

Abstract
Non-technical summary
Long-lasting changes in efficacy of cell–cell communication (long-term potentiation; LTP) at specialized sites (synapses) between neurons in the brain are thought to underlie forms of learning and memory. These forms of LTP can occur at excitatory synapses and inhibitory synapses, thus in- or decreasing the activity of neurons. We provide evidence for a novel form of LTP at inhibitory synapses (LTPi) on a subset of neurons in the amygdala of mice, a brain region involved in fear and anxiety. This LTPi enhances the release of the inhibitory neurotransmitter GABA at synapses between inhibitory interneurons and excitatory principal neurons (PNs) in a sub-region of the amygdala. The described LTPi is heavily dependent on the production and diffusion of the volatile gas nitric oxide (NO), produced by PNs during stages of increased activity. These findings indicate that NO-mediated long-term regulation of inhibitory transmission in the amygdala might contribute to the learning of fear.
Abstract
Long-lasting changes of synaptic efficacy are thought to be a prerequisite for memory formation and maintenance. In the basolateral complex of the amygdala (BLA), one of the main regions for fear and extinction learning of the brain, various forms of long-term potentiation (LTP) have been described for excitatory glutamatergic synapses. In contrast, little is known about the mechanisms of LTP at inhibitory GABAergic synapses. Here we provide evidence that (1) LTP at inhibitory GABAergic synapses (LTPi) between inhibitory interneurons and principal neurons (PNs) can be induced by theta-burst stimulation (TBS), (2) this LTPi is prevented by AMPA- or NMDA-receptor antagonists, and (3) this LTPi is abolished by the NO synthase (NOS) inhibitor l-NAME or the NO scavenger PTIO, and thus is critically dependent on nitric oxide (NO) signalling. These findings are corroborated by immunocytochemical stainings for neuronal (n) NOS, which revealed the existence of nNOS-positive neurons and fibres in the BLA. We conclude that LTP of GABAergic synaptic transmission to PNs is induced by activation of AMPA and NMDA receptors at glutamatergic synapses and subsequent retrograde NO signalling to enhance GABAergic transmission. This form of LTP at GABAergic synapses comprises a novel form of heterosynaptic plasticity within the BLA, apt to shape conditioned fear responses.
Introduction
Long-lasting changes of synaptic transmission in the form of long-term potentiation (LTP) or long-term depression are a basic mechanism for memory formation and storage (Maren & Baudry, 1995). In the basolateral complex of the amygdala (BLA), different forms of homo- and heterosynaptic LTP have been described at excitatory glutamatergic synapses on principal neurons (PNs) and local interneurons (INs; for review see: Ehrlich et al. 2009; Pape & Paré, 2010). In addition, the influence of GABAergic synaptic transmission on the induction of LTP at glutamatergic synapses via activation of pre- or postsynaptic GABAA- and/or GABAB-receptors has been analysed (Royer & Paré, 2002; Shaban et al. 2006). In contrast, a basic mechanism for LTP at IN–PN GABAergic synapses, controlling the excitability of PN, is still lacking in mice. In the BLA, GABA can be released from local and paracapsular interneurons. Paracapsular interneurons appear as clusters of interneurons located at the external capsule or between BLA and the central nucleus of the amygdala (Busti et al. 2011). Released GABA activates GABAA- and GABAB-receptors, which can be expressed pre- and postsynaptically (Szinyei et al. 2000; Shaban et al. 2006), and thus controls the amount of excitation within the BLA and in consequence, influences the form of information processing in the amygdala (e.g. fear and extinction learning).
In various brain regions (e.g. ventral tegmental area (VTA), thalamus and hippocampus) long-term potentiation of GABAergic transmission has been reported (Patenaude et al. 2003; Nugent et al. 2007; Yang & Cox, 2007). One form of GABAergic LTP has been described as depending on activation of nitric oxide synthase (NOS) and production of nitric oxide (NO) from l-arginine (reviewed in Alderton et al. 2001). The volatile gas NO can modulate synaptic efficacy via activation of its canonical pathway, which includes soluble guanylyl cyclase and cGMP, or by nitrosation/S-nitrosylation of various proteins (Stamler et al. 2001; Nugent et al. 2009). In the BLA, NO has been described as modulating glutamatergic synapses and glutamatergic LTP (Schafe et al. 2005).
Here we provide data on a novel mechanism for the induction of LTP at inhibitory GABAergic IN–PN synapses in the BLA (LTPi), which requires activation of NMDA-receptors at glutamatergic synapses and subsequent activation of the neuronal NOS (nNOS).
Methods
The experimental design and animal handling was approved by the ‘Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen’ (reference number: 8.87-51.05.20.10.218).
Electrophysiology
Eight- to twelve-week-old male C57Bl/6J mice were anaesthetized with Forene (isoflurane, 1-chloro-2,2,2-trifluoroethyl-difluoromethylether; 2.5%) and decapit- ated. Coronal slices (300 μm thickness) containing the amygdala were prepared on a vibratome (Leica VT1200S, Germany), allowed to recover for 20 min at 34°C, and stored at room temperature. Single slices were placed in a submersion chamber at 32°C and were perfused with artificial cerebrospinal fluid (ACSF) containing (in mm): NaCl 120, KCl 2.5, NaH2PO4 1.25, MgSO4 2, CaCl2 2, NaHCO3 22 and glucose 20. The pH was set to 7.35 by gassing with carbogen.
Whole-cell patch clamp recordings were performed on morphologically and electrophysiologically identified principal neurons in the lateral nucleus of the amygdala (LA) as described previously (Sah et al. 2003; Sosulina et al. 2010) using an EPC-10 patch-clamp amplifier (HEKA, Germany) at a sampling rate of 10 kHz. Patch-pipettes (2.5–3 MΩ; borosilicate glass; Clark Electromedical Instruments, UK) were filled with an intracellular solution containing (in mm): NaCl 10, potassium gluconate 105, potassium citrate 20, Hepes 10, EGTA 0.5, MgCl2 1, MgATP 3, and NaGTP 0.5. The pH was set to 7.25. The resting membrane potential was determined immediately after accessing the whole-cell configuration. The passive and active membrane properties were recorded in the current-clamp mode at a membrane potential of −70 mV. Hyper- and depolarizing currents were injected (500 ms duration; −40 pA first step; Δ+20 pA) to elicit action potentials. The input resistance was calculated from the steady-state voltage deflection in response to a hyperpolarizing current injection of −40 pA.
To evoke inhibitory postsynaptic currents (IPSCs), a tungsten bipolar stimulation electrode was placed in the LA, and the stimulation strength was set to evoke 50% of the maximal IPSC amplitude (stimulus duration: 500 μs). The monosynaptic nature of the GABAergic transmission was verified by short latencies and its resistance to the bath-application of DNQX. Baseline responses were recorded for 10 min with an interstimulus interval of 20 s at a membrane potential of −5 mV in the whole-cell voltage-clamp mode (mean holding-current: 385 ± 48 pA). The series resistance was controlled during the experiments and remained unchanged. Monosynaptic LTPi was induced by applying four sequences of theta-burst stimuli (TBS) with an interstimulus interval of 25 s at a holding potential of −5 mV. Each sequence consisted of 4 pulses at 100 Hz repeated 25 times at 2 Hz. The TBS was followed by a recovery phase of 1 min. After TBS, IPSCs were monitored for at least 30 min.
In a subset of experiments, evoked IPSCs were recorded at a holding-potential of −70 mV using a high-chloride intracellular solution containing 105 mm KCl instead of potassium gluconate. Evoked IPSCs appeared as inward directed current under these recording conditions. During these experiments the extracellular Mg2+ concentration was reduced to 0.5 mm to relieve NMDA-receptors from the Mg2+ ion block.
For quantification, the amplitudes of the evoked monosynaptic IPSCs of each recording were normalized to the mean amplitude of all evoked responses during baseline stimulation (10 min baseline). LTPi was analysed during the first 10 responses immediately following TBS (time-point I) and during 10 evoked responses at the end of the experiment, at least 30 min after TBS (time-point II). The paired-pulse ratio of evoked IPSCs before and after TBS was calculated by dividing the amplitude of the second response by the amplitude of the first responses at a paired-pulse interval of 50 ms.
AP-5 (d-(–)-2-amino-5-phosphonopentanoic acid; 50 μm; Tocris, UK) or DNQX (6,7-dinitroquinoxaline-2,3-dione disodium salt; 10 μm; Tocris) were bath-applied in a subset of experiments to block NMDA- or AMPA-receptors. The GABAA-receptor antagonist gabazine (6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid hydrobromide; 25 μm; Tocris, UK) was bath-applied to confirm the GABAergic nature of the recorded IPSCs. The NO donor Noc18 (2,2′-(hydroxynitrosohydrazino)bis-ethanamine; 100 μm; Merck, Germany), the NO scavenger PTIO (2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide; 200 μm; Sigma-Aldrich, USA), or the NOS blocker l-NAME (Nω-nitro-l-arginine methyl ester hydrochloride; 200 μm; Sigma-Aldrich, USA) were bath-applied. The reversal potential of the evoked GABAergic IPSCs was measured prior to TBS during baseline recordings, but in the presence of DNQX and AP-5 to block glutamatergic transmission. In a different set of experiments, the reversal potential was monitored after TBS. In these experiments, DNQX and AP-5 were applied after TBS to exclude interference with LTPi induction. GABAergic IPSCs were recorded at holding potentials of −5, −20, −40, −60 and −70 mV, and the reversal potential was calculated by a linear fit of the normalized amplitudes (normalized to the maximal response at −5 mV) plotted against the holding potential.
The number of experiments are given as: (no. of cells/no. of animals). Results are presented as mean ± SEM and significance was determined using Student's t test with a significance level of *P < 0.05 and **P < 0.01. The data sets were tested for statistically significant outliers using the Grubb's test (significance level P < 0.05).
Immunohistochemistry
C57Bl/6J or transgene GAD67-EGFP mice were deeply anaesthetized with an overdose of sodium pentobarbital (100 mg kg−1) and transcardially perfused with 20 ml of ice-cold sodium phosphate-buffered saline (PBS; pH 7.4) followed by 50 ml of 4% paraformaldehyde in PBS (pH 7.4). The brains were removed, postfixed for 2 h, and saturated overnight with 30% sucrose in PBS.
Coronal slices, 50 μm thick, were cut on a freezing-sliding cryotome (Leica Frigomobil, Germany). Free-floating sections were permeabilized with 0.5% Triton-X100 in PBS (20 min at room temperature (RT)) and unspecific binding sites were blocked with 5% BSA, 10% normal goat serum, and 0.2% Triton-X100 in PBS (50 min at RT). The primary antibody (rabbit polyclonal against neuronal NOS; ab1376; 0.5 mg ml−1 stock solution; Abcam, UK) was diluted 1:250 in 2% BSA, 5% normal goat serum, and 0.2% Triton-X100 in PBS. The slices were incubated overnight at 4°C. The slices were washed three times (20 min each) in PBS. The secondary antibodies (goat anti rabbit DyLight488 or goat anti rabbit Cy3-conjugate; Dianova, Germany) were diluted 1:200 in 2% BSA, 5% normal goat serum, and 0.2% Triton-X100 in PBS. The slices were incubated for 50 min at RT. In some experiments the solution additionally contained propidium iodide (1:1000) to stain cell nuclei. After washing, the slices were mounted on glass slides and preserved by ImmuMount (Thermo Scientific, Germany).
The respective slices containing the amygdala were analysed using a laser scanning confocal microscope (Nikon eC1 plus) equipped with a CFI75 LWD 16×/0.8 NA objective (Nikon, Germany). To detect the fluorescence of DyLight488, Cy3 or propidium iodide, we used the 488 nm line of an argon laser and a 543 nm HeNe laser in combination with adequate emission filters (515 nm/30 nm and 605 nm/75 nm).
Results
Long-term potentiation at GABAergic synapses
Principal neurons (PNs) in the BLA were identified by their location, morphology (for schematic representation of the recording site see Fig. 1A), and by their pattern of action potential generation in response to depolarizing current injections during current-clamp recordings (membrane potential: −70 mV; first step: −40 pA; current step size: +20 pA; Fig. 1B). The PNs show a low number of generated action potentials (3.6 ± 0.5 at an injected current of +120 pA) and a typical adaptation of action potential generation as described previously (Fig. 1B; Sah et al. 2003; Sosulina et al., 2006 and 2010), which is clearly distinguishable from modes of action potential generation observed in local inhibitory interneurons (21.3 ± 4.4 action potentials at an injected current of +120 pA; n = 4/4; PN versus IN: P < 0.01; Fig. 1B). These differences in action potential generation were used to electrophysiologically differentiate between recorded PNs and INs. The action potential half-width of the first evoked action potential in PNs was typically 1.7 ± 0.04 ms (n = 22/7), whereas IN showed action potential half-widths of 0.7 ± 0.04 ms (n = 4/4; P < 0.01; Sosulina et al. 2010). The mean resting membrane potential of a representative sample of the recorded PNs was at −66 ± 1 mV (n = 22/7) and the mean input resistance was 281 ± 17 MΩ (n = 22/7). The short latencies of evoked IPSCs (2.5 ± 0.17 ms; n = 22) indicated that a monosynaptic connection was stimulated. TBS evoked a maintained increase of the evoked IPSCs in PNs (LTPi). The induction of LTPi by TBS (Fig. 1C and D) increased the normalized amplitudes by a factor of ∼2 (baseline: 0.92 ± 0.06; postTBS time-point I: 2.3 ± 0.29; P < 0.01; postTBS time-point II: 2.5 ± 0.32; P < 0.01; n = 14/7; Fig. 1D and E) in 14 out of 22 recorded PNs. The mean absolute amplitudes were 142 ± 15 pA during baseline conditions and 314 ± 37 pA and 348 ± 69 pA at time-points I and II (n = 14/7) after TBS, respectively. The recorded IPSCs at a holding potential of −5 mV were sensitive to application of the GABAA-receptor antagonist gabazine (25 μm; n = 19; Fig. 1C). DNQX was bath-applied at the end of the experiments for 10 min (for concentrations see Methods section) to control for the monosynaptic nature of the evoked IPSCs (Fig. 1D). DNQX did not significantly change the amplitudes of evoked GABAergic IPSCs compared to postTBS values measured in the absence of DNQX (postTBS in absence of DNQX: 323 ± 38 pA; postTBS in presence of DNQX: 364 ± 45 pA; n = 10/5). To test whether a specific subset of PNs showed LTPi, we separated the recorded PNs into two groups: those that did show LTPi (LTPi; n = 14) and those that did not (noLTPi; n = 8). No significant differences according to the mean resting membrane potential (LTPi: −67 ± 1 mV vs. noLTPi: −66 ± 2 mV; P = 0.51) and mean input resistance (LTPi: 300 ± 23 MΩvs. noLTPi: 248 ± 24 MΩ; P = 0.2) were found. Surprisingly, the TBS failed to induce GABAergic LTP in PNs in the presence of the AMPA-receptor blocker DNQX. Under these conditions, the normalized amplitudes of evoked IPSCs after TBS were 0.83 ± 0.05 and 1.09 ± 0.07 for time-points I and II (n = 16/6), and were not significantly different from baseline responses (0.96 ± 0.03; time-point I: P = 0.054; time-point II: P = 0.13; Fig. 1D and E). Similarly, interference with NMDA-receptor signalling by applying AP-5 to the bath solution completely abolished the induction of LTPi by TBS. The normalized amplitudes were 0.97 ± 0.04 during baseline conditions and 1.03 ± 0.05 (P = 0.29) and 1.28 ± 0.16 (P = 0.06; n = 16/6) for time-points I and II, respectively (Fig. 1D and E). The normalized amplitudes in the presence of DNQX or AP-5 were significantly different from control conditions at time-point I and time-point II after TBS (Fig. 1E). These data indicate that activation of AMPA- and NMDA-receptors within the neuronal network is a prerequisite for GABAergic LTP at IN–PN synapses.
Figure 1. LTPi in PNs is dependent on NMDA-receptor activation.
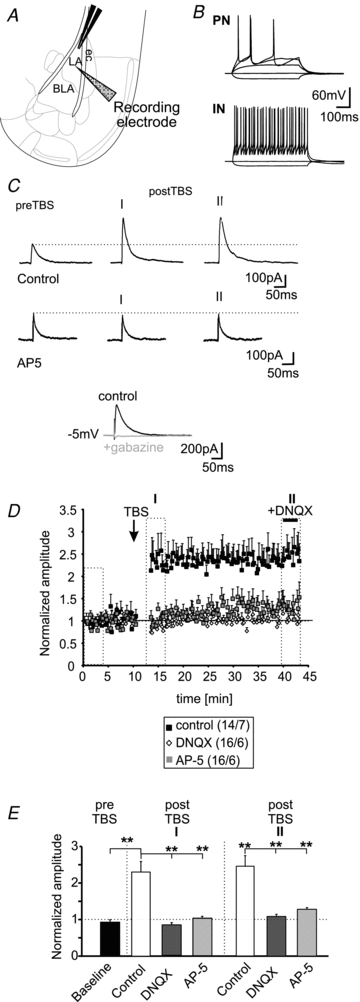
A, schematic representation of the experimental setting. The extracellular stimulation electrode was positioned within the LA. Whole-cell recordings in PNs were done in the adjacent BLA (ec, external capsule). B, examples of current-clamp recordings of a PN and a local IN in the BLA. C, example whole-cell voltage-clamp recordings of PNs at a holding potential of −5 mV under control conditions (upper traces) and in presence of 50 μm AP-5 (middle traces). The current traces show evoked IPSCs during baseline stimulation (preTBS) and IPSCs immediately after theta-burst stimulation (postTBS; I) and IPSCs at the end of the experiment (II; same time-points as in E). The evoked IPSCs could be blocked by application of gabazine (lower traces). Stimulation artifacts have been truncated. D, normalized mean amplitudes of evoked IPSCs show a long-lasting LTP under control conditions after TBS. This form of LTPi was eliminated when recorded in presence of 50 μm AP-5 or 10 μm DNQX. E, quantification of LTPi by TBS under control conditions and in presence of AP-5 or DNQX. (**P < 0.01; t test).
To analyse the involvement of AMPA- and NMDA-receptors in more detail, IPSCs were recorded at a holding-potential of −70 mV using a high-chloride intracellular solution (IPSCs appeared as inward directed currents during these recording conditions; Fig. 2A). In addition, the extracellular Mg2+ concentration was reduced to 0.5 mm, allowing NMDA-receptor activation at this holding potential, and AMPA-receptors were inhibited by DNQX. During these recording conditions, the applied TBS increased the evoked IPSCs significantly compared to baseline responses (baseline: 0.98 ± 0.03; time-point I: 1.8 ± 0.28, P < 0.01; time-point II: 1.7 ± 0.16, P < 0.01; Fig. 2B and C). The absolute amplitudes of the evoked IPSCs were −122 ± 24 pA, −245 ± 70 pA, and −235 ± 51 pA for baseline, time-point I, and time-point II, respectively. At the end of each recording AP-5 was applied to exclude any contamination of the recorded IPSCs by NMDA-receptor-mediated currents. These data show that activation of NMDA-receptors is crucial for LTPi induction, and that AMPA-receptors are needed for the necessary depolarization in the network to allow NMDA-receptor activation.
Figure 2. AMPA-receptor-mediated signalling is dispensable for LTPi induction.
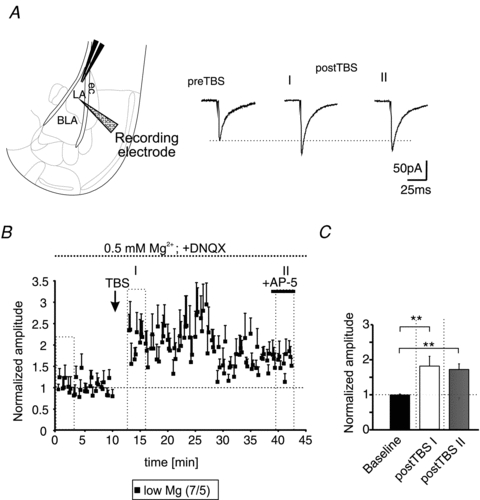
A, scheme of the recording site within the BLA (left). Representative current traces of evoked monosynaptic IPSCs recorded at a holding potential of −70 mV using a high-chloride intracellular solution and a low Mg2+ extracellular solution. Note the increase of the IPSC amplitudes at time-points I and II after TBS (postTBS) compared to baseline (preTBS) in presence of DNQX. B, time-course of LTPi induced by TBS. AP-5 was applied at the end of the experiment (+AP-5) to exclude any contamination by NMDA-receptor-mediated currents. C, quantification of the normalized amplitudes. The TBS significantly increased the normalized IPSC amplitudes compared to baseline stimulation (**P < 0.01; t test).
To test the possible contribution of the IPSC driving force to the observed changes in IPSC amplitude after TBS, the IPSC reversal potential was calculated before and after LTPi induction. The reversal potential of the GABAergic IPSCs was −63.3 ± 1.7 mV (n = 6/3) and −66.9 ± 0.7 mV (n = 4/3) before and after TBS, respectively, displaying no significant difference (P = 0.15; Fig. 3A). Thus, it seems unlikely that the observed increase of the evoked IPSCs after the TBS is due to a shift of the reversal potential of GABAA-mediated currents.
Figure 3. Differentiation between pre- and postsynaptic LTPi.
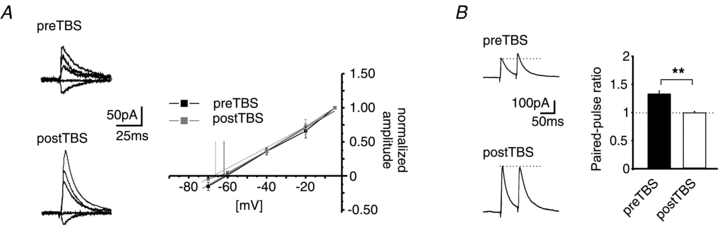
A, example traces of GABAA-receptor-mediated IPSCs at holding potentials of −5, −20, −40, −60 and −70 mV before and after TBS (preTBS and postTBS; left). Plot of normalized amplitudes vs. holding potential. The reversal potential of GABAA-receptor-mediated currents has been calculated by a linear fit of the current–voltage relationship (right). No significant shift of the reversal potential was observed after TBS (postTBS) as compared to baseline (preTBS). B, example traces (left) and quantification (right) of the PPR (PPR = amplitude2/amplitude1) at a paired-pulse interval of 50 ms. The LTPi induction by TBS significantly reduced the PPR as compared to the PPR during baseline conditions, indicating an increase of the release property at GABAergic presynaptic terminals (**P < 0.01; t test).
Next, the paired-pulse ratio (PPR) of evoked responses was examined before and after TBS. The PPR refers to a change in a second synaptic response in a double stimulation protocol, relating to a presynaptically mediated alteration in transmitter release, and changes in PPR properties during synaptic potentiation can signify a presynaptic site of expression. A paired-pulse interval of 50 ms was used, and obtained results are illustrated in Fig. 3B. The PPR of the evoked IPSCs was 1.3 ± 0.05 before application of TBS, and decreased significantly to 1.01 ± 0.03 during stably expressed LTPi upon TBS (n = 5/4; P = 0.001), indicating a contribution of presynaptic mechanisms to LTPi.
nNOS-dependency of LTPi
NO is known to be a multifunctional neuromodulator produced by the neuronal enzyme nNOS, which can act as a retrograde messenger through the presynaptically located soluble guanylyl cyclase, the second messenger cGMP, and the cGMP-dependent protein kinase (cGKI) as canonical signalling pathway (Nugent & Kauer, 2008).
Immunohistochemical stainings revealed nNOS-positive somata and fibres in the BLA in coronal slice sections (Fig. 4A and B). Most nNOS-positive neurons were detected in the ventrolateral part of the LA, where also the most dense immunopositive neuropile was observed. In addition, nNOS-positive neurons were found outside the amygdalar complex. A variety of nNOS-positive somata were detected in the amygdalar/striatal transition zone (AStr) and the dorsal endopiriform cortex (DEn; Fig. 4A). In the clusters of the medial (mpara) and lateral (lpara) paracapsular interneurons some immunopositive fibres, but no nNOS-positive somata were observed (Fig. 4C). The lateral region of the central amygdalar nucleus (CeL) was devoid of nNOS-positive somata and fibres, whereas the medial region of the central amygdalar nucleus (CeM) contained nNOS in somata and neuropile (Fig. 4A and C).
Figure 4. nNOS is present in principal neurons of the BLA.
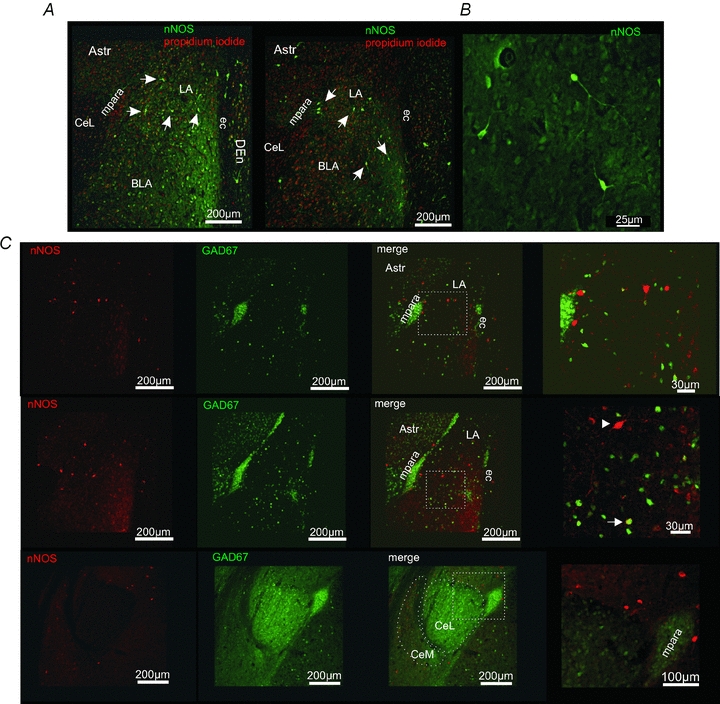
A, nNOS immunoreactivity in the amygdala. nNOS-positive (green) neurons and fibres could be detected in the BLA. Outside the amygdala nNOS-positive neurons were detected in the dorsal endopiriform cortex (DEn) close to the external capsule (ec), and the amygdalar/striatal transition zone (AStr). In the medial paracapsular interneuron clusters (mpara) no nNOS-positive somata could be detected. Propidium iodide (red) was used to stain cell nuclei. B, examples of nNOS-positive neurons within the LA. nNOS-immunoreactivity is visible in the soma and neurites of these neurons. C, immunohistochemical stainings for nNOS (red) in transgene GAD67-EGFP (green) mice. Although in a small subset of neurons a nNOS-GAD67-EGFP co-localization was detected (arrow), most of the nNOS-positive neurons were GAD67-EGFP negative in the BLA (arrowhead). The lateral region of the central amygdalar nucleus (CeL) is virtually devoid of nNOS-positive fibres.
In another set of experiments, transgene GAD67-EGFP-expressing mice were used to differentiate between GAD67-EGFP-positive interneurons and GAD67-EGFP-negative principal neurons in the BLA. Immunohistochemical stainings against nNOS revealed that most nNOS-positive cells in the BLA are lacking GAD67-EGFP and thus probably represent PNs (Fig. 4C). GAD67-EGFP-positive neurons were stained for nNOS in the basal nucleus of the amygdala (Fig. 4C).
The immunohistochemical stainings indicate the presence of nNOS in a subset of neurons and in neurites in the BLA. To test the involvement of nNOS and NO signalling in the induction of LTPi, we applied the nNOS blocker l-NAME to PNs in the BLA. The presence of l-NAME in the extracellular solution did not change the membrane properties of PNs when compared to control recordings. The resting membrane potential was at −64 ± 1 mV (n = 13/5; l-NAME vs. control: P = 0.07) and the membrane input resistance at 314 ± 32 MΩ (n = 13/5; l-NAME vs. control: P = 0.33). However, l-NAME prevented GABAergic LTP in PNs by TBS (normalized amplitudes baseline: 0.99 ± 0.03 and postTBS time-point I: 0.96 ± 0.05; P = 0.58; postTBS time-point II: 1.05 ± 0.07; P = 0.48; n = 13/5; Fig. 5A–C). l-NAME did not significantly change the amplitudes of evoked IPSCs (eIPSCs) during baseline recordings (control: 142 ± 15 pA; l-NAME: 133 ± 17 pA; P = 0.88). In a next set of experiments, the NO scavenger PTIO (200 μm) was used to interfere with NO signalling. In the presence of PTIO, LTPi could not be induced by TBS (Fig. 5B and C). The normalized amplitudes were 1.11 ± 0.11 for time-point I and 0.94 ± 0.15 for time-point II after TBS and were not significantly different compared to baseline amplitudes (P = 0.24 for time-point I and P = 0.79 for time-point II; n = 13/4; Fig. 5C). PTIO did not significantly affect the absolute amplitudes of evoked IPSCs recorded at baseline compared to control recordings (baseline PTIO: 183 ± 19 pA; baseline control: 142 ± 15 pA; P = 0.13). The normalized eIPSC amplitudes retrieved from recordings in the presence of l-NAME or PTIO were significantly smaller compared to control conditions after TBS at the analysed time-points I and II (Fig. 5C).
Figure 5. LTPi is dependent on nNOS activity and NO.
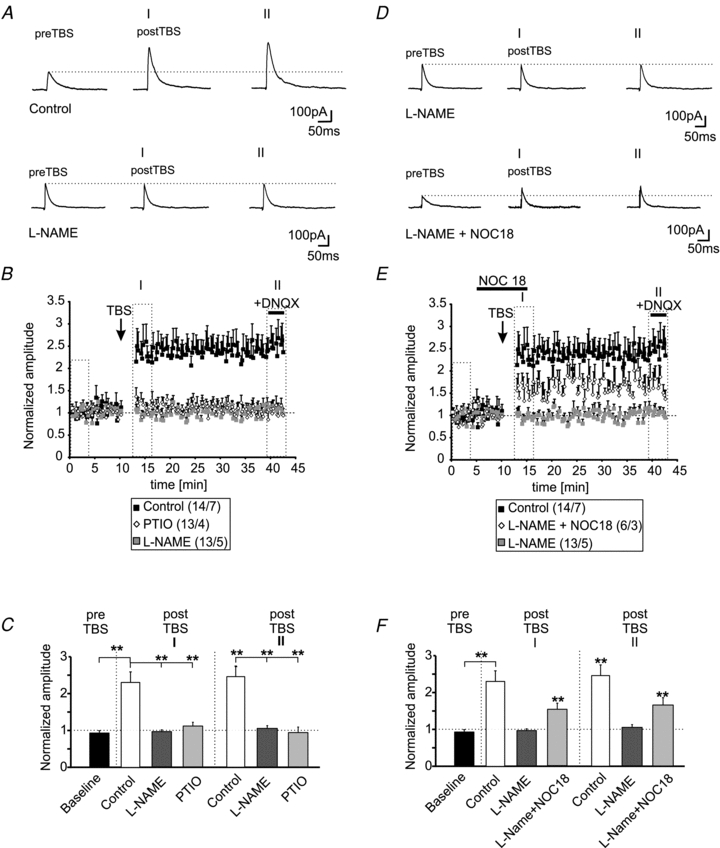
A, example traces of evoked IPSCs under control conditions and in presence of the NOS blocker l-NAME (200 μm) during baseline recordings and during two time-points (I and II) after TBS. B, plot of the normalized amplitudes of evoked IPSCs shows the induction of LTPi during control conditions in presence of the nNOS blocker l-NAME (200 μm) or in presence of the NO scavenger PTIO (100 μm). C, quantification of the normalized IPSC amplitudes during baseline recordings and at two time-points after TBS (I and II; same time-points as in A) under control conditions and in presence of l-NAME or PTIO. (**P < 0.01; t test). D, example traces of evoked IPSCs during baseline recordings and at two time-points after TBS (I and II) in presence of l-NAME alone, and in presence of l-NAME and the NO donor Noc18, added 5 min before TBS for 15 min. E, the normalized IPSC amplitudes show that LTPi is partially restored by addition of the NO donor Noc18 during the TBS. F, quantification of the normalized IPSC amplitudes during baseline recordings and at two time-points after TBS (I and II; same time-points as in A) in presence of l-NAME alone or in combination with Noc18 (*P < 0.05; t test).
To verify the dependency of LTPi on NO synthesis and diffusion, the NO donor Noc18 (100 μm) was bath-applied 5 min before TBS for 15 min in the presence of l-NAME, which has been shown previously to completely abolish LTPi in PN (Fig. 5D). The addition of Noc18 partially restored the induction and maintenance of LTPi in the presence of l-NAME, indicating that LTPi is critically dependent on the presence of NO (Fig. 5D and E). LTPi could be induced in 7 out of 12 recorded PNs (∼58%), when the TBS was applied in the presence of l-NAME and Noc18. The normalized amplitudes of Noc18 +l-NAME were 0.96 ± 0.06 (n = 7/4) under baseline conditions and increased to 1.47 ± 0.15 at time-point I and to 1.68 ± 0.17 at time-point II after TBS (baseline vs. time-point I: P < 0.01; baseline vs. time-point II: P < 0.01; Fig. 5E and F). The recorded IPSCs were sensitive to gabazine, when applied at the end of the experiment (n = 4). The mean absolute amplitudes during baseline stimulation in the presence of l-NAME alone were 119 ± 18 pA and were not significantly different from evoked baseline responses under control conditions (P = 0.66). In the presence of l-NAME and Noc18 prior to TBS, the mean baseline amplitudes were 127 ± 18 pA and not significantly different from either mean amplitudes during control recordings (P = 0.29) or from recordings in the presence of l-NAME alone (P = 0.41). At time-points I and II after TBS, the normalized amplitudes recorded under control conditions were not significantly different from l-NAME + Noc18 (time-point I: P = 0.06; time-point II: P = 0.16). In contrast, the normalized amplitudes of evoked IPSCs in the presence of l-NAME alone were significantly reduced as compared to the normalized amplitudes recorded in the presence of l-NAME + Noc18 at both time-points after TBS (Fig. 5F).
The application of Noc18 (200 μm; 10 min) alone during maintained low frequency stimulation (0.05 Hz) induced a slow-onset LTP at inhibitory IN–PN synapses in 5 out of 10 recorded neurons (Fig. 6A and B). The amplitudes of the evoked IPSCs reached a steady state ∼5 min after the end of the Noc18 application. The normalized amplitudes increased to 1.66 ± 0.17 at time-point I and stayed at an elevated level (1.57 ± 0.14; time-point II) and were significantly different from normalized amplitudes during baseline recordings (0.99 ± 0.07; P < 0.006; n = 5; Fig. 6B and C). These findings indicate that NO alone is able to enhance the GABAergic synaptic transmission at a subset of IN–PN synapses.
Figure 6. NO induces a slow-onset LTP at IN–PN synapses.
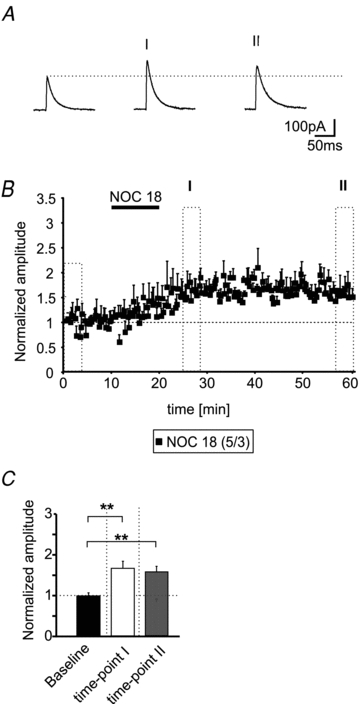
A, example traces of evoked IPSCs during baseline conditions and at two time-points (as indicated in B) after application of the NO donor Noc18 (200 μm). B, time-course of the normalized eIPSC amplitudes. The application of 200 μm Noc18 induces a slow-onset LTP at IN–PN synapses during low frequency stimulation (stimulus frequency: 0.05 Hz). C, quantification of the normalized IPSC amplitudes during baseline conditions and at two time-points after Noc18 application (**P < 0.01; t test).
Overall, these data suggest that nNOS activity and NO signalling mediate the induction of LTPi.
Discussion
The role of NO in GABAergic LTP
Recent publications have described glutamatergic LTP on different subtypes of amygdalar interneurons, the afferent fibres involved, and underlying mechanisms (for review see: Spampanato et al. 2011). In contrast, little is known about mechanisms of monosynaptic plasticity at GABAergic synapses in the BLA. Changes in synaptic efficacy are thought to be critical for information processing and mediation of specific fear responses within the amygdalar complex.
In the present study we provide evidence for a novel mechanism of LTP at GABAergic synapses on PNs in the BLA of the amygdala in mice. The data indicate that LTPi induction by TBS of monosynaptic GABAergic connections involves activation of AMPA- and NMDA-receptors at glutamatergic synapses, and subsequent synthesis of nitric oxide by NOS (Fig. 7A). NO is thought to diffuse into presynaptic terminals and activate soluble guanylate cyclases, which in turn produce cGMP. Downstream cGMP can activate, e.g., protein kinase G, which might modulate the transmitter release machinery. Though this is a widely accepted model, it cannot be excluded that NO is also modulating additional transmitter receptors, for instance in the postsynaptic membrane (Fig. 7A).
Figure 7. Modulation of synaptic transmission in the BLA: schematic diagram of a hypothetical model.
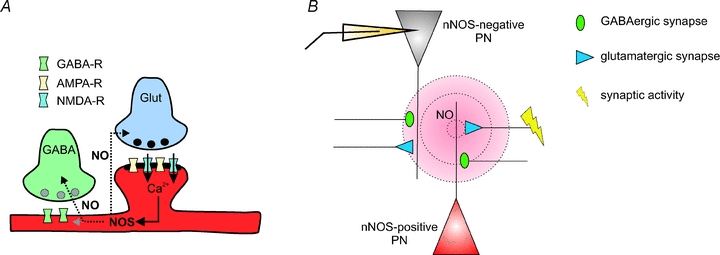
A, synaptic activity at glutamatergic synapses activates AMPA- and NMDA-receptor subtypes. The Ca2+ influx via NMDA-receptors activates the nNOS, producing diffusible NO. NO can modulate the synaptic properties at glutamatergic and GABAergic synapses (in the pre- and/or postsynaptic neuron). The mechanism for LTPi via NO signalling was also suggested in, e.g., VTA neurons (Nugent & Kauer, 2008). B, strong synaptic activity at glutamatergic synapses on nNOS-positive neurons induces the production of NO. Due to its diffusion range (∼80–200 μm; Wood & Garthwaite, 1994; Philippides et al. 2000), NO can influence glutamatergic and GABAergic synapses on nNOS-positive neurons as well as on neighbouring nNOS-negative neurons. Although the overall number of nNOS-positive neurons in the BLA is low, NO modulates a large number of synaptic terminals by ‘volume transmission’.
In ∼60% of recorded PNs, TBS induced robust LTPi at monosynaptic IN–PN connections, whereas LTPi induction failed during the presence of either AMPA- or NMDA-receptor antagonists. These data indicate a novel form of heterosynaptic plasticity at PNs that involves activation of glutamatergic afferent fibres and, in parallel, activation of GABAergic terminals.
We show that NO signalling (either by endogenous NO synthesis due to NOS activity or by addition of NO donors) is a prerequisite for LTPi induction, and that during physiological conditions AMPA- and NMDA-receptors act in concert to allow postsynaptic activation of the NO synthase in NOS-positive neurons. This is confirmed by the loss of LTPi induction in the presence of l-NAME and its recovery upon subsequent administration of the NO donor Noc18. In addition, the application of the NO donor Noc18 alone induced a slow-onset LTPi at a subset of recorded synaptic IN–PN connections (∼50%), which indicates that indeed NO synthesis and signalling is crucial for LTPi in the BLA. A similar form of GABAergic heterosynaptic LTP involving glutamatergic and GABAergic synapses has been previously described in VTA and hippocampal neurons (Zhuo et al. 1993; Nugent et al. 2009). In VTA neurons, NMDA-receptor-mediated Ca2+ influx activates nNOS, and NO in turn modulates the efficacy at GABAergic synapses. Thus, NO modulation of GABAergic transmission and GABAergic plasticity via NMDA-receptor activation might be a mechanism implemented in several brain regions. In our experiments, the reduction of the paired-pulse ratio after TBS indicates that the induced LTPi is of presynaptic origin. NO could directly modulate synaptic transmission via its canonical pathway in the presynaptic terminal, altering the efficacy of transmitter release, or by increasing the excitability of interneurons (for review see Garthwaite, 2008). It has been described earlier that LTP at glutamatergic synapses on BLA interneurons induces an increase in feed-forward inhibition onto the respective target neurons (Szinyei et al. 2000; Polepalli et al. 2010). The mechanism of increasing GABAergic transmission presented here should be regarded as an additional pathway of modulating IN function.
Distribution of nNOS in the LA/BLA-complex
In our experiments, immunohistochemical stainings revealed nNOS-positive neurons and fibres in the BLA. In previous studies, Schafe and colleagues showed that nNOS is co-localized with α-calcium–calmodulin kinase II (αCaMKII) at asymmetrical synapses on spines in the LA (Schafe et al. 2005) indicating that nNOS is associated with excitatory synapses. In the BLA we observed that most nNOS-positive neurons were GAD67 negative, indicating that these neurons are PNs. In the basal nucleus of the amygdala a low number of GAD67-positive neurons were also nNOS positive. As the stimulation sites for the induction of LTPi were situated in the dorsal LA, it seems feasible to conclude that these interneurons have only limited, if any, contribution to the recorded IPSCs. Furthermore, nNOS-positive fibres could be detected in the BLA apparently originating from neurons of the AStr. In contrast, the CeL and the clusters of the medial/lateral paracapsular interneurons were almost devoid of nNOS-positive somata, but nNOS-positive neurites of neurons from adjacent nuclei could be detected. In that case, it seems feasible to conclude that nNOS-positive PNs within the LA as well as nNOS-positive neurites originating from outside the LA (e.g. AStr) might act as NO sources and modulate GABAergic synaptic transmission.
Most likely, the majority of the recorded PNs are nNOS negative, as indicated by the sparse number of nNOS-positive somata in the vicinity of the recording site (Fig. 4A). The success rate of LTPi induction of about 60% and the relatively low number of nNOS-positive neurons suggest that nNOS activity in a low number of neurons/fibres might be sufficient to trigger LTPi at surrounding GABAergic terminals (‘volume transmission’; Fig. 7B), terminating on nNOS-positive or nNOS-negative PNs. Thus, the activation of NMDA-receptors, driven either by AMPA-receptor-mediated depolarization or during low Mg2+ conditions, in nNOS-positive PNs within the network would lead to the production of NO by nNOS. Interestingly, AMPA-receptor signalling is dispensable for LTPi induction when NMDA-receptor signalling is enhanced during low Mg2+ concentrations in the extracellular space. This indicates that AMPA-receptor signalling is needed for a depolarization in the surrounding network during the TBS, strong enough to relieve the Mg2+ block of NMDA-receptors as described for forms of classical LTPs.
In addition, the LTPi induction by Noc18 alone, in the absence of TBS, suggests that NO indeed acts downstream of the glutamate receptor activation (Fig. 7A). These conclusions are in line with the findings of NO pathways in LTPi in the VTA (Nugent et al. 2007; Nugent & Kauer, 2008). It is generally accepted that NO diffuses over wide distances from the site of generation (∼80–200 μm; Wood & Garthwaite, 1994; Philippides et al. 2000) and thereby may act as a retrograde messenger at glutamatergic and GABAergic synapses (for review see Garthwaite & Boulton, 1995; Fig. 6A and B). The findings of the present study indicate that NMDA-receptor-dependent activation of nNOS in a subset of PNs is able to scale the efficacy of GABAergic synapses on nNOS-positive and nNOS-negative PNs during periods of strong network activity within the BLA.
It has been shown previously that NO signalling is important for the induction of LTP at glutamatergic synapses in the LA (Ota et al. 2010) and for fear conditioning and memory consolidation in rats and mice (Schafe et al. 2005; Paul et al., 2008; Kelley et al. 2009, 2010). Using pharmacological interference with NO signalling, Schafe and colleagues (2005) have shown that NO is required for LTP at glutamatergic synapses originating from thalamic afferents in the LA. Furthermore, NO is necessary for memory formation of auditory Pavlovian fear conditioning, but dispensible for retrieval and reconsolidation (Schafe et al. 2005). In addition, nNOS-deficient mice show impaired auditory and visually cued fear conditioning (Ota et al. 2008; Kelley et al. 2011), and, even more severe, contextual fear learning (Kelley et al. 2011). The aforementioned results are discussed as an effect of NO on LTP at glutamatergic synapses in the amygdala almost regardless of the role of the GABAergic system in the formation of conditioned fear memory. GABAergic interactions in amygdaloid networks appear to be specifically involved in generating response specificity to fear-conditioned stimuli (Ehrlich et al. 2009). Furthermore, the inhibitory GABAergic system within the amygdala is crucial for processes of fear extinction learning, and interference with the GABAergic system can lead to generalization phenomena during Pavlovian fear conditioning (Sangha et al. 2009). Therefore it is tempting to speculate that the NO system plays a prominent role in processes of fear generalization inhibition and fear extinction by modulating the GABAergic system.
We suggest a dual function of NO signalling within the BLA. In addition to the previously shown enhancing influence on glutamatergic LTP at thalamic inputs to the LA and the involvement in long-term memory formation, we provide evidence for NO enhancing the efficacy at GABAergic synapses on PNs. In this model, NO could prevent overexcitability in BLA networks, contribute to specific memory formation, and prevent generalization.
Acknowledgments
The authors would like to thank Svetlana Kiesling, Elke Naß, and Petra Berenbrock for excellent technical assistance. We thank Dr Philippe Coulon and Dr Thomas Seidenbecher for critical reading of an earlier version of the manuscript. The project was funded by the German Research Foundation (DFG; SFB-TRR58, TPA03 to H.C.P.), and a Max Planck Research Award (to H.C.P.). The authors have no conflicts of interest to declare.
Glossary
- AStr
amygdalar/striatal transition zone
- CeL/CeM
lateral/medial region of the central amygdalar nucleus
- IN
inhibitory interneuron
- LA
lateral amygdaloid nucleus
- BLA
basolateral amygdaloid complex
- LTP
long-term potentiation
- LTPi
LTP at inhibitory GABAergic synapses
- nNOS
neuronal NOS
- NO
nitric oxide
- NOS
NO synthase
- PN
principal neuron
- PPR
paired-pulse ratio
- TBS
theta-burst stimulation
- VTA
ventral tegmental area
Author contributions
All experiments were done in the Institute of Physiology I at the Westfälische Wilhelms-Universität Münster, Germany. Conception and design of the experiments: K.J., M.D.L., M.D., H.C.P. Collection, analysis and interpretation of data: K.J., M.D.L., M.D., J.L. Drafting the article or revising it critically for important intellectual content: K.J., H.C.P. All authors approved the final version of the manuscript.
References
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busti D, Geracitano R, Whittle N, Dalezios Y, Mańko M, Kaufmann W, Sätzler K, Singewald N, Capogna M, Ferraguti F. Different fear states engage distinct networks within the intercalated cell clusters of the amygdala. J Neurosci. 2011;31:5131–5144. doi: 10.1523/JNEUROSCI.6100-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Lüthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- Kelley JB, Anderson KL, Altmann SL, Itzhak Y. Long-term memory of visually cued fear conditioning: roles of the neuronal nitric oxide synthase gene and cyclic AMP response element-binding protein. Neuroscience. 2011;174:91–103. doi: 10.1016/j.neuroscience.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley JB, Anderson KL, Itzhak Y. Pharmacological modulators of nitric oxide signaling and contextual fear conditioning in mice. Psychopharmacology. 2010;210:65–74. doi: 10.1007/s00213-010-1817-8. [DOI] [PubMed] [Google Scholar]
- Kelley JB, Balda MA, Anderson KL, Itzhak Y. Impairments in fear conditioning in mice lacking the nNOS gene. Learn Mem. 2009;16:371–378. doi: 10.1101/lm.1329209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Baudry M. Properties and mechanisms of long-term synaptic plasticity in the mammalian brain: relationships to learning and memory. Neurobiol Learn Mem. 1995;63:1–18. doi: 10.1006/nlme.1995.1001. [DOI] [PubMed] [Google Scholar]
- Nugent FS, Kauer JA. LTP of GABAergic synapses in the ventral tegmental area and beyond. J Physiol. 2008;586:1487–1493. doi: 10.1113/jphysiol.2007.148098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Niehaus JL, Kauer JA. PKG and PKA signaling in LTP at GABAergic synapses. Neurophsycopharmacology. 2009;34:1829–1842. doi: 10.1038/npp.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature. 2007;446:1086–1090. doi: 10.1038/nature05726. [DOI] [PubMed] [Google Scholar]
- Ota KT, Pierre VJ, Queen K, Schafe GE. The NO-cGMP-PKG signaling pathway regulates synaptic plasticity and fear memory consolidation in the lateral amygdala via activation of ERK/MAP kinase. Learn Mem. 2008;15:792–805. doi: 10.1101/lm.1114808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota KT, Monsey MS, Wu MS, Schafe GE. Synaptic plasticity and NO-cGMP-PKG signaling regulate pre- and postsynaptic alterations at rat lateral amygdala synapses following fear conditioning. PLoS One. 2010;5:e11236. doi: 10.1371/journal.pone.0011236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, Paré D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev. 2010;90:419–463. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patenaude C, Chapman CA, Bertrand S, Congar P, Lacaille JC. GABAB receptor- and metabotropic glutamate receptor-dependent cooperative long-term potentiation of rat hippocampal GABAA synaptic transmission. J Physiol. 2003;553:155–167. doi: 10.1113/jphysiol.2003.049015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul C, Schöberl F, Weinmeister P, Micale V, Wotjak CT, Hofmann F, Kleppisch T. Signaling through cGMP-dependent protein kinase I in the amygdala is critical for auditory-cued fear memory and long-term potentiation. J Neurosci. 2008;28:14202–14212. doi: 10.1523/JNEUROSCI.2216-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippides A, Husbands P, O'Shea M. Four-dimensional neuronal signaling by nitric oxide: a computational analysis. J Neurosci. 2000;20:1199–1207. doi: 10.1523/JNEUROSCI.20-03-01199.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polepalli JS, Sullivan RK, Yanagawa Y, Sah P. A specific class of interneuron mediates inhibitory plasticity in the lateral amygdala. J Neurosci. 2010;30:14619–14629. doi: 10.1523/JNEUROSCI.3252-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Paré D. Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience. 2002;115:455–462. doi: 10.1016/s0306-4522(02)00455-4. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83:803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Sangha S, Narayanan RT, Bergado-Acosta JR, Stork O, Seidenbecher T, Pape HC. Deficiency of the 65 kDa isoform of glutamic acid decarboxylase impairs extinction of cued but not contextual fear memory. J Neurosci. 2009;29:15713–15720. doi: 10.1523/JNEUROSCI.2620-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Bauer EP, Rosis S, Farb CR, Rodrigues SM, LeDoux JE. Memory consolidation of Pavlovian fear conditioning requires nitric oxide signaling in the lateral amygdala. Eur J Neurosci. 2005;22:201–211. doi: 10.1111/j.1460-9568.2005.04209.x. [DOI] [PubMed] [Google Scholar]
- Shaban H, Humeau Y, Herry C, Cassasus G, Shigemoto R, Ciocchi S, Barbieri S, van der Putten H, Kaupmann K, Bettler B, Lüthi A. Generalization of amygdala LTP and conditioned fear in the absence of presynaptic inhibition. Nat Neurosci. 2006;9:1028–1035. doi: 10.1038/nn1732. [DOI] [PubMed] [Google Scholar]
- Sosulina L, Graebenitz S, Pape HC. GABAergic interneurons in the mouse lateral amygdala: a classification study. J Neurophysiol. 2010;104:617–626. doi: 10.1152/jn.00207.2010. [DOI] [PubMed] [Google Scholar]
- Sosulina L, Meis S, Seifert G, Steinhäuser C, Pape HC. Classification of projection neurons and interneurons in the rat lateral amygdala based upon cluster analysis. Mol Cell Neurosci. 2006;33:57–67. doi: 10.1016/j.mcn.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Polepalli J, Sah P. Interneurons in the basolateral amygdala. Neuropharmacology. 2011;60:765–773. doi: 10.1016/j.neuropharm.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Lamas S, Fang FC. Nitrosylation. The prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- Szinyei C, Heinbockel T, Montagne J, Pape HC. Putative cortical and thalamic inputs elicit convergent excitation in a population of GABAergic interneurons of the lateral amygdala. J Neurosci. 2000;20:8909–8915. doi: 10.1523/JNEUROSCI.20-23-08909.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood J, Garthwaite J. Models of the diffusional spread of nitric oxide: implications for neural nitric oxide signalling and its pharmacological properties. Neuropharmacology. 1994;33:1235–1244. doi: 10.1016/0028-3908(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Yang S, Cox CL. Modulation of inhibitory activity by nitric oxide in the thalamus. J Neurophysiol. 2007;97:3386–3395. doi: 10.1152/jn.01270.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M, Small SA, Kandel ER, Hawkins RD. Nitric oxide and carbon monoxide produce activity-dependent long-term synaptic enhancement in hippocampus. Science. 1993;260:1946–50. doi: 10.1126/science.8100368. [DOI] [PubMed] [Google Scholar]