomethyl-4-fluoropiperidine as a T-type Ca2+ channel antagonist. J Med Chem. 2008;51:3692–3695. doi: 10.1021/jm800419w. [DOI] [PubMed] [Google Scholar]
- Shore SE. Multisensory integration in the dorsal cochlear nucleus: unit responses to acoustic and trigeminal ganglion stimulation. Eur J Neurosci. 2005;21:3334–3348. doi: 10.1111/j.1460-9568.2005.04142.x. [DOI] [PubMed] [Google Scholar]
- Stanzione P, Calabresi P, Mercuri N, Bernardi G. Dopamine modulates CA1 hippocampal neurons by elevating the threshold for spike generation: an in vitro study. Neuroscience. 1984;13:1105–1116. doi: 10.1016/0306-4522(84)90291-4. [DOI] [PubMed] [Google Scholar]
- Stojilkovic SS, Murano T, Gonzalez-Iglesias AE, Andric SA, Popovic MA, Van Goor F, Tomic M. Multiple roles of Gi/o protein-coupled receptors in control of action potential secretion coupling in pituitary lactotrophs. Ann N Y Acad Sci. 2009;1152:174–186. doi: 10.1111/j.1749-6632.2008.03994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen AM, Bean BP. Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci. 2003;23:9650–9663. doi: 10.1523/JNEUROSCI.23-29-09650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadics J, Varga C, Molnar G, Olah S, Barzo P, Tamas G. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–235. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- Taddese A, Bean BP. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron. 2002;33:587–600. doi: 10.1016/s0896-6273(02)00574-3. [DOI] [PubMed] [Google Scholar]
- Tscherter A, David F, Ivanova T, Deleuze C, Renger JJ, Uebele VN, Shin HS, Bal T, Leresche N, Lambert RC. Minimal alterations in T-type calcium channel gating markedly modify physiological firing dynamics. J Physiol. 2011;589:1707–1724. doi: 10.1113/jphysiol.2010.203836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, O'Donnell P. Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J Neurosci. 2004;24:5131–5139. doi: 10.1523/JNEUROSCI.1021-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzounopoulos T, Kim Y, Oertel D, Trussell LO. Cell-specific, spike timing-dependent plasticities in the dorsal cochlear nucleus. Nat Neurosci. 2004;7:719–725. doi: 10.1038/nn1272. [DOI] [PubMed] [Google Scholar]
- Walter JT, Alvina K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- Wang S, Polo-Parada L, Landmesser LT. Characterization of rhythmic Ca2+ transients in early embryonic chick motoneurons: Ca2+ sources and effects of altered activation of transmitter receptors. J Neurosci. 2009;29:15232–15244. doi: 10.1523/JNEUROSCI.3809-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K. Dendritic control of spontaneous bursting in cerebellar Purkinje cells. J Neurosci. 2004;24:3511–3521. doi: 10.1523/JNEUROSCI.0290-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Maureira C, Liu X, McCormick D. P/Q and N channels control baseline and spike-triggered calcium levels in neocortical axons and synaptic boutons. J Neurosci. 2010;30:11858–11869. doi: 10.1523/JNEUROSCI.2651-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Tzounopoulos T. Physiological activation of cholinergic inputs controls associative synaptic plasticity via modulation of endocannabinoid signaling. J Neurosci. 2011;31:3158–3168. doi: 10.1523/JNEUROSCI.5303-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]