

Abstract
Non-technical summary
The convulsant t-butylbicyclophosphorothionate (TBPS) is considered a GABAA receptor (GABAAR) open channel blocker. However, the relationship between the functional state of the receptor and TBPS binding remains unclear. Radiolabelled [35S]TBPS binds to GABAARs in the absence of GABA, suggesting that access to the binding site is independent of activation. Furthermore, low concentrations of GABA enhance [35S]TBPS binding, while higher concentrations reduce binding. Using either bicuculline or the α1(K278M) mutant GABAAR subunit to disrupt function, we demonstrate roles for spontaneous gating, GABA-evoked channel activation and desensitization in the three phases of [35S]TBPS binding. These findings provide a framework for using [35S]TBPS binding to identify deficits in GABAAR function.
Abstract
Picrotoxin and t-butylbicyclophosphorothionate (TBPS) are GABAA receptor (GABAAR) open channel blockers. However, picrotoxin displaceable [35S]TBPS binding to α1β2γ2 GABAARs occurs in the absence of GABA, suggesting that access to the binding site is independent of activation. Alternatively, spontaneous gating may provide access to the channel. In the absence of episodic GABA application, picrotoxin and TBPS blocked (by 91 ± 3% and 85 ± 5%, respectively) GABA-evoked currents mediated by α1β2γ2 receptors. We used two approaches to inhibit spontaneous GABAAR gating, bicuculline, which inhibits spontaneous current in the absence of exogenous agonist and the α1(K278M) mutant subunit. Whole-cell patch-clamp recordings demonstrated that α1(K278M)β2γ2 receptors have negligible spontaneous gating. Application of bicuculline to α1β2γ2 receptors in the absence of exogenous GABA caused a 35% reduction of current blockade by TBPS and reduced [35S]TBPS binding by 25%. Consistent with this, in the absence of exogenous GABA, α1(K278M)β2γ2 receptors exhibited reduced blockade by TBPS current compared to wild-type receptors. These data suggest that a decrease in spontaneous gating reduces accessibility of TBPS to its binding site. GABA application during picrotoxin or TBPS administration enhanced α1β2γ2 receptor blockade (to 98% in both cases). The GABA-dependent component of TBPS blockade accounts for the stimulation of [35S]TBPS binding to α1β2γ2 receptors seen with GABA (1 μm) application. Moreover, application of GABA at concentrations that cause significant steady-state desensitization reduced [35S]TBPS binding. The α1(K278M) subunit slowed desensitization kinetics and increased the rate of deactivation of GABA-evoked currents. Furthermore, there was a marked increase in the GABA EC50 for desensitization of α1(K278M)β2γ2 receptors associated with a large increase in the GABA-dependent stimulation of [35S]TBPS binding. These data establish a relationship between GABAAR function and the three phases of [35S]TBPS binding seen in the absence and the presence of GABA.
Introduction
The γ-aminobutyric acid type A receptor (GABAAR) is a member of the Cys-loop receptor family of pentameric ligand-gated ion channels. It shares homology with the glycine, nicotinic-acetylcholine, 5-hydroxytryptamine type 3 (5-HT3) and Zn2+-activated receptors (Connolly & Wafford, 2004). Cys-loop receptors have an extracellular agonist binding pocket, four transmembrane domains (M1–M4), and a large intracellular M3–M4 loop. GABAARs are the major inhibitory receptors in the central nervous system (CNS), formed by several combinations of α1–6, β1–3, γ1–3, δ, ρ1–3, ɛ, θ or π subunits (Whiting et al. 1999; Pirker et al. 2000). The α1β2γ2 combination is the most abundant neuronal GABAA receptor subtype.
GABA binds at the interface between α and β subunits, while allosteric modulation by benzodiazepines occurs through an equivalent site at the interface between α and γ subunits (Whiting et al. 1999). Modulation by most other allosteric GABAAR modulators including general anaesthetics, neurosteroids and convulsant ligands involves interactions with residues within M2, the locus that lines the channel pore (Lambert et al. 2003; Chen et al. 2006; Korpi & Sinkkonen, 2006). The convulsants picrotoxin and t-butylbicyclophosphorothionate (TBPS) inhibit GABAergic transmission by channel blockade. GABAAR activation enhances the rate of blockade by picrotoxin, consistent with a requirement for an open channel for access to the convulsant binding site (Newland & Cull-Candy, 1992; Dillon et al. 1995).
By convention, Cys-loop receptor M2 residues are numbered from intracellular 0′ to extracellular 20′ positions (Miller, 1989). Picrotoxin and TBPS share a common binding motif within the channel pore between the 2′and 9′ residues (Chen et al. 2006; Kalueff, 2007). Mutations that cause non-conservative amino acid substitutions at 2′, 6′ or 9′ positions reduce the potency of picrotoxin in a stoichiometry-dependent manner, suggesting that the convulsant site lies deep within the channel pore (Chang & Weiss, 1999; Buhr et al. 2001, Sedelnikova et al. 2006; Erkkila et al. 2008). The structure of the Caenorhabditis elegans glutamate-gated Cl− channel homomeric α receptor, solved in the presence of picrotoxin, reveals that the molecule interacts at the 2′ level and extends to the level of −2′ (Hibbs & Gouaux, 2011). According to several related structural models, the 2′ residue lies below the proposed Cys-loop receptor gate and therefore it follows that accessibility to the convulsant site requires receptor activation (Wilson & Karlin, 2001; Unwin, 2005; Bali & Akabas, 2007). However, radiolabelled [35S]TBPS binds to recombinant GABAARs in the absence of GABA, in a picrotoxin displaceable fashion, suggesting that the convulsant site is in fact accessible in the absence of receptor activation (Im et al. 1994; Luddens & Korpi, 1995). Thus conflicting findings from electrophysiological studies, structural models and [35S]TBPS binding assays demonstrate a fundamental inconsistency in our understanding of the relationship between convulsant blockade and GABAAR gating.
Autoradiographic analysis of [35S]TBPS binding in the brain and its modulation by exogenous GABA reveals considerable heterogeneity (Edgar & Schwartz, 1990; Sinkkonen et al. 2001; Halonen et al. 2009). While an early study of cortical slices identified a single population of binding sites, more recent analysis in different brain regions reveals components of [35S]TBPS binding that are differentially modulated by GABA. [35S]TBPS binding can be completely displaced by both picrotoxin and GABA in most brain regions, but in the cerebellar granule cell layer and the thalamus a component of the picrotoxin-sensitive [35S]TBPS binding cannot be fully displaced even by supramaximal GABA (Halonen et al. 2009). The capacity to explore [35S]TBPS binding in human post-mortem tissue provides the potential to examine the distribution of GABAAR subtypes and deficits in their function associated with disease (Atack et al. 2007). However, our ability to interpret these data is hampered by a rudimentary knowledge of the relationship between [35S]TBPS binding and GABAAR function.
In this study we explored the relationship between picrotoxin-displaceable [35S]TBPS binding and blockade of GABAARs by both convulsants. We first examined how TBPS accesses its site in the absence of GABAAR activation by GABA. GABAARs exhibit a degree of spontaneous channel activity (Birnir et al. 2001; McCartney et al. 2007). Spontaneous gating may provide access to the convulsant site and contribute to GABA-independent [35S]TBPS binding. To test this possibility, we used bicuculline, which binds at the GABA binding site inhibiting spontaneous gating (McCartney et al. 2007). We also exploited the mutant α1(K278M) subunit, which reduced spontaneous gating of α1β2γ2 receptors, slowed GABA-evoked desensitization and accelerated deactivation. Mutant α1(K278M)β2γ2 receptors also exhibit an increased GABA EC50 for both activation and steady-state desensitization, providing the opportunity to explore the influence of these properties on the profile of [35S]TBPS binding.
Methods
Culturing and transfection
HEK293 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% bovine serum and 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (HEK medium) in a humid environment at 37°C and 5% CO2. Cells were plated onto 35 mm diameter dishes for electrophysiology, or 60 mm diameter dishes for use in binding assays. Twenty-four hours after subculturing, cells were transiently transfected with equimolar concentrations of murine wild-type or mutant GABAA subunit cDNAs and green fluorescent protein (GFP) as a marker for transfection using the Ca2PO4 precipitation method (Chen & Okayama, 1987) and stored at 37°C and 3% CO2 overnight. For comparison, in some experiments cells were transfected in the same way with cDNA encoding either the human 5-HT3A subunit or the rat glycine α1 or α2 subunit. Electrophysiological recordings were obtained 48–96 h after transfection. For binding assays, confluent cells were harvested 72 h after transfection, in room temperature Hanks’ balanced salt solution (HBSS), centrifuged at 3000 g and used immediately for [35S]TBPS binding, or frozen at −80°C for later use. Tissue culture reagents were obtained from Gibco/Invitrogen (Carlsbad, CA, USA and Paisely, UK).
Electrophysiological recordings
For all experiments, the recording electrodes contained the following solution (in mm): 140 CsCl, 2 MgCl2, 10 Hepes, 11 EGTA and 3 ATP at a pH of 7.4 (with CsOH). Individual GFP positive cells were voltage clamped at −60 mV and maintained in an extracellular solution containing (in mm): NaCl 140, KCl 4.7, MgCl2 1.2, CaCl2 2.5, Hepes buffer 10, and glucose 10 at a pH of 7.4 (with NaOH). For experiments measuring the modulation of GABA (100 μm or 10 μm) evoked currents, GABA was applied locally by pressure (10 psi) ejection (Picospritzer II, General Valve Corp., Fairfield, NJ, USA) from a micropipette placed adjacent to the cell, while TBPS, picrotoxin or bicuculline methiodide (referred to from here on as bicuculline) was perfused through the recording chamber at a rate of 5 ml min−1 (Adodra & Hales, 1995). During experiments in which macroscopic current kinetics were examined, drugs were applied rapidly by the three-pipe Perfusion Fast-Step (SF-77B) solution exchange system (Warner Instruments, Hamden, CT, USA). Solution flow (0.3 ml min−1) through the pipes was controlled by a syringe pump (Cole-Parmer, Vernon Hills, IL, USA). Either the entire cell or an excised outside-out patch of membrane was lifted from the base of the recording chamber and placed in front of the stream of control solution. Perfusion pipes were moved rapidly, exposing cells or patches to agonist (GABA, 5-HT or glycine) alone or in combination with either TBPS or picrotoxin. When the three barrelled system was unmodified, the 10–90% rise time for junction currents generated by moving between adjacent perfusion pipes containing dissimilar ionic solutions was approximately 5 ms. Either three barrelled perfusion pipes were modified as described previously (Hinkle et al. 2003), or pulled theta tubes were used for ultra-rapid application of GABA to outside-out patches for the determination of desensitization and deactivation kinetics of wild-type and mutant receptors. The solution flow rate was 0.1 ml min−1. At the end of each of these experiments the 10–90% rise time for junction currents (mean 578 ± 41 μs) was measured after rupturing the outside-out patch.
[35S]TBPS homogenate binding experiments
[35S]t-Butylbicyclophosphorothionate ([35S]TBPS) was obtained from Perkin Elmer (Waltham, PA, USA) and stored at −20°C. In order to obtain a crude membrane fraction, HEK293 cell membranes containing recombinant GABAARs were suspended in 10 ml TEN solution (10 mm Tris-HCl, 1 mm EDTA and 100 mm NaCl, pH 7.5 with HCl) using a Tissue-Tearer hand homogenizer (Biospec Products, Racine, WI, USA) and centrifuged for 30 min at 30,000 g. Membrane pellets (3 to 6 mg ml−1) were suspended in incubation buffer (20 mm Tris-HCl and 1 m NaCl, pH 7.5 with HCl). Aliquots of membrane suspensions were incubated with [35S]TBPS (30 nm) at 25°C for 90 min, either alone or in the presence of various agonists or antagonists. Non-specific binding was determined in the presence of 100 μm picrotoxin. When examining the time course of binding, membranes of HEK293 cells expressing wild-type α1β2γ2 receptors or mutant α1(K278M)β2γ2 receptors were incubated with [35S]TBPS (30 nm) for 30, 90 and 180 min. The binding of [3H]flunitrazepam (50 nm) at 90 min was performed in parallel to determine whether the α1(K278M) mutation caused altered receptor expression. In these experiments non-specific binding was determined using equivalent membranes of non-transfected HEK cells. Incubations were halted by perfusing 5 ml of ice-cold incubation buffer through a Brandel M-48 cell harvester (Gaithersburg, MD, USA). Membranes bound to [35S]TBPS or [3H]flunitrazepam were collected on Whatman GF/B filter paper (Brandel, Gaithersburg, MD or Alphabiotech, Glasgow, UK) pre-soaked with 0.5 m polyethylenimine (Sigma-Aldrich, St Louis, MO, USA). Filters were air dried for 10–15 min, then counted for radioactivity in 4 ml of scintillation fluid using a Beckman LS 6500 scintillation counter (Brea, CA, USA).
Data acquisitions, analysis and statistics
Whole-cell currents recorded using an Axopatch 200B amplifier and low-pass filtered at 2 kHz were digitized at 10 kHz and acquired using a Digidata 1320A interface (Molecular Devices, Sunnyvale, CA, USA) onto the hard drive of a personal computer for off-line analysis. Electrophysiological data were analysed using pCLAMP software (versions 8 and 10, Molecular Devices). For rapid application experiments currents were filtered at 1 kHz and acquired at 10 kHz using a Digidata 1440A and desensitization and deactivation kinetics were determined offline by fitting with the sum of two exponentials. For desensitization kinetics exponentials were fitted between 10 and 90% of the falling phase of the desensitization component of the GABA evoked current recording. For deactivation kinetics solution exchange was determined from the corresponding recording of liquid junction current and exponentials were fitted between the 10% falling phase of the liquid junction current and the 90% falling phase of the deactivation component of the GABA evoked current recording. All experiments were performed at room temperature. Graphs and fitted curves were produced using Slide Write Plus 5.0 (Encinitas, CA, USA). The logistic equation used to fit concentration response relationships has been described previously (Adodra & Hales, 1995). Statistical analysis was performed using Graphpad Prism 5.0 (GraphPad Software Inc., La Jolla, CA, USA). Total protein values were obtained using an ND-1000 spectrophotometer (Nanodrop, Wilmington, DE, USA) or by Bradford standard protein curve programmed on an Eppendorf Biophotometer (Eppendorf International, Cambridge, UK).
Results
Inhibition of GABAARs by TBPS and picrotoxin
We evaluated the relationship between the concentration of picrotoxin or TBPS and inhibition of α1β2γ2 GABAARs using the whole-cell patch-clamp technique (Fig. 1). GABA-evoked currents were recorded from HEK293 cells (voltage-clamped at –60 mV) transiently expressing recombinant α1β2γ2 receptors. Receptors were activated every 60 s by GABA (100 μm for 100 ms) in the absence or presence of bath-applied TBPS (0.01–10 μm, Fig. 1A) or picrotoxin (0.3–100 μm, Fig. 1B). Inhibition was calculated at steady-state by subtracting the current remaining after bath application of picrotoxin or TBPS from the current amplitude recorded in the presence of GABA alone (Fig. 1C). The IC50 values for TBPS and picrotoxin were 0.39 ± 0.07 μm and 2.0 ± 0.2 μm, respectively. Maximal inhibition of GABA-evoked currents occurred in the presence of 10 μm TBPS and 100 μm picrotoxin. Our findings agree with the findings of others who have demonstrated a higher potency of TBPS compared to picrotoxin as an inhibitor of GABAARs (Casida & Lawrence, 1985; Maksay et al. 2003).
Figure 1. Concentration-dependent inhibition of GABAARs by TBPS and picrotoxin.
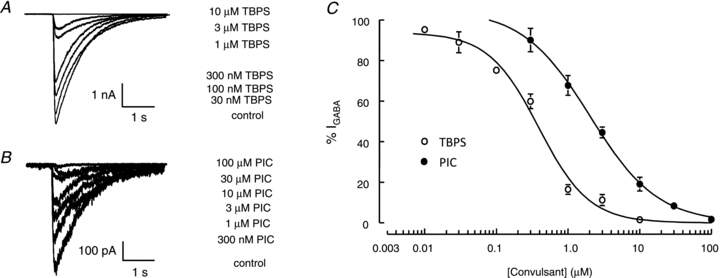
A and B, representative whole-cell currents recorded from α1β2γ2 receptors expressed in HEK293 membranes were activated by GABA (100 μm for 100 ms every 60 s) and inhibited by TBPS (A) and picrotoxin (B). C, concentration–inhibition curves for picrotoxin (•) and TBPS (○). GABA-evoked current amplitudes recorded in the presence of picrotoxin (n≥ 3) or TBPS (n≥ 4) are expressed as a percentage of those recorded under control conditions (%IGABA). TBPS caused a more potent inhibition of GABA-evoked currents. IC50 values for TBPS and picrotoxin were 0.39 ± 0.07 μm and 2.0 ± 0.2 μm, respectively. Data are represented as means ± SEM of 3–10 recordings.
Currents mediated by glycine α1, α2 and 5-HT3A receptors
Picrotoxin inhibits currents mediated by several members of the Cys-loop receptor family. Glycine and 5-HT3A receptors were activated by glycine (α1 receptors by 100 μm and α2 receptors by 1 mm) and 5-HT (100 μm), respectively, while TBPS (10 μm) or picrotoxin (100 μm) were bath applied. TBPS (10 μm) at a concentration sufficient to maximally inhibit α1β2γ2 GABAA receptors, had no effect on either glycine- or 5-HT-evoked currents, which remained at 111 ± 4% (α1 receptors, n = 5, data not shown), 108 ± 20% (α2 receptors, n = 3), and 98 ± 2% (5-HT3A receptors, n = 4) of control current amplitude (Supplementry Fig. 1A). By contrast, the application of picrotoxin (100 μm) to HEK293 cells expressing either glycine α1 (n = 5, data not shown), α2 (n = 4), or 5-HT3A (n = 3) receptors inhibited current amplitudes by 93 ± 1%, 60 ± 13% and 70 ± 1%, respectively (Supplementary Fig. 1B). These data are consistent with previous demonstrations that [35S]TBPS binds selectively to GABAARs in the CNS (Squires et al. 1983).
Blockade by TBPS and picrotoxin occurs independently of GABA activation
We investigated whether blockade by either TBPS or picrotoxin was influenced by GABAAR activation. GABA (100 μm for 100 ms) was episodically (every 60 s) locally applied to HEK293 cells expressing α1β2γ2 receptors and TBPS (10 μm) or picrotoxin (100 μm) was applied to the bath (Fig. 2A). Both picrotoxin and TBPS effectively abolished GABA-evoked currents (98 ± 0.4% and 98 ± 0.6% inhibition, respectively) in the presence of GABA application (Fig. 2C). Other investigators (Newland & Cull-Candy, 1992; Dillon et al. 1995) have observed that the rate of inhibition by picrotoxin is enhanced by episodic GABA application, suggesting a role for an open channel in receptor blockade. We measured the level of inhibition that occurred after a prolonged exposure to either TBPS or picrotoxin in the absence of GABA. The time required to reach maximal inhibition with episodic GABA application was used to determine the duration for bath application of TBPS or picrotoxin to the same cell in the absence of GABA (Fig. 2B). There was a significant (P < 0.05, Student's t test) reduction in the inhibition caused by both TBPS and picrotoxin (86 ± 4.8% and 91 ± 3.1%, respectively) in the absence of episodic GABA application (Fig. 2C). However, the majority of blockade by both antagonists was independent of GABA application. Consistent with a small use-dependent component of blockade, inhibition by TBPS or picrotoxin grew somewhat by the second application of GABA (Fig. 2B). Taken together, these data confirm that while GABA enhances blockade by TBPS and picrotoxin, the majority of blockade is independent of GABA-evoked channel activation.
Figure 2. Inhibition of GABA-evoked currents by TBPS and picrotoxin in the presence and absence of episodic GABA application.
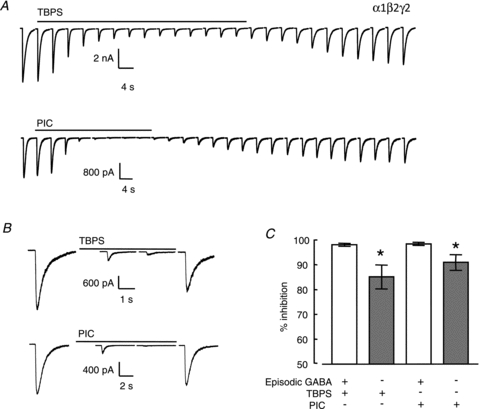
A, top traces, inhibition of episodic GABA (100 μm for 100 ms every 60 s)-evoked currents by TBPS (10 μm) or, bottom traces, picrotoxin (100 μm). The time required to reach maximal inhibition in the presence of GABA was used to determine the duration of bath application of TBPS or picrotoxin to the same cell in the absence of episodic GABA application. B, block occurred after exposure to TBPS or picrotoxin in the absence of episodic GABA. C, graph of % inhibition by TBPS and picrotoxin in the presence and absence of episodic GABA. Data are represented as means ± SEM Open and filled bars represent inhibition in the presence and absence of episodic GABA application, respectively. *P < 0.05 obtained by Student's t test.
Modulation of [35S]TBPS binding by GABA
We further investigated the ability of TBPS and picrotoxin to reach their binding sites in the absence of GABA using [35S]TBPS binding to homogenates of HEK293 cells expressing α1β2γ2 receptors. Our electrophysiological data demonstrate that the majority of blockade by both picrotoxin and TBPS occurs independently of GABA activation of the channel (Fig. 2). Consistent with this [35S]TBPS binds to recombinant receptors in the absence of GABA (Luddens & Korpi, 1995). We observed robust picrotoxin-displaceable [35S]TBPS binding to HEK293 cell membranes containing α1β2γ2 receptors (Supplementary Fig. 2). We examined the time course of [35S]TBPS binding and found that 95% of maximal binding was achieved after 90 min with little increase observed at 180 min. Experiments examining the modulation of [35S]TBPS binding were performed at the 90 min time point. Addition of GABA (30 nm to 100 μm) caused a biphasic modulation of [35S]TBPS binding (Fig. 3). Low concentrations of GABA (30 nm to 1 μm) enhanced [35S]TBPS binding. This enhancement corresponds to the use-dependent component of inhibition by TBPS observed using the electrophysiological assay (Fig. 2). Consistent with previous reports, higher concentrations of GABA (3 μm to 100 μm) reduced [35S]TBPS binding (Fig. 3) presumably through GABAAR desensitization.
Figure 3. Biphasic modulation of [35S]TBPS binding to recombinant α1β2γ2 receptors by GABA.
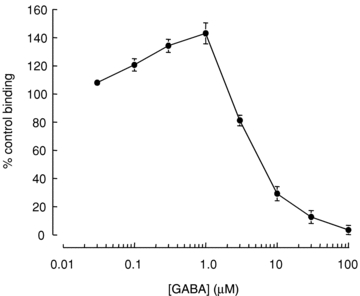
Specific, picrotoxin (100 μm)-displaceable [35S]TBPS binding to α1β2γ2 receptors in HEK293 membrane homogenates was normalized to baseline binding in the absence of GABA (% control binding). Low concentrations of GABA (0.03–1 μm) enhanced baseline binding, while high concentrations of GABA (>1 μm) inhibited binding below baseline. Data are representative of the means ± SEM of 5–8 independent experiments performed in triplicate.
The role of spontaneous gating in GABAAR blockade by TBPS and picrotoxin
Several GABAAR subtypes exhibit a low level of spontaneous gating that can be inhibited by picrotoxin or the inverse agonist bicuculline (McCartney et al. 2007). Consistent with this, local application of picrotoxin (100 μm) to HEK293 cells expressing α1β2γ2 receptors inhibited spontaneous current with a mean amplitude of 11 ± 4 pA (n = 12) corresponding to a mean current density of 0.73 ± 0.19 pA pF−1 (Supplementary Fig. 3). The amplitude of the current inhibited by picrotoxin was 0.27 ± 0.08% of the maximal current evoked in the same cells (n = 12) by locally applied GABA (100 μm). We hypothesized that TBPS and picrotoxin enter spontaneously active channels in the absence of GABA. This mode of entry could account for their apparent use-independent current blockade (Fig. 2). We used bicuculline to inhibit spontaneous gating and examined its effect on [35S]TBPS binding in the absence of GABA.
Bicuculline affects GABA-independent [35S]TBPS binding to α1β2γ2 receptors
Bicuculline (30 nm to 100 μm) reduced picrotoxin-displaceable [35S]TBPS binding in a concentration-dependent manner (Fig. 4). A maximum inhibition of approximately 25% occurred with 3 μm bicuculline (*P < 0.05 compared to 30 nm). The observation that bicuculline reduces, but does not entirely abolish [35S]TBPS binding suggests that spontaneous gating increases accessibility to the convulsant site but is not necessary for blockade in the absence of GABA. These data are consistent with the majority of block by TBPS and picrotoxin occurring independently of channel opening, either spontaneous or induced by GABA. To evaluate this further we examined the ability of bicuculline to prevent current blockade by TBPS or picrotoxin in the presence and absence of GABA.
Figure 4. Bicuculline reduces GABA-independent binding of [35S]TBPS to recombinant α1β2γ2 receptors in a concentration-dependent manner.
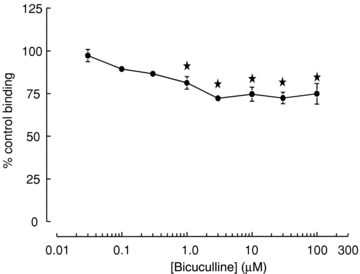
Bicuculline caused a peak inhibition of GABA-independent binding of approximately 25%. Non-specific binding was determined in the presence of 100 μm picrotoxin. Data are represented as mean ± SEM % control binding of 4–10 independent experiments performed in triplicate. *P < 0.05 (one-way ANOVA with post hoc Tukey's test) compared to binding in the presence of 0.03 μm bicuculline.
GABA-independent blockade by TBPS is reduced by bicuculline
We bath applied bicuculline (100 μm) to HEK293 cells expressing α1β2γ2 receptors to inhibit spontaneous agonist-independent channel activity. GABA (10 mm for 100 ms) was locally applied every 60 s in order to activate GABAARs in the presence of bicuculline (Fig. 5A). This high concentration of GABA is sufficient to compete with bath applied bicuculline in the recording chamber, enabling phasic agonist-dependent channel activation in the absence of tonic spontaneous gating. We used this approach to compare the amplitude of activity-dependent and activity-independent blockade of GABAAR-mediated currents by picrotoxin and TBPS. Inhibition in the presence of episodic GABAAR activation was determined using the same approach as described for Fig. 2 with two modifications. First, bicuculline (100 μm) was present in the recording chamber throughout and second, GABA was applied at 10 mm. Bath application of either TBPS or picrotoxin inhibited GABA-evoked current (Fig. 5A). We investigated use-independent GABAAR blockade by picrotoxin or TBPS without episodic GABA application (Fig. 5B). When compared to data from Fig. 2, these experiments reveal that prevention of spontaneous channel gating by bicuculline causes a significant (P < 0.05, ANOVA with post hoc Tukey's test) reduction in the inhibition by TBPS (Fig. 5C). This is most marked using the protocol that lacks episodic GABA application (Fig. 5B and C). The activity-independent inhibition by TBPS in the presence of bicuculline was approximately 40% less than inhibition observed in the presence of both spontaneous and episodic GABA-evoked gating (Fig. 5C). These data suggest that gating provides a significant means of entry for TBPS into the GABAAR channel. By contrast, bicuculline had no significant effect on blockade of GABAARs by picrotoxin (Fig. 5C).
Figure 5. Bicuculline reduces blockade by TBPS.
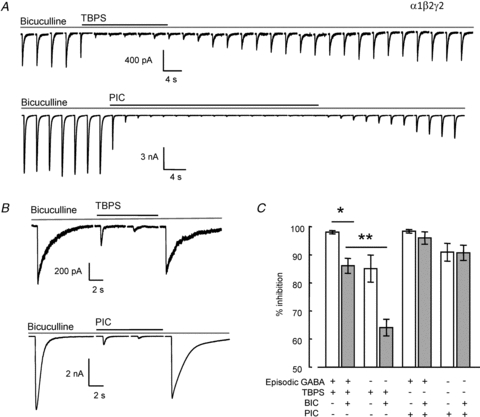
A, top traces, time-dependent inhibition of episodic GABA (10 mm for 100 ms every 60 s)-evoked currents by TBPS (10 μm) or, bottom traces, picrotoxin (100 μm) in the presence of bath applied bicuculline (100 μm). The time required for maximal inhibition with episodic GABA application was used to determine the duration of bath application of TBPS or picrotoxin in the absence of GABA with bicuculline in the bath. B, bicuculline reduced the level of GABA-independent blockade of α1β2γ2 receptors by TBPS, but not by picrotoxin. C, graph of % inhibition by TBPS or picrotoxin in the presence or absence of episodic GABA application. Grey bars indicate the additional presence of bicuculline in the recording chamber. Data are represented as means ± SEM. *P < 0.01, **P < 0.001 (one-way ANOVA with post hoc Tukey's test) comparing inhibition in the presence or absence of bicuculline. Data obtained in the absence of bicuculline are reproduced from Fig. 2C.
A mutation that reduces spontaneous GABAAR gating reduces the GABA-independent convulsant blockade
Replacement of a conserved Lys residue by Met in the M2–M3 loop at position 278 in the α1 subunit reduces the potency and efficacy of GABA (Hales et al. 2006). This mutation also reduced spontaneous GABAAR gating (Supplementary Fig. 3). Local application of picrotoxin (100 μm) to HEK293 cells expressing α1(K278M)β2γ2 receptors in the absence of GABA had little effect on holding current. The mean amplitude of the picrotoxin inhibited holding current was 1.7 ± 0.8 pA (n = 10) corresponding to a mean current density of only 0.10 ± 0.06 pA pF−1. The amplitude of the current inhibited by picrotoxin was 0.07 ± 0.03% of the maximal current evoked in the same cells (n = 10) by locally applied GABA (100 μm). The density of spontaneous current mediated by the mutant receptors was <15% of that mediated by wild-type α1β2γ2 receptors (Supplementary Fig. 3). Logistic fits to the TBPS concentration–inhibition relationships for wild-type (Fig. 1C) and mutant (not shown) GABAARs reveal similar IC50 values for TBPS (0.39 ± 0.07 μm and 0.47 ± 0.04 μm, respectively) suggesting that the mutation does not directly affect binding to the convulsant binding site in the presence of GABA. We investigated the effect of the α1(K278M) mutant on blockade by TBPS and picrotoxin applied using the same protocols for use-dependent and use-independent inhibition described earlier (Fig. 2A and B). Current traces are shown in Fig. 6A and B for recordings made with TBPS (10 μm) and picrotoxin (100 μm) when bath applied in the presence and absence of local episodic GABA application (100 μm), respectively. Although there was no significant difference in the amplitude of block by TBPS of mutant receptors compared to wild-type receptors with or without episodic GABA application, in both cases there was a trend towards a reduction in inhibition of α1(K278M)β2γ2 receptors (Fig. 6C). Furthermore, there was a significant reduction in the level of block by picrotoxin in the absence of episodic GABA application of α1(K278M)β2γ2 receptors compared to wild-type receptors (P < 0.05, ANOVA post hoc Tukey's test). These data suggest that the mutation reduces the accessibility of TBPS and picrotoxin to GABAAR channel particularly in the absence of agonist activation. The observation that the α1(K278M) subunit reduced the blockade by picrotoxin (Fig. 6C), while the application of bicuculline to wild-type GABAARs did not (Fig. 5C), suggests that there are differences between the conformational states of mutant receptors and wild-type receptors bound to bicuculline. It is possible that picrotoxin binds less deeply within the channel than does TBPS (Kalueff, 2007). If this is the case then manipulations that reduce spontaneous gating might be expected to have a greater impact on TBPS compared to picrotoxin binding. As described below, the α1(K278M) subunit also markedly reduced desensitization (Fig. 8), an effect that may adversely affect picrotoxin binding (see Discussion).
Figure 6. Inhibition by TBPS or picrotoxin of GABA-evoked currents mediated by α1(K278M)β2γ2 receptors.
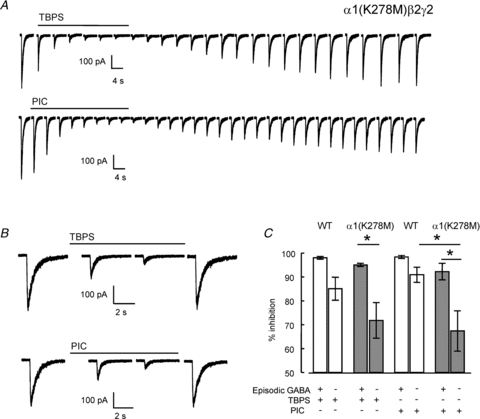
A, top traces, time-dependent inhibition of episodic GABA (100 μm for 100 ms) evoked currents by TBPS (10 μm) or, bottom traces, picrotoxin (100 μm) recorded every 60 s. The time required to reach maximal inhibition in the presence of episodic GABA was used to determine the duration of bath application of TBPS or picrotoxin in the absence of GABA. B, block after exposure to TBPS and picrotoxin in the absence of episodic GABA. C, Graph of % inhibition by TBPS or picrotoxin with or without episodic GABA application. Data from mutant receptors are shown in the filled bars. Open bars are wild-type data reproduced from Fig. 2C. Data are represented as means ± SEM (n≥ 5). Use-independent blockade of α1(K278M)β2γ2 receptors by picrotoxin was significantly lower than that of α1β2γ2 receptors (*P < 0.01, one-way ANOVA with post hoc Tukey's test).
Figure 8. The α1(K278M) mutant slows desensitization kinetics and increases deactivation rates.

A, exemplar traces showing rapid application of GABA (1 mm, 150 ms pulse duration) to outside-out patches containing α1β2γ2 (top trace) or α1(K278M)β2γ2 subunits (middle trace). After each experiment liquid junction currents were obtained by rupturing the patch and stepping the open tip across solutions containing dissimilar ions and are displayed above the corresponding GABA response. B, top graph, combined data demonstrating that the α1(K278M) mutant slows desensitization kinetics (n = 4). Bottom graph, combined data demonstrating that the α1(K278M) mutant accelerates deactivation kinetics (n = 4). Amplitude weighted values are provided here for simplicity. Desensitization and deactivation waveforms were fitted with double exponentials with fast and slow components (see Table 1 for all values). All data are represented as means ± SEM. *P < 0.05 compared to α1β2γ2 by Student's t test.
The mutant α1(K278M) subunit affects [35S]TBPS binding
We examined the effect of the α1(K278M) subunit on [35S]TBPS binding to α1β2γ2 receptors. Receptors containing the α1(K278M) subunit exhibited a reduced level of [35S]TBPS binding compared to wild-type receptors (Supplementary Fig. 2). This coincided with a similar reduction in the level of [3H]flunitrazepam binding suggesting that the mutation causes a reduction in GABAAR expression. Unlike the reduction in [35S]TBPS binding by bicuculline observed for wild-type α1β2γ2 receptors (Fig. 4), bicuculline had no significant effect on [35S]TBPS binding to α1(K278M)β2γ2 receptors (Fig. 7A). These data suggest that the mutation causes a deficit in spontaneous gating and a reduced level of expression of α1(K278M)β2γ2 receptors.
Figure 7. [35S]TBPS binding to mutant α1(K278M)β2γ2 receptors.
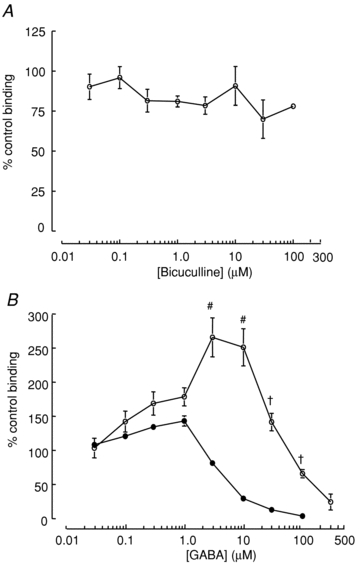
A, bicuculline had no significant concentration-dependent effect on GABA-independent [35S]TBPS binding to mutant α1(K278M)β2γ2 receptors (n≥ 4 experiments performed in triplicate). B, GABA evoked potentiation of [35S]TBPS binding to α1(K278M)β2γ2 receptors was enhanced compared to α1β2γ2 receptors. Wild-type data are included here from Fig. 3 for comparison. GABA (0.1 to 30 μm) enhanced binding to α1(K278M)β2γ2 receptors above baseline levels (n = 3–15 experiments performed in triplicate). Data are represented as means ± SEM. #P < 0.001, *P < 0.0001 compared to α1β2γ2 by Student's t test.
The α1(K278M) subunit enhances GABA-dependent stimulation of [35S]TBPS binding
The α1(K278M) subunit had a dramatic effect on the modulation of [35S]TBPS binding by GABA (Fig. 7B). GABA caused a larger enhancement of [35S]TBPS binding to α1(K278M)β2γ2 receptors compared to wild-type receptors. Furthermore, higher concentrations of GABA (>30 μm) were required to reduce [35S]TBPS binding to α1(K278M)β2γ2 receptors below baseline binding levels compared to wild-type receptors (Fig. 7B). Binding to the latter declined below baseline with GABA concentrations > 1 μm. We speculated that the abolition of [35S]TBPS binding to wild-type α1β2γ2 receptors by 100 μm GABA resulted from receptor desensitization. If this is correct then the failure of GABA to abolish [35S]TBPS binding to α1(K278M)β2γ2 receptors may be due to reduced steady-state desensitization.
The α1(K278M) subunit affects desensitization and deactivation
We previously demonstrated that both the efficacy and apparent potency of GABA are reduced by the α1(K278M) subunit as evidenced by a reduction in single channel mean open time and a dextral shift in the concentration dependence of activation, respectively (Hales et al. 2006). Here we examined the effect of the α1(K278M) subunit on the kinetics of desensitization and deactivation of α1β2γ2 receptors. GABA at 1 mm, a concentration that caused maximal current activation of both mutant and wild-type receptors, was applied rapidly to outside-out patches excised from HEK293 cells expressing either wild-type α1β2γ2 or mutant α1(K278M)β2γ2 receptors (Fig. 8A). After each experiment the patch of membrane was ruptured and the rate of solution exchange was measured by recording the kinetics of the development of junction currents across the open tip of the electrode (see Methods). Junction currents are displayed above the corresponding GABA-evoked current in Fig. 8A. The presence of the α1(K278M) subunit slowed GABA-evoked desensitization compared to that of wild-type receptors. Desensitization in the presence of GABA was fitted with the sum of two exponentials. There was no change in the fast or slow rates of desensitization. However, the amplitude of the fast component was significantly diminished by the presence of the α1(K278M) subunit (Table 1). The mean weighted time constants of desensitization (τW) were 29 ± 5 ms for α1(K278M)β2γ2 receptors and 52 ± 6 ms for α1(K278M)β2γ2 receptors. By contrast, the mutation caused an increase in both the fast and slow rates of deactivation compared to wild-type receptors (Table 1). Wild-type GABA evoked currents deactivated with a τW of 102 ± 30 ms while those mediated by α1(K278M)β2γ2 receptors deactivated with a τW of 9.7 ± 2.7 ms (Fig. 8B).
Table 1.
The α1(K278M) mutation affects the kinetics of GABA-induced desensitization and deactivation
| τfast (ms) | Amplitude τfast (% total) | τslow (ms) | |
|---|---|---|---|
| Desensitization | |||
| α1β2γ2 | 5.8 ± 1.4 | 58 ± 10% | 66 ± 8 |
| α1(K278M)β2γ2 | 5.7 ± 1.6 | 23 ± 6%* | 57 ± 12 |
| Deactivation | |||
| α1β2γ2 | 37 ± 17 | 45 ± 17% | 230 ± 83 |
| α1(K278M)β2γ2 | 4.7 ± 1.1* | 61 ± 9% | 20 ± 5* |
The sum of two exponentials was used to fit the desensitization and the deactivation phases of currents activated by 1 mm GABA (150 ms). Receptors containing the α1(K278M) mutation exhibited slower amplitude weighted time constants (τw) of desensitization (Fig. 8) due to a reduction in the amplitude of the fast component of desensitization (τfast). Receptors containing the α1(K278M) mutation exhibited a faster mean τw for deactivation (Fig. 8), an effect that was due to a reduction in the rate of both τfast and slow component (τslow) of deactivation. All data are represented as means ± SEM.
P < 0.05 compared to α1β2γ2 by Student's t test.
The α1(K278M) subunit reduces steady-state desensitization
In order to approximate steady-state desensitization during whole-cell recording, GABA was bath applied at increasing concentrations (0.1 to 1000 μm) while recording episodic responses to GABA (300 μm or 3 mm for α1β2γ2 or α1(K278M)β2γ2, respectively) locally applied every 180 s for 200 ms to establish fractional current availability (Fig. 9A and B). The concentration of 300 μm GABA was used to maximally activate α1β2γ2 currents (Hales et al. 2006). This concentration was chosen because it appeared to cause less cumulative rundown over the prolonged time course of these whole-cell experiments than was associated with initial attempts to use 1 mm GABA. By contrast, 3 mm GABA achieved maximal α1(K278M)β2γ2 receptor activation with minimal rundown. The concentration of bath applied GABA required for half-maximal steady-state desensitization was higher for α1(K278M)β2γ2 receptors (27 ± 6 μm) compared to α1β2γ2 receptors (0.8 ± 0.3 μm). Interestingly the fractional availability, described by the areas under the curves fitted to plots of activation and desensitization data (indicated in grey), was increased substantially by the presence of the mutant α1(K278M) subunit (Fig. 9C). This increase in fractional availability is likely to contribute to the increased GABA-stimulated [35S]TBPS binding to α1(K278M)β2γ2 receptors compared to wild-type receptors (Fig. 7B).
Figure 9. The mutant α1(K278M) subunit increases the fraction of available GABA-evoked current.
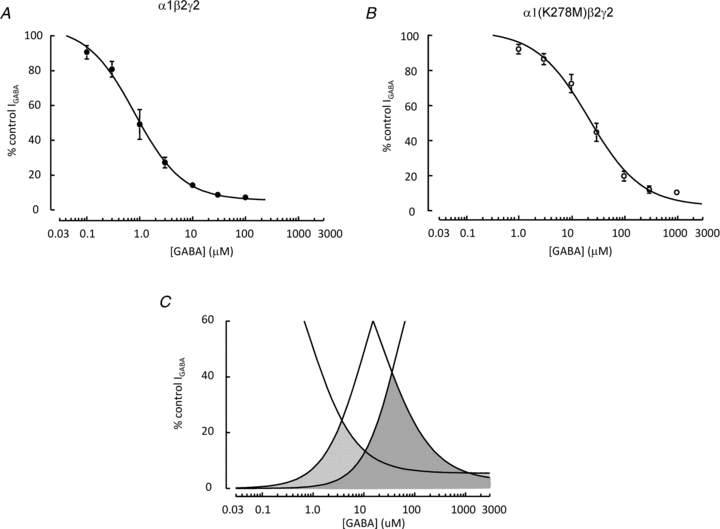
A and B, relationships for GABA concentration and desensitization for wild-type α1β2γ2 receptors (A) and α1(K278M)β2γ2 receptors (B). Current amplitudes are expressed as a function of maximal current evoked by locally applied GABA (300 μm or 3 mm for α1β2γ2 or α1(K278M)β2γ2 respectively). Steady-state desensitization was determined by bath applying GABA at the concentrations indicated. Desensitization (EC50= 26.5 ± 6.2 μm) of α1(K278M)β2γ2 receptors required higher concentrations of GABA compared to desensitization (EC50 0.82 ± 0.3 μm) of wild-type α1β2γ2 receptors. C, the fraction of available current for α1β2γ2 (light shading) and α1(K278M)β2γ2 (dark shading) receptors is represented by the area bounded by fits to the activation and desensitization data. Activation curves were plotted using data from our previous study (Hales et al. 2006). The graph indicates that there is a greater portion of available current for α1(K278M)β2γ2 receptors compared to wild-type receptors.
Discussion
GABAAR activation by episodic GABA application significantly enhances blockade by both TBPS and picrotoxin. This suggests that activation of α1β2γ2 receptors by GABA increases the accessibility of both drugs to their binding sites within the open channel. Consistent with this, low concentrations of GABA enhance [35S]TBPS binding to α1β2γ2 receptors. These findings are in agreement with previous reports of use-dependent facilitation of blockade by picrotoxin (Newland & Cull-Candy, 1992; Dillon et al. 1995) and GABA-evoked elevation of [35S]TBPS binding to recombinant GABAARs (Luddens & Korpi, 1995; Davies et al. 1997). However, the great majority of blockade by picrotoxin and TBPS and picrotoxin-displaceable [35S]TBPS binding occurred in the absence of GABA. We began this study by investigating whether constitutive GABAAR gating provides a mechanism for TBPS entry into the channel pore in the absence of GABA.
Several GABAAR subtypes exhibit infrequent agonist-independent spontaneous gating which can be inhibited by bicuculline and may contribute to tonic inhibition in the CNS (McCartney et al. 2007). We hypothesized that spontaneous gating of the α1β2γ2 receptor provides TBPS access to the open channel in the absence of GABA. Several lines of evidence support this idea. Blockade of α1β2γ2 receptors by TBPS was inhibited by bicuculline. This observation is in keeping with previous findings that bicuculline decreases the rate of both the inhibition of GABA-evoked currents by TBPS and the establishment of equilibrium [35S]TBPS binding (Maksay & Simonyi, 1986; Behrends, 2000). The attenuation by bicuculline of GABAAR blockade was most effective when TBPS was applied in the absence of episodic activation by GABA. Furthermore, in the absence of GABA, bicuculline caused a concentration-dependent inhibition of [35S]TBPS binding to α1β2γ2 receptors. These findings suggest that bicuculline holds the channel in a closed conformation which hinders TBPS from accessing its binding site.
We employed the mutant α1(K278M) subunit which reduces the efficacy of GABA as an agonist at α1β2γ2 receptors (Hales et al. 2006) to further examine the influence of gating on convulsant binding. The α1 subunit K278M substitution caused by the introduction of a synthetic mutation is equivalent to the K289M substitution in the γ2 subunit that is associated with a rare epilepsy mutation. The conserved Lys residue is located within the M2–M3 loop at a position in the α1 subunit thought to be involved in the transduction of GABAAR binding to channel gating (Kash et al. 2003). However, the K to M substitution also reduces the efficacy of activation by propofol (Hales et al. 2006) an allosteric agonist that activates GABAA receptors through an interaction with the transmembrane domains (Bali et al. 2009). Our demonstration that the α1(K278M) subunit also reduces picrotoxin-sensitive spontaneous gating suggests that the energy barrier for channel opening is enhanced by the mutation, an effect that is independent of an agonist interaction at either the orthosteric or allosteric sites. In the absence of episodic GABA, mutant α1(K278M)β2γ2 receptors exhibited reduced blockade by picrotoxin and a trend towards reduced blockade by TBPS also. These observations are consistent with spontaneous gating providing improved access to the convulsant binding site.
The mutant α1(K278M) subunit also had effects on [35S]TBPS binding that were consistent with our electrophysiological observations. In contrast to its effect on wild-type GABAARs, bicuculline did not cause a concentration-dependent inhibition of [35S]TBPS binding to α1(K278M)β2γ2 receptors in the absence of GABA. When considered in the context of the electrophysiological data, this finding is consistent with the idea that the accessibility of TBPS to mutant receptors in the absence of GABA is diminished in part by a reduction in spontaneous GABAAR gating.
[35S]TBPS binding to GABAARs is slow to reach equilibrium (Maksay & Simonyi, 1986; Edgar & Schwartz, 1990). GABAAR activation enhances, while bicuculline decreases the rate of equilibration. In keeping with this, equilibrium binding of [35S]TBPS to α1(K278M)β2γ2 receptors was established more slowly compared to wild-type receptors. Under the conditions of our experiments, an incubation time of 90 min was sufficient to enable 95% and 90% equilibrium [35S]TBPS binding to wild-type and mutant receptors respectively. Taken together, our data and the results of previous studies are consistent with the idea that GABAAR gating, both spontaneous and agonist activated, increases accessibility of TBPS to its binding site.
Low concentrations of GABA enhanced [35S]TBPS binding to wild-type receptors by 40%. By contrast the GABA-evoked enhancement of [35S]TBPS binding to α1(K278M)β2γ2 receptors was 150%, revealing a greater proportion of the agonist-dependent component of binding to mutant receptors. Low GABA concentrations enhance [35S]TBPS binding in part by increasing the rate of achieving equilibrium binding (Maksay & Simonyi, 1986). Our observation of a 150% enhancement of [35S]TBPS binding to α1(K278M)β2γ2 receptors by GABA, under conditions in which 90% of equilibrium binding occurred in its absence, suggests that GABA can substantially enhance equilibrium binding. Furthermore, significant enhancement of [35S]TBPS binding to α1(K278M)β2γ2 receptors occurred at GABA concentrations that we previously demonstrated were insufficient to activate detectable currents (Hales et al. 2006). We hypothesize that [35S]TBPS binding is enhanced by the binding of a single molecule of GABA. Recent evidence suggests that one molecule of GABA is sufficient to cause gating of monoliganded α1β2γ2 receptors (Petrini et al. 2011). It is possible that the α1(K278M) mutation abolishes monoliganded gating induced by the binding of a single GABA molecule without eliminating a conformational change necessary for the binding of TBPS. Future experiments will examine whether α1(K278M)β2γ2 receptors lack monoliganded gating.
Despite their diverse structures several non-competitive GABAAR antagonists including TBPS and picrotoxin appear to bind to a common site involving the 2′, 6′ and 9′ M2 residues (Chen et al. 2006). The 6′ and 9′ M2 residues are above the channel activation gate according to a model of the nicotinic acetylcholine receptor based on substituted cysteine accessibility in open and closed states (Wilson & Karlin, 2001). If this model is applicable to the GABAAR, then binding to these residues may be possible in the closed state and this could explain the significant component of activity-independent inhibition by picrotoxin and TBPS that occurs in the presence of either bicuculline or the α1(K278M) subunit. Binding to the 2′ residue, which is close to the intracellular aspect of M2, and well below the activation gate according to several pentameric ligand-gated ion channel models (Wilson & Karlin, 2001; Unwin, 2005; Bocquet et al. 2009; Corringer et al. 2010), would be likely to be dependent on channel opening. The structure of the C. Elegans glutamate-gated Cl− channel solved in the presence of pricrotoxin reveals that the molecule interacts with the 2′ and −2′ residues (Hibbs & Gouaux, 2011). However, despite all the evidence implicating the 2′ residue the importance of its identity to picrotoxin binding is unclear. The DRL receptor, a pesticide-resistant mutant GABA-gated Cl− channel in Drosophila harbouring a 2′ Ala to Ser substitution, exhibits reduced potency of blockade by picrotoxin and TBPS (Zhang et al. 1994). However, the block by picrotoxin of the mutant channels still occurs at micromolar concentrations. Furthermore, in agreement with prior studies demonstrating block of other Cys-loop receptors (Dibas et al. 2002; Das & Dillon, 2005) we found that picrotoxin blocks 5-HT3A and glycine α1 receptors. These receptors have Ser and Gly residues, respectively, at their M2 2′ positions. By contrast TBPS, at an equivalent effective concentration for block of the α1β2γ2 GABAAR had no effect on glycine α1 or 5-HT3A receptors, the latter observation in agreement with a previous study (Das & Dillon, 2005). We obtained similar results from the glycine α2 receptor which contains 2′ Ser, 6′ Thr and 9′ Leu residues implicated in the binding of both picrotoxin and TBPS. Furthermore, even though the wild-type Drosophila GABA-gated Cl− channel has these same 2′, 6′ and 9′ residues, the order of picrotoxin and TBPS potency is reversed compared to the α1β2γ2 GABAAR. Clearly, these M2 residues do not provide an explanation for the differential potencies of picrotoxin and TBPS. The functional state of the receptor is a major contributing factor.
While the interactions of both TBPS and picrotoxin with the GABAAR are favoured by channel opening, desensitization appears to differentially affect their binding. Previous studies suggest that picrotoxin binds preferentially to desensitized receptors (Newland & Cull-Candy, 1992; Zhang et al. 1994). By contrast, our findings with the α1β2γ2 receptors, in agreement with previous reports involving other GABAAR subtypes (Luddens & Korpi, 1995; Davies et al. 1997), demonstrate that desensitizing concentrations of GABA reduce [35S]TBPS binding.
The α1(K278M) mutation reduced the rate of GABA-evoked desensitization by reducing the contribution of the fast component. It is possible that the mutation slows the transition from the open to the desensitized state. The mutation caused a dextral shift in the concentration dependence of desensitization of α1(K278M)β2γ2 compared to α1β2γ2 receptors. Accumulation of GABAARs in the desensitized state slows the rate of GABA-evoked current deactivation (Jones & Westbrook, 1995). Thus reduced desensitization may contribute to the remarkable acceleration of deactivation of GABA-evoked currents mediated by α1(K278M)β2γ2 compared to α1β2γ2 receptors. However, there are examples of GABAARs that exhibit uncoupling between macroscopic current desensitization and deactivation kinetics (Bianchi et al. 2007). Fast deactivation could reflect a reduction in the affinity of GABA for the mutant receptor. It is also possible that reduced GABA efficacy caused by the mutation could also contribute to fast deactivation.
The combined plots of the concentration dependence of activation and desensitization of mutant compared to wild-type receptors reveal that the fraction of available GABA-evoked current is greater in the former than the latter with GABA concentrations >10 μm. This corresponds to a marked reduction in the GABA-evoked attenuation of [35S]TBPS binding to α1(K278M)β2γ2 receptors. Taken together these data suggest that desensitization reduces accessibility of TBPS to the GABAAR channel. The IC50 for inhibition of TBPS binding by agonist is substantially higher than the agonist's binding affinity (Maksay & Simonyi, 1986). Our data reveal that concentrations of GABA required for 50% displacement of maximal TBPS binding to wild-type and mutant GABAARs are similar to those required for 50% of total desensitization. GABAAR desensitization may reduce TBPS association, increase TBPS dissociation, or both. A desensitization gate located above the TBPS binding site within the channel would provide a mechanism for reducing TBPS association. However, while our interpretation of a relationship between desensitization and displacement of TBPS binding is based on a compelling correlation, it will be important to test the validity of this relationship in future experiments.
[35S]TBPS is an important tool for investigating the distribution of GABAARs. Unlike other GABAAR radioligands, many of which bind either to the orthosteric or the benzodiazepine binding site, [35S]TBPS binding within the channel is dependent on the functional state of the receptor. Different GABAAR subtypes exhibit distinct patterns of GABA modulated [35S]TBPS binding (Luddens & Korpi, 1995; Davies et al. 1997; Sinkkonen et al. 2001). Our study demonstrates that bicuculline-sensitive spontaneous gating contributes to [35S]TBPS binding in the absence of GABA. Furthermore, whether GABA enhances or inhibits TBPS binding depends on the receptor's propensity for desensitization. Mutations that affect GABAAR function can be associated with diseases such as epilepsy (Bianchi & Macdonald, 2002; Macdonald et al. 2003; Hales et al. 2006). Autoradiographic analysis of [35S]TBPS binding can be performed post mortem (Atack et al. 2007) providing an opportunity for examining the functional properties of GABAARs associated with human neurological diseases.
Acknowledgments
We thank Lisa Wright for her expert technical assistance. This work was supported by a grant (GM058037) from the National Institutes of Health.
Glossary
- GABAAR
GABAA receptor
- TBPS
t-butylbicyclophosphorothionate
Author contributions
N.A.O.: Data collection, analysis and interpretation of data; drafting the manuscript. M.G., T.Z.D. and D.T.B-H: Data collection, analysis and interpretation of data. D.C.P.: Design of experiments and interpretation of data. T.G.H: conception and design of the experiments; drafting and revising the article. All authors approved the final version of this manuscript.
Supplementary material
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Adodra S, Hales TG. Potentiation, activation and blockade of GABAA receptors of clonal murine hypothalamic GT1–7 neurones by propofol. Br J Pharmacol. 1995;115:953–960. doi: 10.1111/j.1476-5381.1995.tb15903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atack JR, Ohashi Y, McKernan RM. Characterization of [35S]t-butylbicyclophosphorothionate ([35S]TBPS) binding to GABAA receptors in postmortem human brain. Br J Pharmacol. 2007;150:1066–1074. doi: 10.1038/sj.bjp.0707186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bali M, Akabas MH. The location of a closed channel gate in the GABAA receptor channel. J Gen Physiol. 2007;129:145–159. doi: 10.1085/jgp.200609639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bali M, Jansen M, Akabas MH. GABA-induced intersubunit conformational movement in the GABAA receptor α1M1-β 2M3 transmembrane subunit interface: experimental basis for homology modeling of an intravenous anesthetic binding site. J Neurosci. 2009;29:3083–3092. doi: 10.1523/JNEUROSCI.6090-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrends JC. Modulation by bicuculline and penicillin of the block by t-butyl-bicyclo-phosphorothionate (TBPS) of GABAA-receptor mediated Cl−-current responses in rat striatal neurones. Br J Pharmacol. 2000;129:402–408. doi: 10.1038/sj.bjp.0703063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi MT, Macdonald RL. Slow phases of GABAA receptor desensitization: structural determinants and possible relevance for synaptic function. J Physiol. 2002;544:3–18. doi: 10.1113/jphysiol.2002.020255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi MT, Botzolakis EJ, Haas KF, Fisher JL, Macdonald RL. Microscopic kinetic determinants of macroscopic currents: insights from coupling and uncoupling of GABAA receptor desensitization and deactivation. J Physiol. 2007;584:769–87. doi: 10.1113/jphysiol.2007.142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnir B, Eghbali M, Cox GB, Gage PW. GABA concentration sets the conductance of delayed GABAA channels in outside-out patches from rat hippocampal neurons. J Membr Biol. 2001;181:171–183. doi: 10.1007/s00232-001-0021-5. [DOI] [PubMed] [Google Scholar]
- Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- Buhr A, Wagner C, Fuchs K, Sieghart W, Sigel E. Two novel residues in M2 of the gamma-aminobutyric acid type A receptor affecting gating by GABA and picrotoxin affinity. J Biol Chem. 2001;276:7775–7781. doi: 10.1074/jbc.M008907200. [DOI] [PubMed] [Google Scholar]
- Casida JE, Lawrence LJ. Structure-activity correlations for interactions of bicyclophosphorus esters and some polychlorocycloalkane and pyrethroid insecticides with the brain-specific t-butylbicyclophosphorothionate receptor. Environ Health Perspect. 1985;61:123–132. doi: 10.1289/ehp.8561123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Weiss DS. Allosteric activation mechanism of the α1β2γ2 γ-aminobutyric acid type A receptor revealed by mutation of the conserved M2 leucine. Biophys J. 1999;77:2542–2551. doi: 10.1016/s0006-3495(99)77089-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Durkin KA, Casida JE. Spontaneous mobility of GABAA receptor M2 extracellular half relative to noncompetitive antagonist action. J Biol Chem. 2006;281:38871–38878. doi: 10.1074/jbc.M608301200. [DOI] [PubMed] [Google Scholar]
- Connolly CN, Wafford KA. The Cys-loop superfamily of ligand-gated ion channels: the impact of receptor structure on function. Biochem Soc Trans. 2004;32:529–534. doi: 10.1042/BST0320529. [DOI] [PubMed] [Google Scholar]
- Corringer PJ, Baaden M, Bocquet N, Delarue M, Dufresne V, Nury H, Prevost M, Van Renterghem C. Atomic structure and dynamics of pentameric ligand-gated ion channels: new insight from bacterial homologues. J Physiol. 2010;588:565–572. doi: 10.1113/jphysiol.2009.183160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P, Dillon GH. Molecular determinants of picrotoxin inhibition of 5-hydroxytryptamine type 3 receptors. J Pharmacol Exp Ther. 2005;314:320–328. doi: 10.1124/jpet.104.080325. [DOI] [PubMed] [Google Scholar]
- Davies PA, Kirkness EF, Hales TG. Modulation by general anaesthetics of rat GABAA receptors comprised of α1β3 and β3 subunits expressed in human embryonic kidney 293 cells. Br J Pharmacol. 1997;120:899–909. doi: 10.1038/sj.bjp.0700987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibas MI, Gonzales EB, Das P, Bell-Horner CL, Dillon GH. Identification of a novel residue within the second transmembrane domain that confers use-facilitated block by picrotoxin in glycine α1 receptors. J Biol Chem. 2002;277:9112–9117. doi: 10.1074/jbc.M111356200. [DOI] [PubMed] [Google Scholar]
- Dillon GH, Im WB, Carter DB, McKinley DD. Enhancement by GABA of the association rate of picrotoxin and tert-butylbicyclophosphorothionate to the rat cloned α1β2γ2 GABAA receptor subtype. Br J Pharmacol. 1995;115:539–545. doi: 10.1111/j.1476-5381.1995.tb16368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar PP, Schwartz RD. Localization and characterization of 35S-t-butylbicyclophosphorothionate binding in rat brain: an autoradiographic study. J Neurosci. 1990;10:603–612. doi: 10.1523/JNEUROSCI.10-02-00603.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkkila BE, Sedelnikova AV, Weiss DS. Stoichiometric pore mutations of the GABAAR reveal a pattern of hydrogen bonding with picrotoxin. Biophys J. 2008;94:4299–4306. doi: 10.1529/biophysj.107.118455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales TG, Deeb TZ, Tang H, Bollan KA, King DP, Johnson SJ, Connolly CN. An asymmetric contribution to γ-aminobutyric type A receptor function of a conserved lysine within TM2–3 of α1, β2, and γ2 subunits. J Biol Chem. 2006;281:17034–17043. doi: 10.1074/jbc.M603599200. [DOI] [PubMed] [Google Scholar]
- Halonen LM, Sinkkonen ST, Chandra D, Homanics GE, Korpi ER. Brain regional distribution of GABAA receptors exhibiting atypical GABA agonism: roles of receptor subunits. Neurochem Int. 2009;55:389–396. doi: 10.1016/j.neuint.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle DJ, Bianchi MT, Macdonald RL. Modifications of a commercial perfusion system for use in ultrafast solution exchange during patch clamp recording. Biotechniques. 2003;35:472–474. 476. [PubMed] [Google Scholar]
- Im WB, Pregenzer JF, Thomsen DR. Effects of GABA and various allosteric ligands on TBPS binding to cloned rat GABAA receptor subtypes. Br J Pharmacol. 1994;112:1025–1030. doi: 10.1111/j.1476-5381.1994.tb13185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- Kalueff AV. Mapping convulsants’ binding to the GABAA receptor chloride ionophore: a proposed model for channel binding sites. Neurochem Int. 2007;50:61–68. doi: 10.1016/j.neuint.2006.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash TL, Jenkins A, Kelley JC, Trudell JR, Harrison NL. Coupling of agonist binding to channel gating in the GABAA receptor. Nature. 2003;421:272–275. doi: 10.1038/nature01280. [DOI] [PubMed] [Google Scholar]
- Korpi ER, Sinkkonen ST. GABAA receptor subtypes as targets for neuropsychiatric drug development. Pharmacol Ther. 2006;109:12–32. doi: 10.1016/j.pharmthera.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Lambert JJ, Belelli D, Peden DR, Vardy AW, Peters JA. Neurosteroid modulation of GABAA receptors. Prog Neurobiol. 2003;71:7–80. doi: 10.1016/j.pneurobio.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Luddens H, Korpi ER. GABA antagonists differentiate between recombinant GABAA/benzodiazepine receptor subtypes. J Neurosci. 1995;15:6957–6962. doi: 10.1523/JNEUROSCI.15-10-06957.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Bianchi MT, Feng H. Mutations linked to generalized epilepsy in humans reduce GABAA receptor current. Exp Neurol. 2003;184:S58–67. doi: 10.1016/j.expneurol.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Maksay G, Simonyi M. Kinetic regulation of convulsant (TBPS) binding by GABAergic agents. Mol Pharmacol. 1986;30:321–328. [PubMed] [Google Scholar]
- Maksay G, Thompson SA, Wafford KA. The pharmacology of spontaneously open α1β3ɛ GABAA receptor-ionophores. Neuropharmacology. 2003;44:994–1002. doi: 10.1016/s0028-3908(03)00116-3. [DOI] [PubMed] [Google Scholar]
- McCartney MR, Deeb TZ, Henderson TN, Hales TG. Tonically active GABAA receptors in hippocampal pyramidal neurons exhibit constitutive GABA-independent gating. Mol Pharmacol. 2007;71:539–548. doi: 10.1124/mol.106.028597. [DOI] [PubMed] [Google Scholar]
- Miller C. Genetic manipulation of ion channels: a new approach to structure and mechanism. Neuron. 1989;2:1195–1205. doi: 10.1016/0896-6273(89)90304-8. [DOI] [PubMed] [Google Scholar]
- Newland CF, Cull-Candy SG. On the mechanism of action of picrotoxin on GABA receptor channels in dissociated sympathetic neurones of the rat. J Physiol. 1992;447:191–213. doi: 10.1113/jphysiol.1992.sp018998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrini EM, Nieus T, Ravasenga T, Succol F, Guazzi S, Benfenati F, Barberis A. Influence of GABAAR monoliganded states on GABAergic responses. J Neurosci. 2011;31:1752–1761. doi: 10.1523/JNEUROSCI.1453-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABAA receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000;101:815–850. doi: 10.1016/s0306-4522(00)00442-5. [DOI] [PubMed] [Google Scholar]
- Sedelnikova A, Erkkila BE, Harris H, Zakharkin SO, Weiss DS. Stoichiometry of a pore mutation that abolishes picrotoxin-mediated antagonism of the GABAA receptor. J Physiol. 2006;577:569–577. doi: 10.1113/jphysiol.2006.120287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkkonen ST, Mihalek RM, Homanics GE, Luddens H, Korpi ER. Altered atypical coupling of γ-aminobutyrate type A receptor agonist and convulsant binding sites in subunit-deficient mouse lines. Brain Res Mol Brain Res. 2001;86:179–183. doi: 10.1016/s0169-328x(00)00273-4. [DOI] [PubMed] [Google Scholar]
- Squires RF, Casida JE, Richardson M, Saederup E. [35S]t-Butylbicyclophosphorothionate binds with high affinity to brain-specific sites coupled to γ-aminobutyric acid-A and ion recognition sites. Mol Pharmacol. 1983;23:326–336. [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4Å resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Whiting PJ, Bonnert TP, McKernan RM, Farrar S, Le Bourdellès B, Heavens RP, Smith DW, Hewson L, Rigby MR, Sirinathsinghji DJ, Thompson SA, Wafford KA. Molecular and functional diversity of the expanding GABA-A receptor gene family. Ann N Y Acad Sci. 1999;868:645–653. doi: 10.1111/j.1749-6632.1999.tb11341.x. [DOI] [PubMed] [Google Scholar]
- Wilson G, Karlin A. Acetylcholine receptor channel structure in the resting, open, and desensitized states probed with the substituted-cysteine-accessibility method. Proc Natl Acad Sci U S A. 2001;98:1241–1248. doi: 10.1073/pnas.031567798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HG, ffrench-Constant RH, Jackson MB. A unique amino acid of the Drosophila GABA receptor with influence on drug sensitivity by two mechanisms. J Physiol. 1994;479:65–75. doi: 10.1113/jphysiol.1994.sp020278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.