

Abstract
Non-technical summary
The retina can process over 10 log-units of changes in light intensity allowing us to see in dim starlight to bright daylight. These changes in luminosity are detected first by photoreceptors, rods and cones, and then relayed to second-order neurons, ON and OFF bipolar cells (BCs), before finally being encoded as spikes by ganglion cells (GCs). We found that ON BCs process light signals differently depending on the luminosity of the signal. In response to bright light, ON BCs undergo Ca2+-dependent depression, referred to as desensitization, producing a transient response to a sustained light stimulus; however, no such transformation occurred in dim light. Furthermore, every ON BC was capable of producing desensitization. This desensitization was passed to GCs, resulting in transient spike trains. In conclusion, ON BCs play an important role in retinal processing by converting a sustained light signal into a transient one, in order to prevent saturation.
Abstract
ON bipolar cells invert the sign of light responses from hyperpolarizing to depolarizing before passing them on to ganglion cells. Light responses are generated when a cation channel, recently identified as Trpm1, opens. The amplitude of the light response rapidly decays due to desensitization of Trpm1 current. The role of Trpm1 desensitization in shaping light responses both in bipolar and downstream ganglion cells has not been well characterized. Here we show that two parameters, the amount and the rate of recovery from desensitization, depend on the strength of the presynaptic stimulus. Stimuli that activate less than 20% of the maximum Trpm1 current did not promote any detectable desensitization, even for prolonged periods. Beyond this threshold there was a linear relationship between the amount of desensitization and the fractional Trpm1 current. In response to stimuli that open all available channels, desensitization reduced the response to approximately 40% of the peak, with a time constant of 1 s, and recovery was slow, with a time constant of more than 20 s. In dye-filled bipolar cells classified as transient or sustained using morphological criteria, there were no significant differences in Trpm1 desensitization parameters. Trpm1 activation evoked robust EPSCs in ganglion cells, and removal of Trpm1 desensitization strongly augmented a sustained component of the ganglion cell EPSC irrespective of whether ganglion cells were of the ON or ON/OFF type. We conclude that Trpm1 desensitization impacts the kinetics of ganglion cell EPSCs, but does not underlie the sustained/transient dichotomy of neurons in the ON pathway.
Introduction
Changes in light intensity are detected first by rod and cone photoreceptors, and then relayed to two classes of second-order neurons, ON and OFF bipolar cells (BCs), before finally being encoded as spike rate by ganglion cells (GCs). To generate postsynaptic responses to photoreceptor input, ON BCs express a metabotropic glutamate receptor (mGluR6) (Nakajima et al. 1993), a G protein (Go) (Nawy, 1999; Dhingra et al. 2000, 2002) and at least one type of synaptic channel Trpm1 (Morgans et al. 2009; Shen et al. 2009; Koike et al. 2010). Glutamate released from photoreceptors in darkness binds to mGluR6, activates Go and closes Trpm1. In light, inhibition of Trpm1 is relieved, and the channel opens, depolarizing ON BCs. In all species thus far examined, including tiger salamander (Awatramani & Slaughter, 2000; Nawy, 2004), dogfish (Shiells & Falk, 1999) and mouse (Berntson et al. 2004) retina, Trpm1 undergoes a form of Ca2+-dependent short-term depression during prolonged exposure to light that we have termed desensitization (Nawy, 2004), referring to the decrease in Trpm1 current in the presence of light stimulus.
Desensitization has been proposed to extend the dynamic operating range of the ON BC (Shiells & Falk, 1999; Berntson et al. 2004). Additionally, desensitization could conserve bandwidth in GCs downstream, particularly those that collect input from a large number of BCs. By restoring the membrane potential of illuminated ON BCs and preventing them from continuously releasing transmitter, desensitization would ensure that GCs remain capable of responding to ON BCs in other regions of the GC receptive field. One prediction of this model is that selective elimination of Trpm1 desensitization should increase the sustained component of the GC EPSC and increase spike rate during continuous ON BC depolarization. Here we directly test this prediction by recording GC EPSCs and spike rate both before and during block of Trpm1 desensitization. By locally applying Ca2+-free solution to the region of the ON BC dendrites, we were able to prevent desensitization without compromising synaptic transmission from ON BCs to GCs.
In addition to conserving GC bandwidth, Trpm1 desensitization may also play a role in the refinement of specific ON BC subtypes. Such subtypes can be distinguished based on both morphological and functional characteristics (Euler & Wassle, 1995; Hartveit, 1997; Awatramani & Slaughter, 2000; Wu et al. 2000; Pang et al. 2004). One such characteristic is the shape of the light response, which can be either transient or sustained. An attractive model is that ON BCs may express different components of the mGluR6 cascade such that desensitization of Trpm1 may be more prominent in ON cells with transient light responses than in cells with sustained responses. Accordingly we surveyed a large population of ON BCs and measured properties of desensitization in each cell. We used a pharmacological approach which allowed us to bypass photoreceptors, and measure the kinetics of Trpm1 currents that could be attributed to purely postsynaptic mechanisms. Based on cell morphology and layers of termination, the ON BCs examined formed a heterogeneous population of subtypes, including transient and sustained. Analysis of the distribution of both the desensitization ratios (DRs), a measure of the strength of desensitization, and the rates of desensitization reveal no evidence for differences in the desensitization of Trpm1 current across ON BC subtypes. This suggests that fundamental differences in the mGluR6 cascade may not underlie the formation of transient and sustained channels of information in the retina.
Methods
Ethical approval
All experimental procedures were approved by the Institutional Animal Care and Use Committee, and the Institute for Animal Studies at Albert Einstein College of Medicine. The authors confirm that the experiments comply with the policies and regulations of The Journal of Physiology (Drummond, 2009).
Slice preparation
Larval tiger salamanders (Ambystoma tigrinum) were obtained from Charles Sullivan (Nashville, TN, USA) and kept on a 12 h light–dark cycle at 4–6°C. Retinal slices were prepared as described previously (Nawy, 2004). Briefly, the animal was anaesthetized by immersion in 3-aminobenzoic acid ethyl ester (2 g l-1) (Sigma, St Louis, MO, USA), and killed by decapitation and double-pithing. The eyes were enucleated and the retinas were removed. The isolated retina was placed on a piece of filter paper (0.65 mm nitrocellulose membrane, Millipore) with the vitreal side down and sliced at 150–200 μm thickness using a tissue slicer (Stoelting). Slices were then transferred to the recording chamber and viewed with an upright Olympus microscope equipped with a 40× water immersion lens. Slices were continuously bathed in oxygenated solution containing, unless otherwise noted (in mm): 108 NaCl, 2.5 KCl, 2 CaCl2, 1.2 MgCl2, 5 glucose, 5 Hepes, 0.1 picrotoxin, buffered to pH 7.8 with NaOH.
Electrophysiology and analysis
Patch pipettes were pulled from borosilicate glass (WPI) using a Narishige vertical puller with electrode resistance of 4–7 MΩ. Whole-cell voltage-clamp recordings of ON BCs and GCs were performed with an Axopatch 200A amplifier (Molecular Devices) at room temperature, and data were low-pass filtered at 1 kHz. ON BCs and GCs were held at −40 mV and −70 mV, respectively. The recordings had input and series resistance of 400–1000 MΩ and 5–15 MΩ, respectively. The internal solution contained (in mm): 95 potassium gluconate, 9 KCl, 10 Hepes, 1 MgCl2, 0.5 EGTA, 4 Mg2+-ATP and 1 Li-GTP, buffered to pH 7.4 with KOH. Both internal and external solutions had an osmolarity of 225–230 mosmol l-1. Loose-patch recordings in GCs were made in voltage-clamp mode using extracellular solution in the recording pipette at 0 mV. ON BCs were identified based on their response shape to mGluR6 antagonist and their position in the slice using fluorescence imaging.
Data analysis was performed with Axograph X and Kaleidagraph (Synergy Software, Reading PA, USA). Spontaneous EPSC events were detected and analysed with Axograph, software written by John Clements. We measured the number of quanta released from ON BCs by integrating the entire GC EPSC in 100 ms time intervals and dividing each interval by the integral of the averaged spontaneous EPSC for that cell. This yielded the number of quanta released during each time interval. For each ON BC, the magnitude of desensitization was quantified as desensitization ratio (DR): 1 – (steady statecurrent/peakcurrent).
Drug delivery
An agonist of mGluR6 receptor, 2-amino-4-phosphonobutanoic acid (l-AP4), was added to the bath solution just before the start of experiments to mimic darkness and a simulated light stimulus was generated by application of the mGluR antagonist LY341495. A fast perfusion system or a multi-channel valve system was used to deliver drugs at BC dendrites. The fast perfusion system consisted of two flexible fused silica capillary tubes (ID = 250 μm) containing LY341495 in one and l-AP4 (2–4 μm) in the other. The time required for the solution exchange was estimated by measuring the open-tip current with a solution containing 120 mm or 300 mm NaCl. The 10–90% rise time of the open-tip current ranged between 6 and 20 ms. Stimulus intensity was mimicked by varying the concentration of either LY341495 from 100 μm (bright light) to 2 μm (moderate light) or glutamate from 1 mm (dark) to 100 μm (dim light). For experiments that required exchange of more than two solutions, we fused up to four tubes together. The solution flow was controlled with a pinch valve system (Warner instruments). Flowpipe translations were made by an attached piezo-electric bimorph and driven by a stimulus isolation unit (A.M.P.I.). In other experiments, a picospritzer (Parker) was used to apply K+ to ON BC dendrites by pressure ejection (2–4 psi).
Cell morphology
Bipolar cells were loaded with 100 μm Alexa 488 fluorescent dye (Invitrogen) through the recording pipette. Images were acquired with a cooled CCD camera (Hamamatsu C8484) using μManger (Edelstein et al. 2010). Images were stacked and superimposed in ImageJ (NIH), and further processed in Gimp 2.6.11 to improve the contrast and brightness. Finally, the processed image was superimposed with the DIC image. The layer of axon termination was determined offline in ImageJ by measuring the distance of deepest axon ramification in the inner plexiform layer (IPL).
Results
Desensitization of Trpm1 current can be triggered by multiple sources of calcium
Trpm1 was recently identified as the synaptic transduction channel that mediates synaptic input from photoreceptors to ON BCs in mouse (Morgans et al. 2009; Shen et al. 2009; Koike et al. 2010). Furthermore, Trpm1 was localized to ON BC dendrites in the outer plexiform layer (OPL) using antibodies against Trpm1 in mouse and human retina (Morgans et al. 2009; van Genderen et al. 2009). There is no direct genetic evidence for Trpm1 in salamander, as the genome has not been sequenced yet; however, Trpm1 protein is found or predicted in species ranging from humans to amphibians suggesting the existence of Trpm1 in salamander as well (Table 1) (Li et al. 2009; van Genderen et al. 2009). Furthermore, all components of the mGluR6 signalling cascade have been conserved across species: mouse, rat, mudpuppy, salamander and cat (Slaughter & Miller, 1981; Hirano & MacLeish, 1991; Nakajima et al. 1993; de la Villa et al. 1995; Hartveit et al. 1995; Euler et al. 1996).. Therefore, it is reasonable to assume that Trpm1 and the channel properties are also conserved across species.
Table 1.
Trpm1 protein sequence homology in various species
| Species | Per cent identity to human | Per cent similarity to human | Accession number | Length (van Genderen et al. 2009) |
|---|---|---|---|---|
| Human | 100 | 100 | NP_002411.3 | 1603 |
| Chimpanzee | 99 | 99 | XP_003318097.1 | 1625 |
| Macaque | 98 | 99 | XP_001116180.2 | 1533 |
| Horse | 91 | 95 | XP_001492285.2 | 1655 |
| Mouse | 85 | 91 | NP_001034193.2 | 1622 |
| Rat | 86 | 91 | NP_001032822.1 | 1628 |
| Chicken | 80 | 88 | XP_425066.2 | 1665 |
| Anole | 78 | 86 | XP_003226977.1 | 1631 |
| Frog | 76 | 86 | XP_002934962.1 | 1696 |
| Zebrafish | 67 | 80 | XP_694167.4 | 1694 |
The per cent identity/similarity was identified using BLAST search. Trpm1 protein is predicted for all species except for mouse and human based on sequence homology and alignment.
To elicit Trpm1 currents, we bypass photoreceptors and activate the mGluR6 receptor directly by switching from an mGluR6 agonist (l-AP4) to an antagonist (LY341495, see Methods) (Awatramani & Slaughter, 2000; Snellman & Nawy, 2002). Trpm1 currents activated with a 10–90% rise time of 0.33 ± 0.02 s. After reaching a peak, the current declined exponentially with a time constant of 1.07 ± 0.04 s to a steady-state value that was approximately 42 ± 1.1% of the peak (Fig. 1A). This decline in Trpm1 current has been referred to as desensitization (Nawy, 2004). Under current clamp (Fig. 1B), Trpm1 desensitization resulted in a repolarization of the ON BC membrane to potentials that would not be expected to support transmitter release from BC terminals as L-type voltage-gated Ca2+ channels are not well activated at −50 mV (Protti & Llano, 1998; Singer & Diamond, 2003; Snellman et al. 2009). The desensitization rates in current clamp were slower than in voltage clamp (1.9 ± 0.5 s; n = 3), suggesting that activation or inactivation of voltage-dependent channels may oppose the effect of desensitization on membrane potential. The time course for Trpm1 activation and desensitization were not limited by solution exchange time which was significantly faster when measured in OFF bipolar cells with glutamate. The 10–90% rise time for OFF BCs was 0.14 ± 0.02 s (n = 7, P < 0.0001 compared to ON BCs, Fig. 1A, inset).
Figure 1. Desensitization of Trpm1 current is Ca2+ dependent.
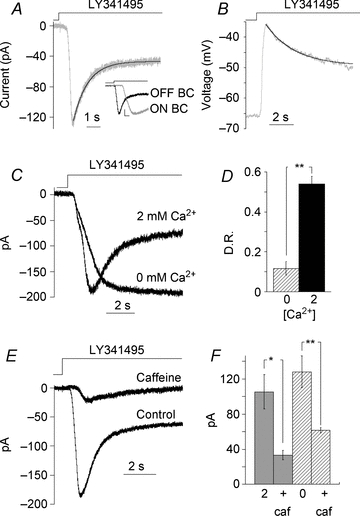
Response of an ON BC to switching from l-AP4 (4 μm) to a solution containing 100 μm LY341495 recorded in voltage (A) and current clamp (B). Records are from different cells. Inset, expanded time scale of response onset for ON and OFF BCs to an application of LY341495 and glutamate, respectively. Response amplitudes were normalized to emphasize the response activation times. The vertical line marks the stimulus onset to trigger flowpipe translation and solution flow. Scale bar: 0.2 and 250 ms. C, response of another ON BC to LY341495 in 2 mm Ca2+ and nominally 0 mm Ca2+. D, summary of the effect of Ca2+ on desensitization ratio (D.R.), defined as 1 – (steady state/peak); n = 9 cells, **P < 0.0001. E, response of an ON BC to LY341495 following a 5 min application of caffeine (10 mm) in the presence of l-AP4. F, summary of the effects of caffeine on peak current amplitude in the presence of 2 mm (n = 8, *P < 0.01) and nominally 0 mm external Ca2+ (n = 10, *P < 0.01).
Trpm1 desensitization has previously been shown to be Ca2+ dependent (Shiells & Falk, 1999; Berntson et al. 2004; Nawy, 2004). In agreement with previous results, removing extracellular Ca2+ strongly reduced desensitization (Fig. 1C). The desensitization ratio (DR, see Methods) was 0.54 ± 0.03 in 2 mm extracellular Ca2+ and 0.11 ± 0.03 in nominally Ca2+-free extracellular solution (P < 0.0001, n = 9, Fig. 1D). These results are consistent with the idea that Ca2+ entry through the channel leads to Trpm1 desensitization, but do not address the possibility that Ca2+ originating from other sources could also drive desensitization.
Ca2+ release from internal stores in dendrites of isolated rod BCs in response to group I mGluR activation has been reported (Koulen et al. 2005). To test the possibility that Ca2+ release from internal stores is capable of desensitizing Trpm1, caffeine was locally applied to BCs and Trpm1 currents were then evoked. Within 1 min of caffeine application, Trpm1 currents were strongly depressed, presumably due to Ca2+-induced Trpm1 desensitization (Fig. 1E). The effect of caffeine persisted in the absence of extracellular Ca2+ (P < 0.001 compared to control, n = 9, Fig. 1F), suggesting that the release of Ca2+ from stores alone can trigger desensitization. While Ca2+ stores are sufficient to induce desensitization, it is unclear whether they are necessary for this process. To address this possibility, we depleted the internal stores prior to the start of the experiments with 10 μm thapsigargin, which is a non-competitive inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. We found that Trpm1 kinetics were unaffected and desensitization was similar to control (DR = 0.56 ± 0.03, n = 6; compared to control 0.58 ± 0.01, n = 94, P = 0.55). Although Ca2+ stores are apparently not required for Trpm1 desensitization, it is possible that Ca2+ store release could act synergistically with Ca2+ entering through the Trpm1 channel to lower the threshold for activation of desensitization.
Threshold behaviour of desensitization prevents loss of synaptic gain following weak stimulation
The experiments described above were carried out using an ‘all-or-none’ protocol in which mGluR6 was alternately activated with a saturating concentration of agonist or inhibited with a high concentration of antagonist. We next examined the impact of desensitization on moderate to weak stimuli. To accomplish this, the maximum possible Trpm1 current was first evoked by switching from 1 mm glutamate to a test solution containing 100 μm LY341495. Progressively weaker stimuli were then presented to the same cell by switching from 1 mm glutamate to test solutions containing either 2 μm LY341495, 10 μm glutamate or 100 μm glutamate (Fig. 2A). For these experiments we used glutamate as the mGluR6 agonist because it was easier to work with than the high-affinity agonist l-AP4. For each cell, the peak Trpm1 response and DR obtained with each test solution were expressed as a fraction of the maximum peak response and DR (i.e. in the presence of 100 μm LY341495). For stimuli that activated less than 20% of the total Trpm1 current, most points fell to the right of the regression line, indicating a sublinear relationship between Trpm1 activation and desensitization. However, for stronger stimuli, desensitization was highly correlated with the size of the current (correlation coefficient 0.93, P < 0.0001, Fig. 2B, n = 42 cells). These findings are consistent with the findings of an earlier study that moderate to bright light intensities induce stronger desensitization compared to dim light (Berntson et al. 2004).
Figure 2. Trpm1 desensitization initiates when Ca2+ is elevated above threshold.
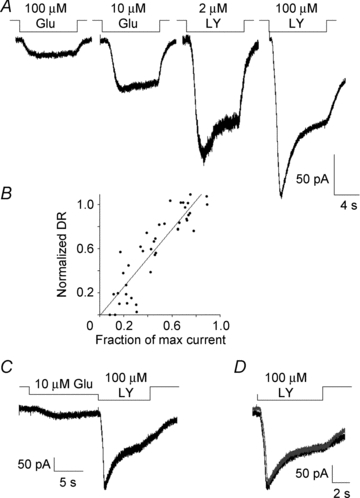
A, response of an On BC to varied stimulus intensity by changing concentrations of mGluR6 ligands. Records are from the same cell. B, summary data showing the relationship between DR and the fraction of maximum current elicited by 100 μm LY341495. For each cell, the response to 100 μm LY341495 and one other concentration of agonist or antagonist (test stimulus) was measured. The DR of the test stimulus was normalized to the maximum to eliminate cell-to-cell variability and plotted. The correlation coefficient for the data was 0.93 and r2 of the fit was 0.81. C, response from an ON BC to a prolonged weak stimulus, 10 μm glutamate, followed by a strong stimulus, 100 μm LY341495. D, an overlay of the response to 100 μm LY341495 application before and after pairing it with the weak stimulus. Per cent response evoked by 100 μm LY341495 when pre-treated with 10 μm glutamate was 91.5 ± 5.2% of control (n = 7).
To further investigate the relationship between weak stimuli and desensitization, we measured the amplitude of the peak Trpm1 current evoked by 100 μm LY341495 before and after a long (typically 60 s) application of a test stimulus that evoked about 20% of the maximum Trpm1 current (Fig. 2C). In this example, the amplitude of the peak response was unchanged following incubation in low glutamate (Fig. 2D). Overall, exposure to low glutamate resulted in a less than 10% depression of the peak response (91.5 ± 5.2% of control response, n = 7). This finding implies that a continuous background light stimulus that evokes 20% or less of the total Trpm1 current in ON BCs will not substantially activate the desensitization mechanism and therefore will not compromise synaptic gain.
The response to repetitive stimulation in ON BCs is limited by Trpm1 desensitization
The rate of recovery from Trpm1 desensitization could impose limitations on the frequency of ON BC activation. To address this possibility, we gave paired applications of 100 μm LY341495 to open Trpm1 channels (Fig. 3A). Data were pooled from eight cells, and the mean recovery time was fitted by a single exponential with a time constant of 25 ± 3.5 s (Fig. 3C), much longer than the value of 375 ms obtained from mouse rod BCs (Berntson et al. 2004). We repeated these experiments using a lower concentration of LY341495 (5 μm), which elicited a Trpm1 current that was 75 ± 2.1% (n = 16) of the maximum current obtained in 100 μm of the antagonist. Under these conditions, recovery from desensitization was much faster (Fig. 3B), with a time constant of 5.6 ± 1.8 s (Fig. 3C). In both conditions, the peak amplitude varied with paired application of LY341495, but the steady-state current remained unaltered and did not display paired-pulse depression. When Ca2+ was absent in the bath solution, the second pulse of LY341495 produced a response that was as large as the first (Fig. 3D) and the paired-pulse depression was minimal compared to control, supporting a role for Ca2+-dependent desensitization of Trpm1. The time course of recovery from desensitization may reflect the rate of removal of residual Ca2+, or a separate Ca2+-dependent process that is required for the channel to return to the fully conducting state, such as phosphorylation or dephosphorylation.
Figure 3. Paired-pulse experiments reveal the rate that Trpm1 recovers from desensitization.
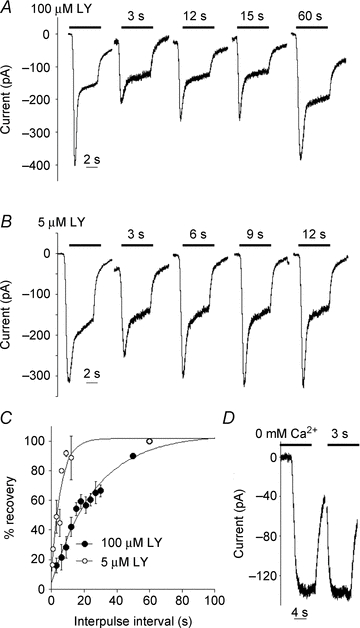
A and B, responses of an ON BC to paired applications of 100 μm or 5 μm LY341495 in the OPL; inter-pulse intervals are indicated by a bar above the traces. C, plot of rates of recovery, calculated as the ratio of 2nd peak amplitude with respect to the 1st peak. Data were fitted with an exponential function yielding a time constant (τ) of 25 and 6 s for 100 and 5 μm LY341495, respectively. D, response to paired applications of 100 μm LY341495 in nominally Ca2+-free solution.
Trpm1 desensitization cannot explain transient versus sustained classes of ON bipolar cell
ON BCs in the tiger salamander retina are thought to be divided into two classes, based on the kinetics of their response to light, one class responding to light transiently, and the other with a more sustained response (Awatramani & Slaughter, 2000). According to this model, each class of BCs contributes to the kinetics of ON–OFF and sustained ON GCs by selective wiring to the appropriate GC type. Transient ON BCs terminate in the middle of the inner plexiform layer, near the boundary of sublaminae a and b, while sustained ON cells terminate more proximally in sublamina b (Awatramani & Slaughter, 2000). One mechanism that could account for the difference in ON BC kinetics would be differences in the properties of Trpm1 expressed in these two cell types, specifically the rate and/or magnitude of desensitization. To test this possibility, we recorded from a large number of ON BCs, targeting cell bodies with a variety of shapes and positions within the inner nuclear layer to enhance the probability of targeting different classes of ON BC. For each cell, both the magnitude and the rate of Trpm1 desensitization were measured, and the results are plotted in Fig. 4A and B. Both parameters appear to form a single distribution, and there is no evidence for a secondary peak that might indicate the presence of a second population of Trpm1 channels or a presence of a distinct sustained/transient ON BC subtypes.
Figure 4. Trpm1-mediated responses are uniform across ON BC subtypes.
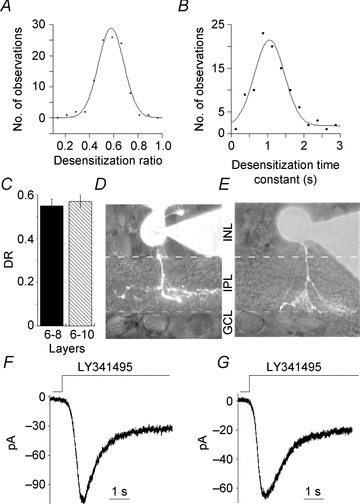
A and B, distributions of DR and time constant of desensitization for ON BCs. The smooth curve is a fit to a single Gaussian function yielding the maxima at 0.58 and 1.0 s for DR and time constant for desensitization, respectively (R = 0.99 and 0.97). C, DRs were sorted into two groups based on the location of axon terminals in sublamina b. Cells that ramified in layers 6–8 had a mean DR of 0.55 ± 0.03 (n = 11) and 0.57 ± 0.03 for those in layers 8–10 (n = 13). There was no significant difference in Trpm1 desensitization parameters in these two groups (P = 0.64). D–G, an example current trace with cell morphology from each group is shown in d-g. An ON BC terminating in layer 7 and its response is in D and F (DR = 0.69; time constant = 1.1 s); another cell in layer 10 with the current trace is shown in E and G (DR = 0.69; time constant = 0.82 s). Cells were filled with 100 μm Alexa 488 dye through the recording pipette.
In addition, cells were filled with Alexa 488 to identify the stratification layer of axon termination. Wu and coworkers (2000) established the widely accepted criteria to distinguish sustained and transient ON BCs based on the ramification of deepest axon terminals. Layers 6–8 which lie just proximal to the border of sublamina b contain transient ON BCs, and layers 8–10, which are more proximal, closer to the GC layer, include sustained ON BCs (Wu et al. 2000). Following these criteria, cells were assigned to one of these two groups. Figure 4D and E shows an example of two cells and the corresponding Trpm1 currents from each category (Fig. 4F and G). There were no detectable differences in desensitization parameters in these, or any of the ON BCs identified by layer of axon termination (Fig. 4C). In a recent study of a Trpm1 knock out mouse, Morgans et al. (2009) provided evidence for the presence of a novel, yet unidentified channel in cone ON BCs, downstream of mGluR6 signalling cascade. The response kinetics were transient in the knock-out, however in WT mouse, cone ON BC responses were sustained. It is possible that the unidentified channel is functional only in the knock out, or that the differences are difficult to observe in a normal WT mouse. We did not find the existence of that novel channel in our study, perhaps due to differences in species or because salamander ON BCs receive mixed rod–cone input making it difficult to activate purely cone pathway in the ON BCs. Based on our findings, we suggest that if ON BCs can be classified as either transient or sustained, that such a distinction is unlikely to be accounted for by the differential expression of Trpm1 isoforms.
Recovery from desensitization of Trpm1 imposes limits on ganglion cell response frequency
Our results described earlier suggest that strong activation of Trpm1 receptors results in a period of ON BC quiescence, and that during this period, the ON BC membrane potential is sufficiently hyperpolarized to prevent further signalling to downstream GCs. To test this idea directly, we recorded from a GC and delivered LY341495 to the dendrites of an ON BC that was vertically aligned with the GC. A similar strategy has been used previously to generate synaptic responses in cells that are postsynaptic to ON BCs (Chavez et al. 2006; Snellman et al. 2009). The first application of LY341495 elicited a robust GC EPSC, while the response to the second application was dramatically reduced (Fig. 5A). The responses of GC recovered as the duration between applications was increased. The rate of recovery could be fitted by a single exponential with a time constant (τ) of 18.1 ± 2.4 s (n = 5) (Fig. 5D), in good agreement with the time constant of recovery from Trpm1 desensitization directly recorded in ON BCs. It is possible that the first pulse was sufficient to deplete the RRP underlying the fast component of release, and that the slow recovery rate reflects the time required for replenishment of this pool, measured to be 8–10 s in goldfish BCs (Mennerick & Matthews, 1996; von Gersdorff & Matthews, 1997). To address this, we bypassed the mGluR6/Trpm1 pathway and depolarized ON BCs directly by applying isotonic K+ onto their dendrites. EPSCs elicited in this way recovered quickly (Fig. 5B), indicating that vesicle depletion does not impose a limit on the rate of recovery in response to paired applications of LY341495. Application of K+ generated synaptic responses rather than a direct depolarization of GC since addition of Co2+ to the bathing solution completely blocked synaptic transmission abolishing the responses (data not shown). Although we did not apply both K+ and LY341495 to the same cells, the duration of K+ application was adjusted to yield EPSCs with similar charge transfers under both conditions (K+; 147.0 ± 32.6 pA.s, n = 6, LY341495; 138 ± 36.0 pA.s, n = 6, P = 0.86) so that depolarization by either manipulation would be equally likely to result in depletion of synaptic vesicles.
Figure 5. Recovery of Trpm1 from desensitization limits the response frequency in ganglion cells.
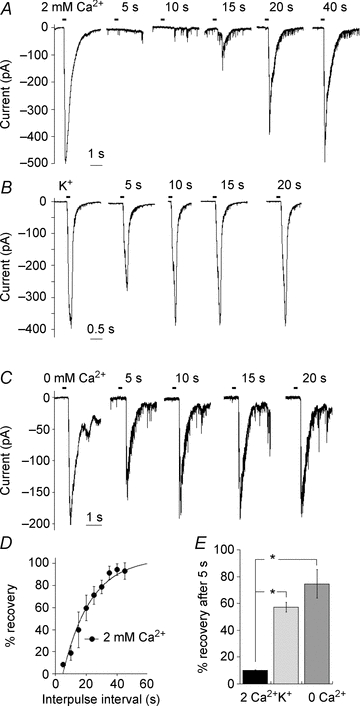
A–C, examples of the response of three different GCs to paired applications of LY341495 and normal Ca2+ (A), isotonic K+ (B) and LY341495 in Ca2+-free solution (C) to the OPL. The timing of drug application is indicated by dashes above each trace. Drugs were applied from a puffer pipette. In each case, the response to the first drug application is the average of all first application responses, while the second responses are individual traces. Paired-pulse depression (PPD) in GCs is observed when a presynaptic ON BC is depolarized with LY341495 in normal Ca2+, but not by K+ or LY341495 in Ca2+-free solution. D, summary of the downstream effect of Trpm1 desensitization on the GC responses to paired ON BC stimulation. Data were fitted with a single exponential with a time constant of 18.1 ± 2.4 s (n = 5 cells). E, summary data indicating that the recovery from PPD after 5 s in ganglion cell is significantly faster when BC desensitization is minimal (0 Ca2+), and when mGluR6 pathway involving Trpm1 is not activated (K+) (n = 6, P < 0.01).
The Ca2+-dependent Trpm1 desensitization in ON BCs was abolished by locally applying Ca2+-free solution to the region of the BC dendrites, to prevent desensitization without compromising synaptic transmission from ON BC to GCs. Application of LY341495 in a nominally Ca2+-free solution failed to induce depression of the second pulse (Fig. 5C), consistent with a role for Ca2+-dependent Trpm1 desensitization. A minor depression of the second pulse of both K+ and LY341495 was often observed, as is shown in the raw records and in the summary (Fig. 5E), and vesicle depletion or another inhibitory mechanism may play a role in this. Nevertheless, the difference in the time constant of paired pulse depression in normal Ca2+, compared with either K+ or LY341495 in low Ca2+, was highly significant (P < 0.01).
Trpm1 desensitization reduces the sustained component of release from both transient and sustained GCs
Data presented here and in previous studies strongly support the idea that Trpm1 desensitization shapes the response of ON BCs to a sustained light stimulus. However, none of the previous studies address whether Trpm1 desensitization significantly shapes the downstream GC response. Because the effect of amacrine cell inhibitory feedback on GC EPSCs has been characterized extensively elsewhere (Lukasiewicz & Werblin, 1994; Zhang & Slaughter, 1995; Dong & Werblin, 1998; Roska et al. 1998), picrotoxin and strychnine were applied in these experiments to block feedback and isolate the effects of Trpm1 desensitization. We voltage clamped single GCs while applying LY341495 with a fast-flow apparatus to the dendrites of BCs that were vertically aligned with the targeted GC.
EPSCs generated by Trpm1 activation in ON BCs were typically composed of a transient and clearly defined steady-state component (Fig. 6A). The transient component of the GC EPSC decayed with a time constant of 323 ± 46 ms (n = 7 cells) to a steady-state component whose amplitude was approximately 25% of the peak (Fig. 6A). To test for a role of Trpm1 desensitization in shaping the EPSC, we exploited the requirement for extracellular Ca2+ to support Trpm1 desensitization. Figure 6A compares the response to LY341495 delivered in normal Ca2+ (black trace) with the response to LY341495 delivered to the same cell in a nominally Ca2+-free solution (grey trace).
Figure 6. Trpm1 desensitization decreases transmitter release at the synapse.
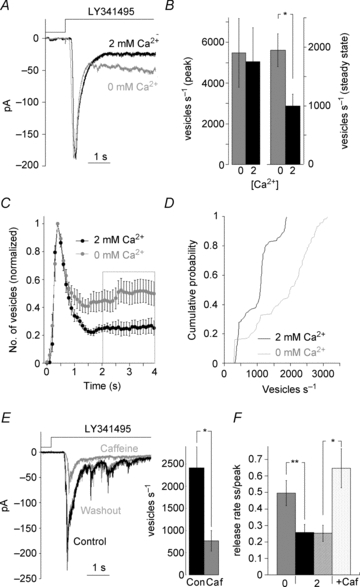
A, response of a GC to ON BC depolarization with LY341495 in control (black trace) and in nominally Ca2+-free solution (grey trace). B, summary of the rate of vesicle release at peak and steady state in control and nominally Ca2+-free solution (P = 0.28, n = 6 left; P = 0.01, n = 8 right). C, analysis of the transmitter release when ON BC desensitization is reduced in 7 cells. Data are binned in 100 ms intervals and normalized to the peak; rate of release is significantly increased in nominally Ca2+-free solution (n = 7, P = 0.01). D, cumulative distribution of vesicles released in each 100 ms bin at the steady state (2–4 s after the peak) in the presence and absence of Ca2+ (n = 7, P < 0.001). E, GC EPSC after ON BC was exposed to caffeine in the presence of l-AP4 and the response recovery after caffeine washout (grey trace). The rate of vesicle release by ON BCs was significantly reduced after caffeine application (n = 6, P < 0.02). F, summary graph comparing the ratio of vesicles released at steady state to peak when Ca2+ concentration in BC was low and high, nominally free Ca2+ and caffeine, respectively. Two groups of control experiments are plotted as each paired with a different experimental condition.
The rate of vesicle release during the peak response was not significantly different in 0 versus 2 mm Ca2+ (P = 0.28; n = 6 cells; Fig. 6B, left). However, the average steady state release rate measured 2–4 s after the onset of the EPSC was significantly increased when the extracellular Ca2+ concentration at the BC dendrites was reduced (P = 0.01; n = 7 cells; Fig. 6B, right). When each 100 ms response interval is normalized to the peak response (Fig. 6C), it is clear that the rate of release was increased in low Ca2+ at every time point beyond 1 s, consistent with the time course of Trpm1 desensitization. This is illustrated in the cumulative distribution of the number of vesicles released in each 100 ms interval during the 2–4 s window for seven cells, which indicated a highly significant difference between 2 mm and 0 mm Ca2+ (P < 0.001, Kolmogorov–Smirnov test; Fig. 6D). In conclusion, desensitization of Trpm1 limits neurotransmitter release on to GCs. It should be pointed out that the overall shape of the GC EPSC was not as dramatically altered by removing Ca2+ as was the BC Trpm1 current itself. A likely explanation is that vesicle depletion also contributes to a reduction of the steady-state release rate.
Conversely, pretreatment of BC dendrites with caffeine should initiate Trpm1 desensitization even before the channels open, leading to an overall reduction in transmitter release during LY341495 application. To test this possibility, a control EPSC was elicited in a GC, and then caffeine was applied to ON BC dendrites in the presence of L-AP4. After 60 s, re-application of LY341495 revealed that caffeine strongly reduced vesicle release rate during both the peak and steady state components of the EPSC (P < 0.01; n = 6 cells; Fig. 6E). The effects of caffeine were reversible (Fig. 6E). Caffeine and low external Ca2+ both significantly increased the steady state to peak ratio of release compared to 2 mm Ca2+ alone (0 mm Ca2+; P < 0.001, n = 6, caffeine; P < 0.02, n = 6, Fig. 6F). Comparison of release rates measured under conditions designed to produce the maximum and minimum amount of Trpm1 desensitization reveals an approximately fourfold change in the rate of steady state transmitter release (caffeine; 463 ± 118 quanta s−1, n = 6, 0 Ca2+; 1712 ± 332 quanta s−1, n = 7, P < 0.01). These results indicate that Ca2+-dependent desensitization of Trpm1 has a profound influence on transmitter release from ON BC terminals, particularly at times greater than 1 second following initiation of the depolarizing stimulus.
The sustained component of release revealed upon abolishing Trpm1 desensitization is sufficient to trigger GC action potentials
Our results imply that desensitization of Trpm1 contributes to a more transient EPSC in GCs. Ultimately, however, it is the rate of action potential firing of GCs that is transmitted to the brain, and it is unclear whether a change in the amplitude of the steady state component of the EPSC will modulate action potential frequency. To examine this question more directly, we recorded from GCs in the loose patch configuration and monitored spike activity while applying LY341495 to the dendrites of ON BCs. Picrotoxin and strychnine were added to the bath to isolate the effect of Trpm1 desensitization on spike rate. GC spike rate increased markedly in response to inactivation of the ON BC mGluR6 pathway, but the increase was transient, as the spike rate nearly returned to baseline firing rate within 1–2 s after the start of the LY341495 application (Fig. 7A). This is illustrated further in a raster plot of the number of spikes in nine successive trials (Fig. 7B), and in a plot of the mean number of spikes in those trials as a function of time (Fig. 7C). For each trial, the arrival of the first spike following the application of LY34145 was set to time (t) = 0. Subsequent spikes were plotted relative to the timing of the first spike and binned in 500 ms intervals. Lowering Ca2+ in the region of the ON BC dendrites resulted in a more uniform GC firing rate throughout the application of LY341495 (Fig. 7D, E and F). The GC firing rate during the first 500 ms of the response increased only slightly compared with normal Ca2+ (2 Ca2+; 16.0 ± 2.2 spikes s−1, in 0 Ca2+; 19.4 ± 2.8 spikes s−1) and this increase was not statistically significant. The primary effect of lowering Ca2+ on spike rate was observed approximately 1 s after the initiation of spiking (P < 0.01, n = 15, Fig. 7G), mirroring the time course of the increase in the amplitude of the EPSC described above. These results support the hypothesis that Trpm1 desensitization reduces the generation of action potentials in GCs in response to maintained illumination by decreasing the sustained component of the GC EPSC.
Figure 7. Trpm1 desensitization generates transient spike trains in ganglion cells.
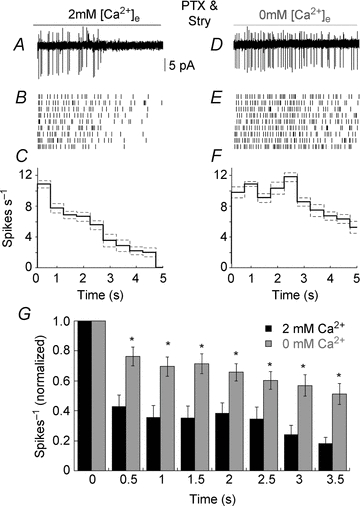
A, example of a loose-patch recording from a GC in response to activation of the mGluR6 signal cascade in ON BCs with LY341495. Feedback inhibition was blocked in all of the experiments summarized in this figure. B, raster plot of spike number evoked by LY341495 from nine consecutive trials. C, plot of the mean number of spikes (black continuous line) and SEM (grey dashed line) versus time for the cell shown in B. The data in A–C are shown on the same x axis. D–F, same cell as in A–C but with reduced extracellular Ca2+ in the OPL. G, summary histogram displaying spikes observed in each 500 ms bins in normal (black) and when Ca2+ in the OPL was reduced (grey) (n = 15, P < 0.01). For each cell, spike rate was normalized to the bin with the maximum number of spikes, which occurred at the initiation of spike train activity.
Discussion
Is there pathway specific desensitization in ON bipolar cells?
One goal of this study was to measure and compare desensitization parameters in functionally distinct populations of ON BCs. There is a general consensus that Trpm1 is required for ON BC function (Morgans et al. 2009; Shen et al. 2009; Koike et al. 2010), but whether ON BCs utilize different Trpm1 isoforms, depending on specific function, is not clear. In the salamander retina, there is strong evidence that the type of photoreceptor input that an ON BC receives is well correlated with the layer of termination of its axon, with rod-dominated cells terminating in layers 9–10, and cone-dominated cells in layers 6–7 (Wu et al. 2000; Pang et al. 2004; Zhang & Wu, 2009). Awatramani & Slaughter (2000) reported that ON BCs in these layers have strikingly different light response kinetics. The differences in kinetics may originate not from distinct properties of the postsynaptic cell, but rather from the kinetics of the presynaptic photoreceptor light response. Rods respond transiently to maintained illumination of moderate intensity (Fain et al. 1978; Attwell et al. 1987), and decreases in transmitter release rate during the light response would complicate interpretation of the kinetic behaviour of Trpm1. To avoid this concern, we have examined the kinetics of ON BC and GC EPSCs using a pharmacological approach rather than measuring light responses as others have done. In this way, we can measure synaptic Trpm1 currents using the same protocol in both rod and cone-driven ON BCs, bypassing the need for activation of the presynaptic cell. We observed desensitization in virtually every ON BC that was tested using this protocol, supporting the idea that differences in response kinetics of ON BCs are not due to intrinsic differences in the mGluR6/Trpm1 cascade, but are likely to be presynaptic.
Our results also suggest that desensitization is present in ON BCs regardless of whether they receive input from rods or cones. This would seem to contradict the work of Berntson et al. (2004), who reported that desensitization of Trpm1 current is present in ON BCs that carry information from rod, but not cone pathways in mouse. However, in amphibian retina most, if not all ON BCs receive rod input (Lasansky, 1973; Hensley et al. 1993) and so even ON BCs that receive input predominantly from cones would therefore possess a desensitizing rod-driven component of the pharmacologically evoked response. Alternatively if Ca2+ domains in mixed input ON BC dendrites are sufficiently large to allow for spread of Ca2+ from dendrites contacting rods to nearby cone-contacting sites, then cross desensitization of rod and cone signals might occur. In summary, our experiments fail to observe a difference in the kinetic behaviour of Trpm1 currents between transient, predominantly rod-driven and sustained, predominantly cone-driven ON BCs.
Microdomain organization of Ca2+ signal to induce desensitization
The dependence of desensitization upon recording conditions provide insight into the size of the domain associated with Ca2+-dependent Trpm1 desensitization. A Ca2+ signal associated with a single channel, within a few nanometers of the channel pore operates within a nanodomain (Chad & Eckert, 1984; Simon & Llinas, 1985; Oheim et al. 2006), while diffusion of Ca2+ that extends beyond a few microns from the mouth of the channel is referred to as a microdomain (Llinas et al. 1995; Neher, 1998a,b; Parekh, 2008). Four independent lines of evidence strongly suggest that Ca2+ required for Trpm1 desensitization is organized into microdomains rather than nanodomains. First, desensitization of the peak current is not observed under all conditions. As reported above, activation of approximately 20% or less of the maximum Trpm1 current produces little or no desensitization. The lack of desensitization under conditions when only a small fraction of channels are opened allows for the ON BC to reliably transmit the signal to GCs, when L-type Ca2+ channels in axon terminals are at the foot of activation curve (Protti & Llano, 1998). If desensitization occurred at the level of single Trpm1 channel, then it should be proportional to the size of the current for all measurable current amplitudes. This conclusion assumes that varying mGluR ligand concentration does not alter Trpm1 single channel conductance or open time but rather the mean open probability of the entire channel population. Second, keeping the open probability of Trpm1 constant with a saturating concentration of mGluR antagonist, while varying the Ca2+ driving force by clamping the cell at different voltages produced different amounts of desensitization (Nawy, 2004). Desensitization was greatest at −70 mV and progressively decreased as the holding potential was made more positive. Within the Trpm1 nanodomain, the Ca2+ concentration should reach steady state within a few microseconds of channel opening (Llinas et al. 1992; Oheim et al. 2006). Therefore, if Ca2+ was organized into nano-domain then desensitization should not vary regardless of the spatial extent of Ca2+ flux. This finding further demonstrates the role of Ca2+ is spatially localized to microdomains. Third, the Ca2+ chelators EGTA and BAPTA bind Ca2+ equally at equilibrium, but EGTA binds ∼100-fold more slowly than BAPTA (Marty & Neher, 1985; Neher, 1998a). At equivalent concentrations, BAPTA and EGTA were found to be equally effective at blocking desensitization (DRs: 3 mm BAPTA, 0.29 ± 0.06, n = 12, versus 3 mm EGTA, 0.28 ± 0.04, n = 8, P = 0.9; 5 mm BAPTA, 0.17 ± 0.11, n = 3, versus 5 mm EGTA, 0.16 ± 0.06, n = 5, P = 0.11; 10 mm BAPTA, 0.10 ± 0.06, n = 5, versus 10 mm EGTA, 0.11 ± 0.03, n = 5, P = 0.11). DRs are progressively smaller as the buffering capacity increased which indicates that Ca2+ ions must diffuse away from the channel pore to its target site to induce desensitization.
Last, the slow rate at which desensitization proceeds following Trpm1 channel opening (τ= 1 s) is also consistent with the idea that the Ca2+ binding site is not within the nanodomain of the Trpm1 channel pore, as Ca2+-dependent activation of processes within a nano-domain proceed very rapidly. Furthermore, we find that the time course of recovery from Trpm1 desensitization is dependent on the Trpm1 open channel probability generated by application of different concentrations of LY314159 (Fig. 3). A nano-domain model would predict that the rate of recovery from desensitization for a given Trpm1 channel should be independent of the total number of open channels, and instead should be governed by the kinetic properties of the channel itself. Instead, recovery from desensitization seems to reflect more global changes in Ca2+ concentration, perhaps indicating the presence of a Ca2+ sensor that is separate from the channel.
In conclusion, both spatial and temporal properties of desensitization strongly indicate that the Ca2+ binding site to trigger desensitization is not the channel itself, but a site which extends along the membrane within a microdomain. However, it is quite possible that the final target for desensitization is the channel itself (e.g. recruitment and activation of a kinase or phosphatase directly affecting the channel).
Desensitization may prevent saturation caused by spatial summation
Recovery from desensitization on the part of Trpm1 receptors imposes a limit on the rate of signalling between bipolar and ganglion cells. This would seem to be particularly advantageous for ON BCs that sample from a large number of photoreceptors, such as the tiger salamander (Lasansky, 1978). If the light stimulus impinging on the presynaptic photoreceptor pool is not homogenous, but instead covers only a portion of the pool, then strong stimulation of the Trpm1 channels that are postsynaptic to the stimulated photoreceptors could be sufficient to saturate the synaptic output of that of ON BCs. Desensitization should extend the operating range of the ON BC by preventing saturation due to spatial summation of inputs, essentially silencing the stimulated pool after the initial light signal has been passed downstream to the GC. It is reasonable for BCs from salamander retina, where there are multiple inputs impinging on the cell, but this seems to be a less effective strategy for cells where spatial summation is not a concern, such as midget BCs in primate retina, which receive input from only a single photoreceptor (Dowling & Boycott, 1966).
We measured the time constant of recovery from desensitization to a saturating stimulus to be approximately 20 s, which seems like a very long time to reduce or eliminate synaptic input from photoreceptors. In lower vertebrates, BCs receive input from both rod and cone photoreceptors. The time required for rods to recover from bright light is also very long, in the order of 30 s or more, and this prolonged response to light is passed on to second-order horizontal cells (Attwell et al. 1987; Wu, 1987; Belgum & Copenhagen, 1988; Shiells & Falk, 1999). Prolonged depolarization in response to bright light could obscure responses from cone input to mixed ON BCs. Provided that the rise in Ca2+ is localized sufficiently to desensitize rod input, rather than spreading to neighbouring cone contacts, as discussed above, desensitization could selectively reduce rod input, allowing the BC to respond to cone input. In mouse, rod and cone input are segregated, allowing for a separate channel of information flow when the rod pathway becomes saturated. Interestingly, desensitization is present in rod-driven BCs, but both onset and recovery from desensitization are rapid, in the order of milliseconds. Hence, it is possible that desensitization of Trpm1 is a mechanism used by lower vertebrates to separate rod and cone input.
Acknowledgments
We would like to thank R. Carroll and the other members of the Nawy lab for helpful discussions that contributed to this study. This work was supported by a grant from the National Institutes of Health (EY010254) and an unrestricted grant from the Research to Prevent Blindness foundation to S.N.
Glossary
- BC
bipolar cell
- DR
desensitization ratio
- GC
ganglion cell
- IPL
inner plexiform layer
- l-AP4
2-amino-4-phosphonobutanoic acid
- OPL
outer plexiform layer
Author contributions
Both authors designed the experiments; data was collected by T.K. and analysed and interpreted by T.K. and S.N. Manuscript was prepared by T.K. and S.N. All authors approved the final version of the manuscript submitted for publication.
References
- Attwell D, Borges S, Wu SM, Wilson M. Signal clipping by the rod output synapse. Nature. 1987;328:522–524. doi: 10.1038/328522a0. [DOI] [PubMed] [Google Scholar]
- Awatramani GB, Slaughter MM. Origin of transient and sustained responses in ganglion cells of the retina. J Neurosci. 2000;20:7087–7095. doi: 10.1523/JNEUROSCI.20-18-07087.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgum JH, Copenhagen DR. Synaptic transfer of rod signals to horizontal and bipolar cells in the retina of the toad (Bufo marinus. J Physiol. 1988;396:225–245. doi: 10.1113/jphysiol.1988.sp016960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berntson A, Smith RG, Taylor WR. Postsynaptic calcium feedback between rods and rod bipolar cells in the mouse retina. Vis Neurosci. 2004;21:913–924. doi: 10.1017/S095252380421611X. [DOI] [PubMed] [Google Scholar]
- Chad JE, Eckert R. Calcium domains associated with individual channels can account for anomalous voltage relations of CA-dependent responses. Biophys J. 1984;45:993–999. doi: 10.1016/S0006-3495(84)84244-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez AE, Singer JH, Diamond JS. Fast neurotransmitter release triggered by Ca influx through AMPA-type glutamate receptors. Nature. 2006;443:705–708. doi: 10.1038/nature05123. [DOI] [PubMed] [Google Scholar]
- de la Villa P, Kurahashi T, Kaneko A. L-glutamate-induced responses and cGMP-activated channels in three subtypes of retinal bipolar cells dissociated from the cat. J Neurosci. 1995;15:3571–3582. doi: 10.1523/JNEUROSCI.15-05-03571.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra A, Jiang M, Wang TL, Lyubarsky A, Savchenko A, Bar-Yehuda T, Sterling P, Birnbaumer L, Vardi N. Light response of retinal ON bipolar cells requires a specific splice variant of Gαo. J Neurosci. 2002;22:4878–4884. doi: 10.1523/JNEUROSCI.22-12-04878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra A, Lyubarsky A, Jiang M, Pugh EN, Birnbaumer L, Sterling P, Vardi N. The light response of ON bipolar neurons requires Gαo. J Neurosci. 2000;20:9053–9058. doi: 10.1523/JNEUROSCI.20-24-09053.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C-J, Werblin FS. Temporal contrast enhancement via GABAC feedback at bipolar terminals in the tiger salamander retina. J Neurophysiol. 1998;79:2171–2180. doi: 10.1152/jn.1998.79.4.2171. [DOI] [PubMed] [Google Scholar]
- Dowling JE, Boycott BB. Organization of the primate retina: electron microscopy. Proc R Soc Lond B Biol Sci. 1966;166:80–111. doi: 10.1098/rspb.1966.0086. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. Computer control of microscopes using microManager. Curr Protoc Mol Biol. 2010 doi: 10.1002/0471142727.mb1420s92. Chapter 14, Unit14 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euler T, Schneider H, Wassle H. Glutamate responses of bipolar cells in a slice preparation of the rat retina. J Neurosci. 1996;16:2934–2944. doi: 10.1523/JNEUROSCI.16-09-02934.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euler T, Wassle H. Immunocytochemical identification of cone bipolar cells in the rat retina. J Comp Neurol. 1995;361:461–478. doi: 10.1002/cne.903610310. [DOI] [PubMed] [Google Scholar]
- Fain GL, Quandt FN, Bastian BL, Gerschenfeld HM. Contribution of a caesium-sensitive conductance increase to the rod photoresponse. Nature. 1978;272:467–469. doi: 10.1038/272467a0. [DOI] [PubMed] [Google Scholar]
- Hartveit E. Functional organization of cone bipolar cells in the rat retina. J Neurophysiol. 1997;77:1716–1730. doi: 10.1152/jn.1997.77.4.1716. [DOI] [PubMed] [Google Scholar]
- Hartveit E, Brandstatter JH, Enz R, Wassle H. Expression of the mRNA of seven metabotropic glutamate receptors (mGluR1 to 7) in the rat retina. An in situ hybridization study on tissue sections and isolated cells. Eur J Neurosci. 1995;7:1472–1483. doi: 10.1111/j.1460-9568.1995.tb01142.x. [DOI] [PubMed] [Google Scholar]
- Hensley SH, Yang XL, Wu SM. Relative contribution of rod and cone inputs to bipolar cells and ganglion cells in the tiger salamander retina. J Neurophysiol. 1993;69:2086–2098. doi: 10.1152/jn.1993.69.6.2086. [DOI] [PubMed] [Google Scholar]
- Hirano AA, MacLeish PR. Glutamate and 2-amino-4-phosphonobutyrate evoke an increase in potassium conductance in retinal bipolar cells. Proc Natl Acad Sci U S A. 1991;88:805–809. doi: 10.1073/pnas.88.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike C, Obara T, Uriu Y, Numata T, Sanuki R, Miyata K, Koyasu T, Ueno S, Funabiki K, Tani A, Ueda H, Kondo M, Mori Y, Tachibana M, Furukawa T. TRPM1 is a component of the retinal ON bipolar cell transduction channel in the mGluR6 cascade. Proc Natl Acad Sci U S A. 2010;107:332–337. doi: 10.1073/pnas.0912730107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulen P, Wei J, Madry C, Liu J, Nixon E. Differentially distributed IP3 receptors and Ca2+ signalling in rod bipolar cells. Invest Ophthalmol Vis Sci. 2005;46:292–298. doi: 10.1167/iovs.04-0939. [DOI] [PubMed] [Google Scholar]
- Lasansky A. Organization of the outer synaptic layer in the retina of the larval tiger salamander. Philos Trans R Soc Lond B Biol Sci. 1973;265:471–489. doi: 10.1098/rstb.1973.0033. [DOI] [PubMed] [Google Scholar]
- Lasansky A. Contacts between receptors and electrophysiologically identified neurones in the retina of the larval tiger salamander. J Physiol. 1978;285:531–542. doi: 10.1113/jphysiol.1978.sp012587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Sergouniotis PI, Michaelides M, Mackay DS, Wright GA, Devery S, Moore AT, Holder GE, Robson AG, Webster AR. Recessive mutations of the gene TRPM1 abrogate ON bipolar cell function and cause complete congenital stationary night blindness in humans. Am J Hum Genet. 2009;85:711–719. doi: 10.1016/j.ajhg.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Sugimori M, Silver RB. Microdomains of high calcium concentration in a presynaptic terminal. Science. 1992;256:677–679. doi: 10.1126/science.1350109. [DOI] [PubMed] [Google Scholar]
- Llinas R, Sugimori M, Silver RB. Time resolved calcium microdomains and synaptic transmission. J Physiol Paris. 1995;89:77–81. doi: 10.1016/0928-4257(96)80554-7. [DOI] [PubMed] [Google Scholar]
- Lukasiewicz P, Werblin F. A novel GABA receptor modulates synaptic transmission from bipolar to ganglion and amacrine cells in the tiger salamander retina. J Neurosci. 1994;14:1213–1223. doi: 10.1523/JNEUROSCI.14-03-01213.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty A, Neher E. Potassium channels in cultured bovine adrenal chromaffin cells. J Physiol. 1985;367:117–141. doi: 10.1113/jphysiol.1985.sp015817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Matthews G. Ultrafast exocytosis elicited by calcium current in synaptic terminals of retinal bipolar neurons. Neuron. 1996;17:1241–1249. doi: 10.1016/s0896-6273(00)80254-8. [DOI] [PubMed] [Google Scholar]
- Morgans CW, Zhang J, Jeffrey BG, Nelson SM, Burke NS, Duvoisin RM, Brown RL. TRPM1 is required for the depolarizing light response in retinal ON-bipolar cells. Proc Natl Acad Sci U S A. 2009;106:19174–19178. doi: 10.1073/pnas.0908711106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima Y, Iwakabe H, Akazawa C, Nawa H, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a novel retinal metabotropic glutamate receptor mGluR6 with a high agonist selectivity for L-2-amino-4-phosphonobutyrate. J Biol Chem. 1993;268:11868–11873. [PubMed] [Google Scholar]
- Nawy S. The metabotropic receptor mGluR6 may signal through Go, but not phosphodiesterase, in retinal bipolar cells. J Neurosci. 1999;19:2938–2944. doi: 10.1523/JNEUROSCI.19-08-02938.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawy S. Desensitization of the mGluR6 transduction current in tiger salamander On bipolar cells. J Physiol. 2004;558:137–146. doi: 10.1113/jphysiol.2004.064980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. Usefulness and limitations of linear approximations to the understanding of Ca2+ signals. Cell Calcium. 1998a;24:345–357. doi: 10.1016/s0143-4160(98)90058-6. [DOI] [PubMed] [Google Scholar]
- Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998b;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Oheim M, Kirchhoff F, Stuhmer W. Calcium microdomains in regulated exocytosis. Cell Calcium. 2006;40:423–439. doi: 10.1016/j.ceca.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Pang J-J, Gao F, Wu SM. Stratum-by-stratum projection of light response attributes by retinal bipolar cells of Ambystoma. J Physiol. 2004;558:249–262. doi: 10.1113/jphysiol.2004.063503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB. Ca2+ microdomains near plasma membrane Ca2+ channels: impact on cell function. J Physiol. 2008;586:3043–3054. doi: 10.1113/jphysiol.2008.153460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protti DA, Llano I. Calcium currents and calcium signalling in rod bipolar cells of rat retinal slices. J Neurosci. 1998;18:3715–3724. doi: 10.1523/JNEUROSCI.18-10-03715.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roska B, Nemeth E, Werblin FS. Response to change is facilitated by a three-neuron disinhibitory pathway in the tiger salamander retina. J Neurosci. 1998;18:3451–3459. doi: 10.1523/JNEUROSCI.18-09-03451.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Heimel JA, Kamermans M, Peachey NS, Gregg RG, Nawy S. A transient receptor potential-like channel mediates synaptic transmission in rod bipolar cells. J Neurosci. 2009;29:6088–6093. doi: 10.1523/JNEUROSCI.0132-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiells RA, Falk G. Ca2+-induced light adaptation in retinal ON-bipolar cells. Keio J Med. 1999;48:140–146. doi: 10.2302/kjm.48.140. [DOI] [PubMed] [Google Scholar]
- Simon SM, Llinas RR. Compartmentalization of the submembrane calcium activity during calcium influx and its significance in transmitter release. Biophys J. 1985;48:485–498. doi: 10.1016/S0006-3495(85)83804-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer JH, Diamond JS. Sustained Ca2+ entry elicits transient postsynaptic currents at a retinal ribbon synapse. J Neurosci. 2003;23:10923–10933. doi: 10.1523/JNEUROSCI.23-34-10923.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaughter MM, Miller RF. 2-Amino-4-phosphonobutyric acid: a new pharmacological tool for retina research. Science. 1981;211:182–185. doi: 10.1126/science.6255566. [DOI] [PubMed] [Google Scholar]
- Snellman J, Nawy S. Regulation of the retinal bipolar cell mGluR6 pathway by calcineurin. J Neurophysiol. 2002;88:1088–1096. doi: 10.1152/jn.2002.88.3.1088. [DOI] [PubMed] [Google Scholar]
- Snellman J, Zenisek D, Nawy S. Switching between transient and sustained signalling at the rod bipolar–AII amacrine cell synapse of the mouse retina. J Physiol. 2009;587:2443–2455. doi: 10.1113/jphysiol.2008.165241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Genderen MM, Bijveld MM, Claassen YB, Florijn RJ, Pearring JN, Meire FM, McCall MA, Riemslag FC, Gregg RG, Bergen AA, Kamermans M. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am J Hum Genet. 2009;85:730–736. doi: 10.1016/j.ajhg.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G. Depletion and replenishment of vesicle pools at a ribbon-type synaptic terminal. J Neurosci. 1997;17:1919–1927. doi: 10.1523/JNEUROSCI.17-06-01919.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SM. Light-dependent synaptic delay between photoreceptors and horizontal cells in the tiger salamander retina. Vision Res. 1987;27:363–367. doi: 10.1016/0042-6989(87)90085-x. [DOI] [PubMed] [Google Scholar]
- Wu SM, Gao F, Maple BR. Functional architecture of synapses in the inner retina: segregation of visual signals by stratification of bipolar cell axon terminals. J Neurosci. 2000;20:4462–4470. doi: 10.1523/JNEUROSCI.20-12-04462.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A-J, Wu SM. Receptive fields of retinal bipolar cells are mediated by heterogeneous synaptic circuitry. J Neurosci. 2009;29:789–797. doi: 10.1523/JNEUROSCI.4984-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Slaughter MM. Preferential suppression of the ON pathway by GABAC receptors in the amphibian retina. J Neurophysiol. 1995;74:1583–1592. doi: 10.1152/jn.1995.74.4.1583. [DOI] [PubMed] [Google Scholar]