

Abstract
Non technical summary
Muscle cells have receptors that are activated by the neurotransmitter acetylcholine. The probability that these channels conduct ions across cell membranes increases dramatically when transmitter molecules are present at two binding sites, which are far from the region that regulates ionic conductance. In order to understand the molecular basis of receptor ‘gating’ we seek to learn how neurotransmitters and other small molecules activate this protein. We used single-channel electrophysiology of receptors expressed in tissue-cultured cells to study the effects of mutations at the C-terminus of loop 9, a region near intra-protein interfaces that are known to be important with regard to gating and assembly. We found that the mutation had modest but measurable effects on channel gating (mainly those in the epsilon subunit). We also found that mutations of loop 9 in the alpha subunit increase the kinetic heterogeneity of gating, which suggests that they alter the stability of the extracellular transmembrane domain interface.
Abstract
Acetylcholine receptor-channels (AChRs) mediate fast synaptic transmission between nerve and muscle. In order to better-understand the mechanism by which this protein assembles and isomerizes between closed- and open-channel conformations we measured changes in the diliganded gating equilibrium constant (E2) consequent to mutations of residues at the C-terminus of loop 9 (L9) in the α and ε subunits of mouse neuromuscular AChRs. These amino acids are close to two interesting interfaces, between the extracellular and transmembrane domain within a subunit (E–T interface) and between primary and complementary subunits (P–C interface). Most α subunit mutations modestly decreased E2 (mainly by slowing the channel-opening rate constant) and sometimes produced AChRs that had heterogeneous gating kinetic properties. Mutations in the ε subunit had a larger effect and could either increase or decrease E2, but did not induce kinetic heterogeneity. There are broad-but-weak energetic interactions between αL9 residues and others at the αE–T interface, as well as between the εL9 residue and others at the P–C interface (in particular, the M2–M3 linker). These interactions serve, in part, to maintain the structural integrity of the AChR assembly at the E–T interface. Overall, the energy changes of L9 residues are significant but smaller than in other regions of the protein.
Introduction
Ligand-gated ion channels are allosteric proteins that spontaneously isomerize between closed- (R) and open-channel (R*) conformations. The five subunits of the nicotinic acetylcholine receptor-channel (AChR) are arranged symmetrically around a central ion permeation pathway that changes shape (R↔R*) to regulate the flow of ions across the cell membrane (Unwin, 2005; Hilf & Dutzler, 2008; Bocquet et al. 2009; Hibbs & Gouaux, 2011). The probability that the AChR adopts the active, ion-permeable conformation increases in the presence of agonists because these bind more tightly to R* compared to R (Monod et al. 1965). In order to understand the molecular mechanism of gating it is essential to know the magnitudes and timing of structural and energy changes that take place in different regions and residues in the protein. In neuromuscular AChRs, such energy changes are widespread (Auerbach, 2010). Here, we address the gating energy changes associated with residues at the C-terminus of loop 9 (also known as loop F).
The adult neuromuscular AChR is composed of two α1 subunits and one each of β, δ and ε. The two transmitter binding sites are in the extracellular domain and at the α–ε or α–δ subunit interfaces, ∼30 Å above the level of the membrane. The transmembrane domain of each subunit is composed of four helices, with the M2 helix from each subunit coming together to form the lining of the ion permeation pathway. Loop 9 (L9) is in the extracellular domain and spans the entire region between the transmitter binding site (N-terminus) and the membrane (C-terminus) (Fig. 1).
Figure 1. Location of the C-terminus of loop 9 of AChRs.
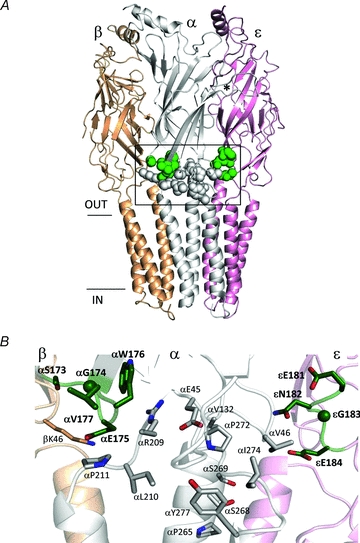
A, side view of the Torpedo AChR (accession number 2bg9.pdb). The mutated loop 9 positions in the α and ε subunits are shown as dark grey spheres (green spheres online); the residues tested for gating interactions with the loop 9 residues are shown as white spheres. *marks approximately the αε transmitter binding site and the horizontal lines mark approximately the membrane. The M4 transmembrane helices have been removed for clarity. B, higher resolution view of the boxed area in A. The residues are labelled according to their numbers and side chains in mouse AChRs (loop 9 residues, dark grey (green online) and bold).
The C-terminus of L9 is located near two interesting interfaces. One is an inter-subunit interface between the primary and complementary subunits (the P–C interface), for example between α and ε. Mutations of residues in the α subunit along this interface result in large changes in the gating equilibrium constant, suggesting that some residues here move (change energy) in the channel-opening process (Chakrapani et al. 2004; Purohit & Auerbach, 2007a,b). It has been suggested that molecular motions of amino acids along the P–C interface constitute the main pathway for the propagation of the AChR gating conformational change from the transmitter binding site to the gate (Mukhtasimova & Sine, 2007; Auerbach, 2010; Cadugan & Auerbach, 2010). In the open-channel form of the C. elegans homologue GluCl, C-terminal L9 residues in the complementary subunit are in contact with those in the M2–M3 linker of the adjacent, primary subunit (Hibbs & Gouaux, 2011).
The second interesting interface near the C-terminus of L9 is an intra-subunit one, between the extracellular and transmembrane domains (the E–T interface). Here, a network of charged amino acids plays important roles in both receptor expression and in R↔R* gating (Kash et al. 2004; Lee & Sine, 2005; Xiu et al. 2005; Mercado & Czajkowski, 2006; Purohit & Auerbach, 2007a; Bruhova & Auerbach, 2010). The close proximity of the C-terminus of L9 to these key inter- and intra-subunit interfaces makes this region a good candidate for probing the effects of mutations on AChR function.
Many previous studies of AChRs and related receptors suggest that structural rearrangements in L9 occur in both binding and gating processes (Leite et al. 2003; Lyford et al. 2003; Newell & Czajkowski, 2003; Bouzat et al. 2004; Hansen et al. 2005; Cheng et al. 2006; Thompson et al. 2006; Szarecka et al. 2007; Khatri et al. 2009; Law & Lightstone, 2009; Pless & Lynch, 2009). In order to clarify the role of the C-terminus of L9 we have examined the consequences of mutations of residues here in the α and ε subunits of adult mouse neuromuscular AChRs with regard to the channel-gating process. We also measured the degrees to which these mutations interact energetically with others located at the nearby inter- and intra-subunit interfaces, and the extents to which they induce heterogeneity in the gating rates and equilibrium constants.
Methods
Mutagenesis and expression
Mutations were made in the α or ε subunits in loop 9 (αL9 or εL9), and elsewhere (Fig. 1). Mutants were made in mouse AChR subunit cDNAs by using the QuikChange site-directed mutagenesis kit (Stratagene), and were verified by nucleotide sequencing. Cells of the human embryonic kidney cell line HEK 293 were transiently transfected using the calcium phosphate precipitation method. Cells were treated with 3.5–5.5 μg DNA per 35 mm culture dish in the ratio of 2:1:1:1 (α:β:δ:ε) for ∼16 h at 37°C. Most electrophysiological recordings were made ∼24 h later. For hybrid experiments, HEK cells were treated with cDNAs in the ratio 1:1:1:1:1 (αmut:αwt:β:δ:ε).
Electrophysiology
More detailed accounts of mutation, expression, recording and analysis methods can be found in Jha et al. (2007) and Jadey et al. (2011). Briefly, recordings were performed in the cell-attached patch configuration at 23°C with agonist only in the patch pipette. Except where noted, the agonist was dissolved in Dulbecco's phosphate-buffered saline containing (mm): 137 NaCl, 0.9 CaCl2, 2.7 KCl, 1.5 KH2PO4, 0.5 MgCl2 and 8.1 Na2HPO4 (pH 7.3). Pipettes made from borosilicate capillaries were coated with Sylgard (Dow Corning Corp.). The average pipette resistance was ∼10 MΩ. Single-channel currents were recorded using a PC-505 amplifier (Warner Instrument Corp.) with low-pass filtering at 20 kHz. The currents were digitized at a sampling frequency of 50 kHz using a SCB-68 acquisition board (National Instruments Corp.) and QUB software (http://www.qub.buffalo.edu).
We recently developed new methods for compensating for the effects of sub-saturation of the binding site and channel block on the kinetic analyses (Jadey et al. 2011). Consequently, different experimental conditions were use to examine the α and ε L9 mutants. For the αL9 mutants, the bath solution was identical to the pipette solution (but without agonist). The pipette potential was +70 mV (which corresponds to a membrane potential of approximately ∼−100 mV) and the agonist concentration was either 20 mm choline or 500 μm acetylcholine (ACh). These concentrations are ∼5 times the equilibrium dissociation constant of the R conformation (Kd), so the transmitter binding sites were almost continuously occupied by agonist molecules. Channel block by the agonist was apparent in these experiments, so the observed backward, channel-closing rate constant (b2) underestimated the true value. To correct for block, single-channel current amplitudes were estimated both at a low agonist concentration (30 μm ACh or 200 μm choline) where there was essentially no channel block by the agonist (i0∼7 pA), and also at a high agonist concentration (500 μm ACh or 20 mm choline) where fast channel block reduced the current amplitude (iB). We assumed that channel block slows closing and reduced the amplitude to the same extent, so for each construct the observed b2 value was corrected as follows (Neher & Steinbach, 1978): assume the block kinetic scheme is C(losed)–O(pen)–B(locked), with forward and backward rate constants f2, b2 (C–O) and p, q (O–B). With fast channel block, b2observed is the inverse lifetime of the aggregate {O–B}: b2observed=b2/(1 +p/q). In the presence of fast block the single-channel current amplitude is iB=i0/(1 +p/q). Therefore b2corr=b2observed(i0/iB). This correction assumes that the rate constant for channel closing is negligible when the pore is occupied by the blocker (Purohit & Grosman, 2006). For some constructs a second estimate of b2 was obtained from the inverse of the apparent open time at low agonist concentration. The two estimates were in good agreement (see Supplemental material, Table 1S, available online only). No correction was necessary for the forward, channel-opening rate constant (f2).
For the more recent εL9 experiments, the bath NaCl was replaced with KCl and the pipette potential was –70 mV (which corresponds to a membrane potential of exactly +70 mV). Also, the agonist concentration was 100 mm choline (which fully saturates the binding sites) and no NaCl was included in the pipette solution. At a membrane potential of +70 mV the current is outward and there is essentially no channel block by the agonist. In order to compensate for the effect of depolarization on AChR gating kinetics (Jadey et al. 2011) the εL9 mutants were expressed with a distant background mutation in the αM3 helix (αV283W) (Cadugan & Auerbach, 2007). As described below, the rate and equilibrium constant estimates for the εL9 mutations were corrected for the combined effects of depolarization and the αV283W mutation.
For rate constant analysis, clusters of individual channel activity were selected by eye. After filtering digitally (12 kHz), current clusters were idealized into noise-free intervals by using the segmental k-means algorithm (Qin, 2004) with a C(losed)↔O(pen) model. The gating rate constants were estimated from the idealized interval durations by using a maximum-interval likelihood algorithm (Qin et al. 1997) after imposing a dead time correction of 50 μs. In some patches the rate constants were estimated by using a simple C↔O model (because the log likelihood of the fit did not increase upon the addition of more C or O), while in others a second C state was connected to the O state to accommodate a relatively rare and short-lived non-conducting state associated with desensitization (Salamone et al. 1999; Elenes & Auerbach, 2002).
Φ was estimated as the slope of the rate equilibrium (R–E) relationship, which is a plot of log f2 vs. log diliganded equilibrium constant (E2=f2/b2). For residues that showed both an increase and a decrease in E2 upon mutation, the rate constants for AChRs activated by choline or by ACh were combined into the same R–E plot after normalizing both f2 and E2 by the corresponding wild-type (wt) values. In the R–E plots the wt values used for normalization were 120 s−1 and 0.046 for choline (Mitra et al. 2005) and 48,000 s−1 and 28.2 for ACh (Chakrapani & Auerbach, 2005). The change in relative end-state energy (kcal mol−1) caused by a mutation was calculated as: ΔG =–0.59 × ln(E2mutant/E2wt). For each residue, a ‘range energy’ was calculated as 0.59 × ln(E2maximum/E2minimum) for all tested mutations at that position (Jha et al. 2009).
The Kd for acetylcholine was estimated only for αE175W. Open and shut interval durations were measured at three different ACh concentrations (30, 50 and 100 μm). The two agonist binding sites were assumed to be equivalent and independent (Salamone et al. 1999; Jha & Auerbach, 2010), and the interval durations at all three concentrations were fitted together by using a C↔AC↔A2C↔A2O kinetic model (A is the agonist; dead time was 75 μs). Three rate constants were free parameters: single-site association (k+, scaled by [A]), single-site dissociation (k−) and b2 (f2 was fixed to the value obtained at 5 ×Kd agonist concentration) (Kd=k−/k+).
Sequence and structure
The location of L9 in the Torpedo AChR (Unwin, 2005) is shown in Fig. 1. In the α1 subunit of the mouse AChR the C-terminal residues of L9 are (N-to-C) FMESGEW. The GEW sub-sequence is conserved in all mouse AChR subunits except β1 (GQW) and α6 (SEW). The L9 residues we studied are dark grey (green online) in the figure and were positions 173–177 in the α subunit (SGEWV) and 181–184 in the ε subunit (ENGE). We measured the coupling energy between different combinations of L9 residues and the white residues in Fig. 1B (Supplemental Table 2S).
Results
Point mutations of L9 in the α subunit
Example agonist-activated currents from AChRs having a side-chain substitution in αL9 (both α subunits mutated) are shown in Supplemental Fig. 1S. None of the mutations significantly altered the single-channel current amplitude relative to the wild-type (wt), which suggests that they did not alter channel conductance, selectivity or block by the agonist.
To estimate the magnitude and relative timing of the energy changes experienced by αL9 residues in the gating isomerization we measured the forward, channel-opening rate constant (f2), the backward channel-closing rate constant (b2) and the gating equilibrium constant (E2=f2/b2) in the mutant AChRs. These values are shown as rate–equilibrium (R–E) plots for each position in Fig. 2 (Supplemental Table 1S). Mutation of αS173 (to A, D, G, P or K) and the β9-strand residue αV177 (to A, D, E, F, N or Q) yielded AChRs having nearly wt gating properties. That is, the range of gating equilibrium constants for the entire mutant series was small (<1 kcal mol−1). The fact that so many diverse side chain substitutions minimally altered the diliganded gating equilibrium constant indicates that these two positions are nearly iso-energetic between the R and R* conformations. Most mutations at positions αG174, αE175 and αW176 modestly decreased E2 (increased the relative stability of the closed-channel conformation of the protein).
Figure 2. Rate–equilibrium (R–E) plots for mutations of loop 9 in the α subunit.
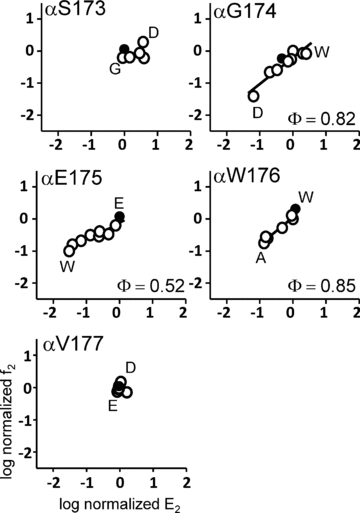
f2 is the forward, channel-opening rate constant (s−1) and E2 is the diliganded gating equilibrium constant. Values have been normalized by the wt value (filled circles). In each panel, the x-axis range reflects the mutational sensitivity of the position with regard to E2. Only positions 174, 175 and 176 show significant changes in gating upon mutation.
The slope of the R–E plot (Φ) quantifies the extent to which a change in E2 was caused by a change in the opening rate constant vs. the closing rate constant on a scale from 1 (all opening; an ‘early’ energy change) to 0 (all closing; a ‘late’ energy change). The Φ values for αG174 and αW176 were high (0.82 ± 0.01 and 0.85 ± 0.09; mean ± SD), whereas that for αE175 was lower (0.52 ± 0.06; see below). Since Φ cannot be estimated accurately when the range in E2 is very low, these calculations were not made for positions αS173 and αV177.
Although the currents with most αL9 mutations could be described by a simple two-state gating scheme, the F, Y, C and Q mutations of αE175 generated current clusters that were heterogeneous with regard to their cluster open probability (Fig. 3A). For these mutations no clearly predominant population of cluster could be identified. For αE175C, we estimated f2 and E2 for each kinetic mode separately (open probability 0.38, 0.68 or 0.95). The resulting R–E plot is shown in Fig. 3B. The Φ value for the unknown structural perturbation that gives rise to the distinct kinetic modes of αE175C was 0.62 ± 0.03. The Φ values for the modes of the F and Y substitutions were 0.66 and 0.71 (not shown). A change in open probability implies a change in E2 and, hence, a structural difference that alters the relative free energy of R vs. R*. Many residues at the E–T interface have similar Φ values (Jha et al. 2007; Purohit & Auerbach, 2007b; Bruhova & Auerbach, 2010), so we speculate that the structural perturbation that gives rise to kinetic heterogeneity is located in this region of the protein. The heterogeneity in E2 could reflect multiple populations of AChRs that have one (or more) E–T residues in slightly different positions, or a slow isomerization of an E–T side chain(s) in a homogeneous AChR population.
Figure 3. Some mutations of αE175 give rise to currents showing heterogeneous kinetic properties.
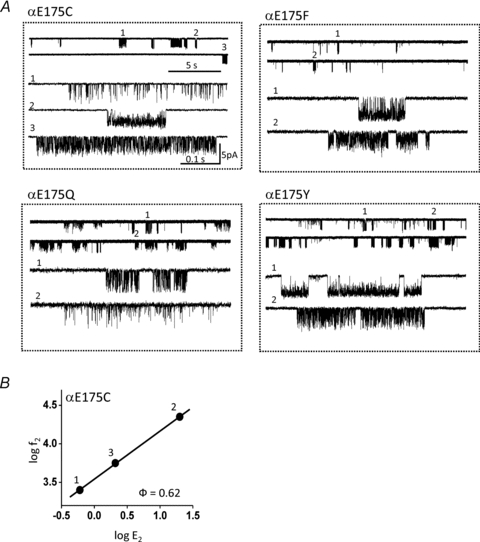
A, example currents from αE175 mutant AChRs exhibiting heterogeneous kinetics. Three populations of current clusters are apparent in αE175C. B, R–E analysis of the αE175C populations shows that both the forward and backward gating rate constants change between populations. The spontaneous structural perturbation(s) that generates the heterogeneity has a Φ value of 0.62.
The AChR is composed of two α subunits. To address the question of whether or not the αL9 mutation in each subunit contributes equally to the total fold-change in E2 we added both mutant αE175R and wt α subunits to the transfection cocktail. Accordingly, ‘pure’ AChRs were expressed that had either 2 wt or 2 mutant α subunits, along with two ‘hybrid’ populations having one wt and one mutant αε(or αδ) subunit. The results are shown in Fig. 4. In each patch we could clearly identify clusters arising from the wt and double-mutant populations. In addition, we observed a single, novel population of clusters that we attribute to the αE175 +αE175R hybrids. In this population the fold-change in E2 was almost exactly half of the fold-change of the double-mutant (compared to the wt). The simplest interpretation is that the energetic consequence of the αE175R mutation was equal and independent in the two α subunits. This result is similar to that found for several other hybrid constructs, including αS269I (Mitra et al. 2005), αP265K (Bafna et al. 2008) and αC418W (Mitra et al. 2004). In addition, the αE175R hybrid and double-mutant constructs had similar Φ values (Fig. 4B). These results suggest that the magnitude and relative timing of the gating energy changes at position αE175 are similar in the two α subunits.
Figure 4. The αE175R mutation has equivalent effects in each α subunit.
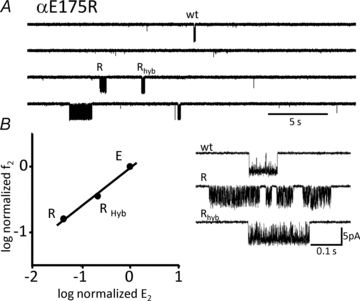
AChRs were expressed having 0 (wt), 1 (Rhyb) or 2 (R) mutated α subunits. A, low time resolution view of current clusters. B, left, R–E analysis of cluster populations. Right, example currents at higher resolution. Only a single hybrid population was apparent, with a fold-change in E2 (relative to the wt) that was half of that for the double-mutant.
To test if there is an energetic connection between αL9 and the transmitter binding sites we measured the ACh binding rate and equilibrium constants for one αL9 mutant construct, αE175W (Supplemental Fig. 2S). From the single-site association (k+) and dissociation (k−) rate constants to the resting conformation, we calculated the equilibrium dissociation constant (Kd=k−/k+). In this mutant, k+, k− and Kd were all similar to wt values (Chakrapani et al. 2004).
Point mutations of L9 in the ε subunit
We also quantified the effects on gating of C-terminal L9 point mutations in the ε subunit. εL9 lies along the critically important α–ε interface, whereas αL9 projects into the less energetically sensitive β–α (or ε–α) inter-subunit interface.
The R–E plots for εL9 mutants are shown in Fig. 5. In contrast to αL9 mutations, which mostly decreased E2, εL9 mutations either decreased or increased E2. Interestingly, the ranges of E2 values were larger in εL9 (0.78–3.3 kcal mol−1) compared to αL9 (0.39–2.2 kcal mol−1) even though there are two α subunits and only one ε subunit. The ε amino acid showing the largest range energy was εE184 (3.3 kcal mol−1). This value, on a per residue basis, is about three times larger than that for the homologous αE175 position.
Figure 5. Rate–equilibrium (R–E) plots for mutations of loop 9 in the ε subunit.
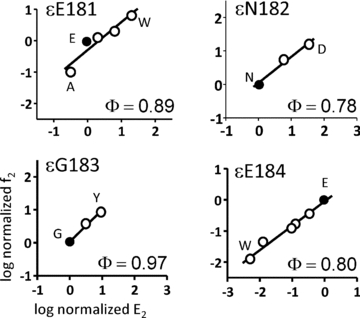
f2 is the forward gating rate constant (s−1) and E2 is the diliganded gating equilibrium constant. Values have been normalized by the wt value (filled circles). In each panel, the x-axis range reflects the sensitivity of the position with regard to E2. All positions show significant changes in gating upon mutation.
The Φ values for all of the εL9 residues were high (0.86 ± 0.09), and on average, modestly higher than for αL9 residues (0.73 ± 0.18; Table 1). In contrast to αE175, the Φ value for εE184 was about the same as for its neighbours.
Table 1.
Φ and range energy values for loop 9
| Residue | Φ(± SD) | Range energy (kcal mol−1) |
|---|---|---|
| αS173 | — | 0.9 |
| αG174 | 0.82 ± 0.10 | 2.2 |
| αE175 | 0.52 ± 0.06 | 2.1 |
| αW176 | 0.85 ± 0.09 | 1.2 |
| αV177 | — | 0.4 |
| εE181 | 0.89 ± 0.10 | 2.5 |
| εN182 | 0.78 ± 0.09 | 2.1 |
| εG183 | 0.97 ± 0.08 | 1.3 |
| εE184 | 0.80 ± 0.06 | 3.3 |
None of the εL9 mutations generated the heterogeneous gating behaviour apparent in some αL9 mutants.
Interaction energies (‘coupling’)
We sought to measure the extents to which L9 mutations interact energetically in the gating process with other nearby amino acids. We probed the degree of such coupling between αE175 and other α subunit residues at the E–T interface, and between εE184, εG183 and εN182 in combination with α subunit residues at the P–C interface (Figs 1B and 6; see Supplemental material, Fig. 5S and Table 2S).
Figure 6. Loop 9 residue coupling energies.
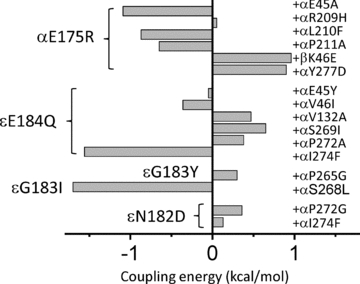
The interaction (‘coupling’) energy between mutations was estimated by comparing the fold-changes in E2 caused by single vs. mutation pairs (see Fig. 1B). At the α E–T interface the strongest interaction was between αE175R and αE45A, but there was not coupling with αR209H. At the α–ε P–C interface the strongest interaction was between εE184Q and αI274F (in the M2–M3 linker). At the β–α P–C interface, βK46E and αE175R interact.
The first E–T interfacial residue we probed in combination with αE175 was loop 2 residue αE45 (Fig. 1B). This important amino acid experiences a very large energy change, early (Φ= 0.80) in the channel-opening process (Lee & Sine, 2005; Purohit & Auerbach, 2007a). For the double-mutant α(E175R + E45A), the observed fold-change in E2 was only ∼6.4-fold greater than the value predicted assuming energy independence, which corresponds to a modest coupling energy for this mutant pair of –1.1 kcal mol−1.
The next residue we examined in combination with αE175 was αR209, which is in the pre-M1 linker that joins the extracellular domain (ECD) to the transmembrane domain (TMD) (Fig. 1B). Mutations of this residue can affect gating and also reduce the expression of functional channels (Lee & Sine, 2005; Purohit & Auerbach, 2007a). The α(E175R + R209H) combination behaved as if the two residues were energetically independent (coupling energy, +0.05 kcal mol−1). We tested four other αE175 +αR209 combinations (R + E, R + K, F + Q, H + K, G + Q), but no single-channel currents were observed for any of these constructs (3–10 patches mutant−1, 20 min patch−1). We conclude that these constructs either fail to express AChRs or express AChRs that fail to function in a manner consistent with the time resolution of the patch clamp. Interestingly, αR209K and Q mutations alone allow the expression of functional AChRs but fail to do so with the αE175 mutant background. This suggests that there are interactions between these two positions, but with regard to receptor assembly and expression rather than gating.
We also made coupling energy estimates for αE175 and three other α subunit E–T interfacial residues (αL210, αP211 and αY277). We chose αL210 and αY277 because these have low Φ values (∼0.3; Cadugan & Auerbach, 2007; Purohit & Auerbach, 2007a), and, hence, might be responsible for the anomalously low Φ value for αE175. There was a modest degree of coupling energy for the α(E175R/K + Y277D) and α(E175R + L210F) combinations (∼+0.9 kcal mol−1). However, the Φ value for the αE175R and K mutants was similar on the wt vs. the αY277D background (Supplemental Fig. 3S). Similar absolute degrees of coupling energy were observed for the constructs α(E175R + L210F) (+0.9 kcal mol−1) and α(E175R + P211S) (–0.7 kcal mol−1). We also tested for αL9 coupling across the α–β subunit interface. A modest coupling energy (+1.0 kcal mol−1) was apparent for the αE175R +βK46E (in loop 2) combination.
Some of these double-mutant constructs gave rise to clusters with heterogeneous kinetic properties. Example currents from the α(E175R + E45L) and some α(E175 + L210) combinations are shown in Supplemental Fig. 4S. Distinct from the single-mutant αE175 constructs, in these combinations there was no clear separation of clusters into modes. In each patch there was a broad range of cluster kinetic properties, where the open probability within a cluster could span nearly the entire range, 0–1. Therefore, we were unable to estimate either the coupling energies for these mutant combinations or Φ values for the unknown perturbation(s) that generated the different cluster kinetics.
We next measured coupling energies between εL9 residues 182–184 and several nearby residues in the α subunit (Fig. 1B). This region is located along the α–ε P–C interface and experiences large gating energy changes when subject to mutation. The structure of GluCl, apparently in the open conformation, shows that L9 from a complementary subunit is near the M2–M3 linker from a primary subunit (Hibbs & Gouaux, 2011), so we included in our experiments residues from the αM2–M3 linker. In contrast to some αL9 mutations, none of the εL9 mutations (either alone or in combination) generated heterogeneous gating behaviour. The interaction energy was small for most of the combinations (Fig. 6; Supplemental Table 2S). Only two combinations showed an interaction energy >1.4 kcal mol−1 (a ∼10-fold deviation from independence), εE184Q +αI274F (in the M2–M3 linker) and εG183I +αP265G (in the M2 ‘cap’).
Discussion
Mutations of the amino acids at the C-terminus of L9 had modest-but-measurable effects on the diliganded AChR gating equilibrium constant. The changes in E2 caused by L9 mutations in the ε subunit were larger than in the α subunit. Considering all mutations, the range energy (kcal mol−1) was 5.3 kcal mol−1 for εL9 but only 2.9 kcal mol−1 for αL9. Assuming that for all mutants the gating energy changes were divided equally between the two α subunits (Fig. 4), εL9 was more than three times more sensitive to mutation than αL9.
A change in E2 caused by a mutation indicates that the side chain substitution changed the free energy difference between the R and R* end states compared to the wt. This suggests that the mutated amino acid (and, possibly, water) sustains a motion, strain or change in dynamics during the gating isomerization. The results regarding the effects of L9 mutations on E2 are thus consistent with previous reports showing that this region moves during the gating isomerization. Our results further suggest that the energetic consequence of this motion is greater in ε compared to α. The basis for the greater sensitivity of εL9 is not known, but it may be relevant that its C-terminus is adjacent to the α–ε subunit interface and thus near a pathway for conformational change that may link the transmitter binding site with gate. Overall, the energetic effects of mutations at the C-terminus of L9 were modest compared to those in other regions of the protein (Cadugan & Auerbach, 2010).
We observed no effect of the αE175W mutation on ACh binding. This is not surprising given the distance between the C-terminus of αL9 and the α–ε transmitter binding site. A mutation of an εL9 amino acid (εN182Y) has been shown to influence agonist binding as well as gating (Sine et al. 2002). Thus, there may be energetic interactions between εL9 and the binding site that are transmitted along the α–ε P–C interface.
Φ values provide information on the relative timing of the energy changes that occur within R↔R* gating (Auerbach, 2007). One interpretation of Φ is that it reflects a perturbation in the energy of a microwell that is part of the gating transition state ensemble (Auerbach, 2005). Recently a brief, non-conducting state (‘flip’) has been detected that may be a signal from sojourns in one (or more) of such microwells, possibly associated with a configuration in which the binding site has undergone its low-to-high affinity transformation but the pore is still non-conducting (Lape et al. 2008). The rate constants, equilibrium constants and Φ values we measured for loop 9 residues pertain to the overall R↔R* isomerization and incorporate sojourns in this and other intermediate states of gating.
With one exception, Φ values for L9 residues in both the α and ε subunits were similar and high, ∼0.85. This suggests that the energy (structure) of the C-terminus of L9 is mostly ‘open-like’ at the gating transition state, and, hence, that the motion of L9 occurs relatively early in the channel-opening process. Other amino acids at the ECD–TMD and inter-subunit interfaces have somewhat lower Φ values, ∼0.7 (Jha et al. 2007; Purohit & Auerbach, 2007a). Perhaps the fact that the N-terminus of L9 approaches a transmitter binding site, a region where many residues have Φ values ∼1, is related to the high Φ values of L9, overall.
The exceptional, low-Φ position was αE175. We considered the possibility that this residue might experience multiple energy perturbations in the opening process, one at the onset (in synch with its high-Φ neighbours) and again, later in the reaction, by virtue of energy coupling with some nearby, low-Φ residue(s). However, we found that the Φ value for the αE175R perturbation was the same when secondary mutations were made at two such low-Φ positions, αL210 and αY277 (both of which have Φ∼0.3). There may be other nearby (and uncharted) low-Φ neighbours, so our speculation has not been disproved. However, the anomalously low Φ value of residue αE175 remains unexplained. There are other examples of residues having Φ values that are distinct from their neighbours (Bafna et al. 2008; Jha et al. 2009). Without a clear theory for Φ, these observations remain unexplained.
We tested for interactions between αE175 and other residues at the α subunit E–T interface. For six different mutant combinations the average absolute value of the coupling energy was ∼0.9 kcal mol−1, which represents only a ∼5-fold deviation of the value of E2 expected given independent energy changes at the two positions. Although this amount of interaction energy is small (compared to, for example, the ∼6 kcal mol−1 coupling between αA96C and αY127C; Cadugan & Auerbach, 2010), it is easily measured and may be significant with regard to the assembly and physiology of AChRs. It appears that αE175 is part of a broad network of electrostatic interactions that prevails at the E–T interface that serves, in part, to maintain the stability of the receptor assembly. With regard to the α–ε P–C interface, we found even smaller degrees of energy coupling with εE184Q and five different α-subunit amino acids. However, two εL9 combinations (between the αM2–M3 linker and αM2 ‘cap’) produced somewhat larger coupling energies. Coupling energies depend on the specific side chains, so the estimates of interaction energy are likely to increase as more mutant combinations are probed. Also, adjacent side chains may show very different interaction energies, so it is still possible that stronger energetic interactions exist between εL9 and residues in the α subunit.
αE175 (but not εE184) mutations could result in the expression of AChR showing heterogeneous gating kinetics. We noticed this behaviour both with some single mutations (in both α subunits; F, C, Y and Q) and especially for paired mutations with residues at the α subunit E–T interface. We attribute the kinetic heterogeneity to non-local conformational differences caused by these αL9 mutations that, based on their Φ values, are located in the α subunit E–T interfacial region. For example, mutations of AChR M2–M3 linker amino acids change E2 substantially and with approximately the same Φ value as the source of the perturbation that changes cluster open probability (Jha et al. 2007). We speculate that a network of interacting residues at the E–T interface serves not only to regulate channel gating but also the stability of this interface and, more generally, the assembly and expression of AChRs.
Acknowledgments
We thank M. Merritt, M. Shero and M. Teeling for technical assistance. This work was supported by the National Institutes of Health (NS-23513).
Glossary
- AChR
acetylcholine receptor
- ECD
extracellular domain
- E–T
extracellular and transmembrane
- L9
loop 9
- P–C
primary and complementary subunits
- R–E
rate equilibrium plot
- TMD
transmembrane domain
- wt
wild-type
Author contributions
A.J, S.G. and S.N.Z. performed the experiments and analysed the data. A.J. and A.A. wrote the paper. All the experiments in this study were carried out at the Department of Physiology and Biophysics, State University of New York at Buffalo, Buffalo, USA.
Supplementary material
Supplementary Fig 1S
Supplementary Fig 2S
Supplementary Fig 3S
Supplementary Fig 4S
Supplementary Fig 5S
Supplementary Table 1S
Supplementary Table 2S
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Auerbach A. Gating of acetylcholine receptor channels: brownian motion across a broad transition state. Proc Natl Acad Sci U S A. 2005;102:1408–1412. doi: 10.1073/pnas.0406787102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A. How to turn the reaction coordinate into time. J Gen Physiol. 2007;130:543–546. doi: 10.1085/jgp.200709898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A. The gating isomerization of neuromuscular acetylcholine receptors. J Physiol. 2010;588:573–586. doi: 10.1113/jphysiol.2009.182774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bafna PA, Purohit PG, Auerbach A. Gating at the mouth of the acetylcholine receptor channel: energetic consequences of mutations in the αM2-cap. PLoS One. 2008;3:e2515. doi: 10.1371/journal.pone.0002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Gumilar F, Spitzmaul G, Wang HL, Rayes D, Hansen SB, Taylor P, Sine SM. Coupling of agonist binding to channel gating in an ACh-binding protein linked to an ion channel. Nature. 2004;430:896–900. doi: 10.1038/nature02753. [DOI] [PubMed] [Google Scholar]
- Bruhova I, Auerbach A. Subunit symmetry at the extracellular domain-transmembrane domain interface in acetylcholine receptor channel gating. J Biol Chem. 2010;285:38898–38904. doi: 10.1074/jbc.M110.169110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadugan DJ, Auerbach A. Conformational dynamics of the αM3 transmembrane helix during acetylcholine receptor channel gating. Biophys J. 2007;93:859–865. doi: 10.1529/biophysj.107.105171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadugan DJ, Auerbach A. Linking the acetylcholine receptor-channel agonist-binding sites with the gate. Biophys J. 2010;99:798–807. doi: 10.1016/j.bpj.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrapani S, Auerbach A. A speed limit for conformational change of an allosteric membrane protein. Proc Natl Acad Sci U S A. 2005;102:87–92. doi: 10.1073/pnas.0406777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrapani S, Bailey TD, Auerbach A. Gating dynamics of the acetylcholine receptor extracellular domain. J Gen Physiol. 2004;123:341–356. doi: 10.1085/jgp.200309004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Wang H, Grant B, Sine SM, McCammon JA. Targeted molecular dynamics study of C-loop closure and channel gating in nicotinic receptors. PLoS Comput Biol. 2006;2:e134. doi: 10.1371/journal.pcbi.0020134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenes S, Auerbach A. Desensitization of diliganded mouse muscle nicotinic acetylcholine receptor channels. J Physiol. 2002;541:367–383. doi: 10.1113/jphysiol.2001.016022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SB, Sulzenbacher G, Huxford T, Marchot P, Taylor P, Bourne Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005;24:3635–3646. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilf RJ, Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- Jadey SV, Purohit P, Bruhova I, Gregg TM, Auerbach A. Design and control of acetylcholine receptor conformational change. Proc Natl Acad Sci U S A. 2011;108:4328–4333. doi: 10.1073/pnas.1016617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A, Auerbach A. Acetylcholine receptor channels activated by a single agonist molecule. Biophys J. 2010;98:1840–1846. doi: 10.1016/j.bpj.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A, Cadugan DJ, Purohit P, Auerbach A. Acetylcholine receptor gating at extracellular transmembrane domain interface: the cys-loop and M2-M3 linker. J Gen Physiol. 2007;130:547–558. doi: 10.1085/jgp.200709856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A, Purohit P, Auerbach A. Energy and structure of the M2 helix in acetylcholine receptor-channel gating. Biophys J. 2009;96:4075–4084. doi: 10.1016/j.bpj.2009.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash TL, Dizon MJ, Trudell JR, Harrison NL. Charged residues in the β2 subunit involved in GABAA receptor activation. J Biol Chem. 2004;279:4887–4893. doi: 10.1074/jbc.M311441200. [DOI] [PubMed] [Google Scholar]
- Khatri A, Sedelnikova A, Weiss DS. Structural rearrangements in loop F of the GABA receptor signal ligand binding, not channel activation. Biophys J. 2009;96:45–55. doi: 10.1016/j.bpj.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lape R, Colquhoun D, Sivilotti LG. On the nature of partial agonism in the nicotinic receptor superfamily. Nature. 2008;454:722–727. doi: 10.1038/nature07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law RJ, Lightstone FC. Modeling neuronal nicotinic and GABA receptors: important interface salt-links and protein dynamics. Biophys J. 2009;97:1586–1594. doi: 10.1016/j.bpj.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WY, Sine SM. Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature. 2005;438:243–247. doi: 10.1038/nature04156. [DOI] [PubMed] [Google Scholar]
- Leite JF, Blanton MP, Shahgholi M, Dougherty DA, Lester HA. Conformation-dependent hydrophobic photolabeling of the nicotinic receptor: electrophysiology-coordinated photochemistry and mass spectrometry. Proc Natl Acad Sci U S A. 2003;100:13054–13059. doi: 10.1073/pnas.2133028100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyford LK, Sproul AD, Eddins D, McLaughlin JT, Rosenberg RL. Agonist-induced conformational changes in the extracellular domain of α7 nicotinic acetylcholine receptors. Mol Pharmacol. 2003;64:650–658. doi: 10.1124/mol.64.3.650. [DOI] [PubMed] [Google Scholar]
- Mercado J, Czajkowski C. Charged residues in the α1 and β2 pre-M1 regions involved in GABAA receptor activation. J Neurosci. 2006;26:2031–2040. doi: 10.1523/JNEUROSCI.4555-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A, Bailey TD, Auerbach AL. Structural dynamics of the M4 transmembrane segment during acetylcholine receptor gating. Structure. 2004;12:1909–1918. doi: 10.1016/j.str.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Mitra A, Cymes GD, Auerbach A. Dynamics of the acetylcholine receptor pore at the gating transition state. Proc Natl Acad Sci U S A. 2005;102:15069–15074. doi: 10.1073/pnas.0505090102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions : A plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- Mukhtasimova N, Sine SM. An intersubunit trigger of channel gating in the muscle nicotinic receptor. J Neurosci. 2007;27:4110–4119. doi: 10.1523/JNEUROSCI.0025-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Steinbach JH. Local anaesthetics transiently block currents through single acetylcholine-receptor channels. J Physiol. 1978;277:153–176. doi: 10.1113/jphysiol.1978.sp012267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell JG, Czajkowski C. The GABAA receptor α1 subunit Pro174–Asp191 segment is involved in GABA binding and channel gating. J Biol Chem. 2003;278:13166–13172. doi: 10.1074/jbc.M211905200. [DOI] [PubMed] [Google Scholar]
- Pless SA, Lynch JW. Ligand-specific conformational changes in the α1 glycine receptor ligand-binding domain. J Biol Chem. 2009;284:15847–15856. doi: 10.1074/jbc.M809343200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P, Auerbach A. Acetylcholine receptor gating at extracellular transmembrane domain interface: the “pre-M1” linker. J Gen Physiol. 2007a;130:559–568. doi: 10.1085/jgp.200709857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P, Auerbach A. Acetylcholine receptor gating: movement in the α-subunit extracellular domain. J Gen Physiol. 2007b;130:569–579. doi: 10.1085/jgp.200709858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit Y, Grosman C. Block of muscle nicotinic receptors by choline suggests that the activation and desensitization gates act as distinct molecular entities. J Gen Physiol. 2006;127:703–717. doi: 10.1085/jgp.200509437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F. Restoration of single-channel currents using the segmental k-means method based on hidden Markov modeling. Biophys J. 2004;86:1488. doi: 10.1016/S0006-3495(04)74217-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F, Auerbach A, Sachs F. Maximum likelihood estimation of aggregated Markov processes. Proc Biol Sci. 1997;264:375–383. doi: 10.1098/rspb.1997.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone FN, Zhou M, Auerbach A. A re-examination of adult mouse nicotinic acetylcholine receptor channel activation kinetics. J Physiol. 1999;516:315–330. doi: 10.1111/j.1469-7793.1999.0315v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, Shen XM, Wang HL, Ohno K, Lee WY, Tsujino A, Brengmann J, Bren N, Vajsar J, Engel AG. Naturally occurring mutations at the acetylcholine receptor binding site independently alter ACh binding and channel gating. J Gen Physiol. 2002;120:483–496. doi: 10.1085/jgp.20028568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szarecka A, Xu Y, Tang P. Dynamics of heteropentameric nicotinic acetylcholine receptor: implications of the gating mechanism. Proteins. 2007;68:948–960. doi: 10.1002/prot.21462. [DOI] [PubMed] [Google Scholar]
- Thompson AJ, Padgett CL, Lummis SC. Mutagenesis and molecular modeling reveal the importance of the 5-HT3 receptor F-loop. J Biol Chem. 2006;281:16576–16582. doi: 10.1074/jbc.M601265200. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4Å resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Xiu X, Hanek AP, Wang J, Lester HA, Dougherty DA. A unified view of the role of electrostatic interactions in modulating the gating of Cys loop receptors. J Biol Chem. 2005;280:41655–41666. doi: 10.1074/jbc.M508635200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.