

Abstract
Multiple myeloma plasma cells home and expand in the bone marrow where cause an unbalanced bone remodelling with increased bone resorption and low bone formation that represent the typical feature in the majority of patients. A clinically relevant aspect of the interactions of multiple myeloma plasma cells in the bone marrow microenvironment is neovascularization, a constant hallmark of disease progression. This process is only partially supported by factors such as vascular endothelial growth factor, fibroblast growth factor-2 and metalloproteinases, which are directly secreted by the tumor cells. In fact, the presence in the bone marrow microenvironment of cytokines, in particular interleukin-6, as a consequence of plasma cell-stromal cell interactions, induces the production and secretion of angiogenic factors by other cells present in the bone microenvironment, thus contributing to the angiogenic switch during the progression of the disease. Near angiogenesis vasculogenesis occur in the bone marrow of myeloma patients and contribute to the vascular three formation. In the bone marrow of myeloma patients haematopoietic stem cells are recruited and induced to differentiate into endothelial cells by the angiogenic cytokines present in the microenvironment. Myeloma plasma cells also induce angiogenesis indirectly via recruitment and activation of stromal inflammatory cells (i.e.: macrophages and mast cells) to secrete their own angiogenic factors. They are recruited and activated by tumor plasma cells through the secretion of fibroblast growth factor-2, interleukin-8, and other chemokines, such as ITAC, Mig, IP-10. When macrophages and mast cells are activated they secrete their angiogenic factors: fibroblast growth factor-2, vascular endothelial growth factor, granulocyte-colony stimulating factor, granulocyte macrophage-colony stimulating factor, which contribute to enhance the tumor neovascularization. Finally, myeloma macrophages when exposed to vascular endothelial growth factor and fibroblast growth factor-2 secreted by plasma cells shows vasculogenic ability and acquire endothelial cell markers and transform into cells functionally and phenotypically similar to paired bone marrow endothelial cells. So they participate to the formation of the bone marrow capillary network (vasculogenic mimicry).
Keywords: Angiogenesis, endothelial cells, fibroblast growth factor-2, multiple myeloma, tumor progression, vasculogenesis, vascular endothelial growth factor
Introduction
In the past decades, the major focus of cancer research has been the malignant cell itself. This has led to the identification of oncogenes and tumor suppressor genes and associated signalling pathways by which they modulate growth, survival and proliferation of tumor cells [1].
Multiple Myeloma (MM) is a debilitating malignancy that is part of a spectrum of diseases ranging from monoclonal gammopathy of unknown significance (MGUS) to plasma cell leukemia. First described in 1848, multiple myeloma is a disease characterized by a proliferation of malignant antibody-forming cells (ie, plasma cells) and a subsequent overabundance of monoclonal (M) paraprotein. They cause an unbalanced bone remodelling with increased bone resorption and low bone formation that represent the typical feature in the majority of patients [2]. MM plasma cells home to and expand in the bone marrow [2]. Evidence is accumulating that myeloma plasma cells interact with surrounding host cells and extracellular matrix (ECM) [3], this crosstalk affecting the most important aspects of the malignant pheno-type. The role of host cells or the niche microen-vironment and ECM is becoming an intense area of research, finalized at a better understanding of the pathophysiological modifications of the complete tumor entity, i.e., malignant cells and microenvironment.
Pathophysiological interactions of myeloma cells in the bone marrow microenvironment are highlighted by the progression-associated bone disease and neovascularization, and are witnessed by autocrine/paracrine circuits that activate multiple signalling pathways and affect the most important aspects of malignant pheno-type, i.e., apoptosis/survival, proliferation, invasion, and angiogenesis [4].
The IMWG has defined the criteria for progressive disease [5], and the American Society of Haematology/Food and Drug Administration (ASH-FDA) has defined specific criteria for disease progression to active myeloma in patients with smouldering myeloma [6]. The cellular and molecular basis of disease progression are fovourited by mechanisms involving the bone marrow micoenvironment: a) bone disease mediated by interactions between plasma cells, osteoblasts, osteoclasts and macrophages; b) neovascularization, that represents the main feature of disease progression and that is supported by all the cellular and extracellular elements of the bone marrow microenvironment; c) bone marrow microenvironment and inflammatory cells as real protagonists.
Bone marrow angiogenesis in Multiple Myeloma
Angiogenesis is the sprouting of new blood vessels from a pre-existing vasculature and it is a tightly regulated process [7].
During embryogenesis two major processes of blood vessel formation are implicated in the development of the vascular system: vasculogenesis and angiogenesis [8,9]. Vasculogenesis starts from mesodermal-derived cells, the hemangioblasts, which differentiate both into angioblasts-endothelial cells and into hemato-poieticstem cells [9]. Vasculogenesis prevails in the embryo but it may have physiological roles in health and disease in adults [10]. Both mechanisms, angiogenesis and vasculogenesis, occur in ischemic and tumor tissues in response to growth factors, such as VEGF and bFGF, produced by tumor and stromal cells [11]. In MM, as well as in other aggressive tumors, the vessel wall is lined with only cancer cells as a mosaic of cancer cells and endothelial cells (Figure 1). This phenomenon is called “vasculogenesis mimicry” [12].
Figure 1.
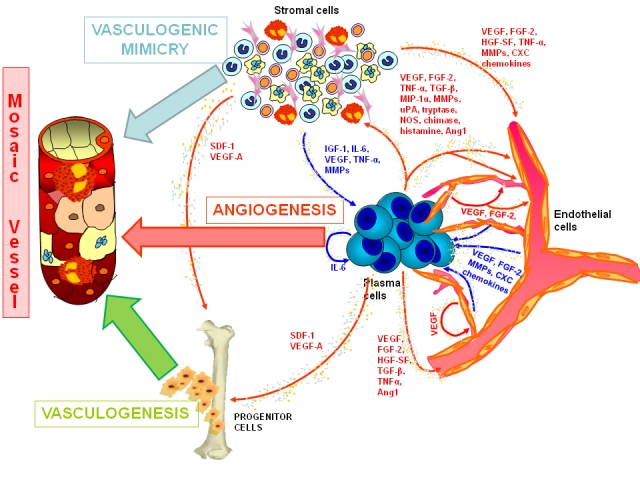
The vascular three formation in the bone marrow of multiple myeloma patients.
Angiogenesis is uncontrolled and unlimited in time, and essential for tumor growth, invasion and metastasis during the transition from the avascular to the vascular phase [4]. The angiogenic switch is preceded by the expression of oncogenes (c-myc, c-fos, c-jun, ets-1) coding for angiogenic factors, and activated as a consequence of immunoglobulin translocations and genetic instability of plasma cells [13]. So tumor plasma cells acquire an angiogenic phenotype due to clonal expansion and epigenetic modifications (hypoxia, shear stress) [14]. There is a shift from CD45-positive to CD45-negative plasma cells that produce VEGF [15]. VEGF stimulates proliferation and chemotaxis in both endothelial cells and stromal cells [14]. These cells are rapidly phosphorylated by the interaction with VEGF, and signal via extracellular signal-related kinase-2 [16]. VEGF acts as an autocrine inducer of growth and chemotaxis via VEGER-1 [17]. It increases IL-6 (a major growth and survival factor for MM plasma cells) production by bone marrow stromal cells via VEGFR-2 and thus forming a paracrine loop for tumor growth [18] and angiogenesis. Moreover, adhesion of plasma cells to bone marrow stromal cells increases VEGF secretion by both cell types [19], and so enhances angiogenesis. VEGF production by plasma cells is also regulated by TNF-α of bone marrow stromal cells [20]. TNF- α mediates upregulation of adhesion molecules of plasma cells and bone marrow stromal cells, and thus enhances heterotypic adhesions and activates IL-6 secretion by bone marrow stromal cells [21]. TNF-α secreted by plasma cells induces upregulation of adhesion molecules on both MM plasma cells and bone marrow stromal cells [22], thereby increasing the binding of MM plasma cells to bone marrow stromal cells with associated cell adhesion mediated-drug resistence and induction of IL-6 and VEGF secretion by bone marrow stromal cells [23,24], which mediates MM cell homing and migration, as well as angiogenesis [25]. VEGF signalling also contributes to inhibit antiangiogenic signals such as semaphorin3A (SEMA3A) whose autocrine loops are usually activated to self-limit physiologic angiogenesis [26].
FGF-2 is an other important angiogenic growth factor, and it represent a potent activator of endothelial proliferation and can thus stimulate angiogenesis, promote stromal fibroblast proliferation and extracellular matrix formation leading to excessive bone marrow fibrosis and can directly affect neoplastic cells by acting on their high affinity FGFRs [27]. FGF-2 increases IL-6 secretion; conversely IL-6 enhances FGF-2 expression and secretion by MM plasma cells [27], thus forming a paracrine IL-6/FGF-2 crosstalk between MM plasma cells and bone marrow stromal cells that triggers neovascularisation as well as MM cell growth and survival [28].
miRNA and angiogenesis
The recent discovery of microRNA (miRNA) genes, encoding for a class of small non-coding RNAs involved in the regulation of cell cycle, survival and differentiation programmes has added a further level of complexity to normal and cancer cell biology. Through complementary base pairing to specific protein-coding transcripts, miRNAs direct mRNA silencing by message degradation and translational repression [29]. Impaired miRNA expression has already been demonstrated in a number of solid tumors and, more recently, in some hematological disorders [30-32]. To date, only little evidence of miRNA expression/deregulation in MM has been reported: recently, it has been demonstrated that miR-21 can be induced by STAT3 and mediate IL-6-dependent human myeloma cell lines (HMCLs) survival [33]. Successively, Pichiorri et al reported a miRNA microarrays and quantitative PCR (Q-RT-PCR) analysis of HMCLs and PCs from patients with MM, MGUS and normal controls, showing a set of differentially expressed miRNAs that can be associated with neoplastic transformation and progression [34]. Recently, an integrative genomic approach that revealed coordinated expression of some intronic miRNAs with their deregulated host genes has been performed [35]. In particular, it has been monitored host transcript expression values generated on Affymetrix oligonucleotide microarrays in a panel of 20 HMCLs and identified miRNA host genes whose expression varied significantly across the dataset. Moreover, the expression levels of the corresponding intronic miRNAs by Q-RT-PCR has been evaluated, and it has been identified a significant correlation between the expression levels of MEST, EVL, and GULP1 genes and those of the corresponding miRNAs miR-335, miR-342-3p, and miR-561, respectively. Notably, miRNAs and their host genes were overexpressed in a fraction of primary tumors with respect to normal plasma cells, and interestingly, the predicted putative miRNA targets and the transcriptional profiles associated with the primary tumors suggested that MEST/miR-335 and EVL/miR-342-3p may play a role in plasma cell homing and/or interactions with the bone marrow microenvironment.
These first evidences suggest that, as already extensively observed in other tumours, miRNAs could play a critical role also in MM, and their expression profiling could add a further level to our understanding of its pathogenesis. Important evidences have been shown by Roccaro et al, who has identified a MM-specific microRNA signature characterized by down-expression of microRNA-15a/-16 and overexpression of microRNA-222/-221/-382/-181a/-181b. MicroRNA-15a and -16 regulate proliferation and growth of MM cells by inhibiting AKT serine/ threonineprotein-kinase (AKT3), ribosomal-protein-S6, MAP-kinases, and NF-κB-activator MAP3KIP3. Moreover, miRNA-15a and -16 exert their anti-MM activity even in the context of the bone marrow microenvironment. They reduce VEGF secretion from MM cells at the protein level, thereby reducing MM cell-induced proangiogenic activity on endothelial cells. So miRNA-15a and -16 are critical regulators of MM pathogenesis both directly by targeting clonal plasma cells, and indirectly by reducing BM neoangiogenesis and the interaction between tumor cells and BM milieu [36].
Bone marrow microenvironment
The MM microenvironment is characterized by the presence of plasma cells, ECM proteins, hematopoietic stem cells and bone marrow stromal cells, including fibroblasts, osteoblasts, osteoclasts, chondrocytes, endothelial cells, endothelial progenitor cells, T lymphocytes, neutrophils, macrophages and mast cells [37]. We have already explained some of the interactions between these components, which determine the proliferation, migration and survival of plasma cells, as well as drug resistance and formation of bone disease [38]. So a permissive stromal environment is important in supporting tumor progression in combination with genetic alterations [39].
Endothelial cells
Tumor endothelial cells differ greatly from those of quiescent healthy vessels [40]. They proliferate rapidly in keeping with the enhanced angiogenesis that accompanies tumor progression [41]. Their intercellular adhesion and to the ECM during sprouting (that implies cell proliferation and migration) is greatly reduced since they have different profile and level of cell adhesion molecules [42]. Their survival is markedly dependent on growth factors secreted by the tumor and its microenvironment, and on their expression of specific receptors for these factors [43]. They are abnormal in shape and highly permeable due to the presence of fenestrae, vesicles, transcellular holes, widened intercellular junctions, and a discontinuous basement membrane [44]. They share the lining of new vessels with tumor cells able to mimic vessels [45]. The fast growth of endothelial cells and tumour cells, coupled with their structural and functional abnormalities make tumor vessels thin, tortuous, and arborized [8]. As a consequence, tumor blood flow is chaotic and variable and leads to hypoxic and acidic environment that stimulate further angiogenesis [46].
MM endothelial cells intensely express markers of vivid angiogenesis such as VEGFR-2 and Tie/ Tek. This implies synergistic activity of VEGF and Ang-2, produced by plasma cells, in the induction of sprouts from existing vessels [40]. MM endothelial cells sizably express CD133, a marker of the progenitor endothelial cells involved in pre-natal vasculogenesis [55]. It has been proved that some CD133+ hematopoietic stem and progenitor cells contribute to the formation of the vessel wall of newly forming blood vessels together with FVIII-RA+, VEGFR-2+, and VE-cadherin+ MM endothelial cells [47]. MM plasma cells and inflammatory cells secrete high levels of VEGF, FGF-2, and insulin-like growth factor (IGF), which recruit bone marrow and circulating hematopoietic stem and progenitor cells into the tumor microenvironment [3], where they differentiate into MM endothelial cells and participate to the formation of the new vessel wall. High expression of β3-integrin, which prevents apoptosis of endothelial cells and favours their adhesion to the ECM, proliferation, migration, and capillarogenesis [14], also implies vivid neovascularization. Overexpression of endoglin, that enhances the expression of the adhesion molecule CD31, which is the ligand of the plasma cell CD38, by endothelial cells suggests enhanced opportunities for plasma cells to interact with the new-formed blood vessels, enter circulation and disseminate [40]. Frequent interactions between plasma cells and new-formed blood vessels are also mediated by the high expression of E-selectin by endothelial cells [48]. Moreover, MM endothelial cells intensely express a water transporter, namely aquaporin 1, which enhances vascular permeability, facilitates plasma extravasation, increases interstitial pressure, induces hypoxia, and upregulates hypoxia inducible factor-1 alpha (HIF-1α) and VEGF [49].
A paracrine loop for tumor angiogenesis and growth has been demonstrated in MM patients, mediated by VEGF-A and FGF-2 [50,51]. Plasma cells secrete VEGF-A and this induces endothelial cell proliferation and chemotaxis through VEGFR-2, prevalently expressed on these cells, which display constitutive autophosphorylation of VEGFR-2 and the associated kinase ERK-2 [50, 52].
Another important role is played by the paracrine loop existing between MM endothelial cells and plasma cells involving CXC-chemokines and their cognate receptors, which mediate plasma cell proliferation and chemotaxis [53]. Bone marrow endothelial cells express and secrete high amounts of the CXC-chemokines CXCL8/IL-8, CXCL11/interferon-inducible T-cell alpha chemoattractant (I-TAC), CXCL12/stromal cell-derived factor (SDF)-1α, and CCL2/monocyte chemotactic protein (MPC)-1 [64]. Several MM cell lines display a complex expression pattern of chemokine receptors (CXCR, CCR) [64], some of which also mediate the interactions between plasma cells and stromal cells in the bone marrow microenvironment [54].
To summarize, MM endothelial cells show constitutively ultrastructural features of enhanced metabolic activation, an high expression of typical endothelial markers (Tie2/Tek, VEGFR-2, FGFR-2, CD105-endoglin, and VE-cadherin), an high secretion of matrix metalloproteinases-2 and -9, and up-regulation of angiogenic genes (VEGF, FGF-2, Gro-α chemokine, transforming growth factor beta (TGF-β), Tie2/Tek, HIF-1α, ETS-1, and osteopontin) [40]
Progenitor cells
Vasculogenesis, i.e. the in situ differentiation of the primitive endothelial progenitors known as angioblasts from groups of mesodermal cells into endothelial cells that aggregate into a primitive capillary plexus, is responsible for the primary development of the vascular system during embryogenesis [55].
This process is particularly important for vascular development, e.g., the formation of the yolk sac vasculature, of the heart, and of the dorsal aortae [56]. Important evidences suggest how the bone marrow neovascularization is partly formed by postnatal vasculogenesis [57].
Various studies have suggested that endothelial stem cells may persist into adult life, where they contribute to the formation of new blood vessels [58], and that in the post-natal life vasculogenesis may also occur [57]. Isolation of putative endothelial progenitor cells from peripheral blood was initially suggested by Asahara et al [59]. Peripheral blood CD34+ cells expressing VEGFR-2 were cultured on fibronectin-coated plates for 4 weeks, and the attached cells sotein couplehowed a typical spindle-shaped morphology, the uptake of acetylated low-density lipoproteins (Ac-LDL) and the expression of several endothelial cell markers (CD34, CD31, Flk-1, Tie-2 and E-selectins). More recently, it has been confirmed that 01-0-5% of circulating CD34+ cells express VEGFR-2/KDR receptors and that pluripotent haematopoietic stem cells are restricted to this fraction [60].
It has been demonstrated that angiogenesis is an important process in MM progression and represents an important prognostic factor [3,61]. Moreover, the in vitro generation of endothelial cells from haematopoietic stem cells mobilized in MM patients and their expansion and differentiation into endothelial cells in the presence of angiogenic cytokines has been obtained [47]. These data also demonstrate that in the bone marrow of MM patients, but not of MGUS patients, some isolated endothelial cells express on their surface the typical endothelial cell markers, such as factor VIII-related antigen (FVIII-RA), vascular endothelial-cadherin (VE-cadherin), VEGFR-2, and TIE/Tek, as well as the CD133 staminal antigen whose expression was found in the microvascular wall together with FVIII-RA or VE-cadherin in some active MM patients.
Monocyte/macrophages
Macrophages contribute to tumor angiogenesis, and there are several reports describing an association between macrophage infiltration, vascularity and prognosis [39]. Tumor-associated macrophages accumulate in poorly vascularised hypoxic or necrotic areas [62] and respond to experimental hypoxia by increasing the release of VEGF and FGF-2 and a broad range of other factors, such as tumor necrosis factor alpha (TNF-α), urokinase and matrix metalloproteinases [63]. Moreover, activated macrophages synthesize and release inducible nitric oxide synthase, which increases blood flow and promotes angiogenesis [64]. Lastly, macrophages recruit mast cells [65].
We have demonstrated that bone marrow macrophages in patients with active MM contribute to build neovessels through vasculogenic mimicry, in parallel to progression of plasma cell tumors [66]. Macrophages display oblong and spindle shape with thin cytoplasmic expansions, some of which are either arranged to form microvessel-like lumen or anastomosed with each other and with those of nearby macrophages to form tubular-like structures [66]. Macrophages retain their own CD14 and CD68 lineage markers, indicating that they do not transdifferentiate into endothelial cells, but only adapt functionally, phenotypically and morphologically [66].
Under a synergistic stimulation by VEGF-FGF-2, macrophages undergo a phenotypic and functional adaptation [67], starting to behave like MM endothelial cells. VEGF and FGF-2 bind to VEGFR-1 and FGFR-1, -2 and -3 expressed on monocytes/macrophages surface [68]. VEGFR-1 is involved in macrophage chemotaxis [68] and vasculogenesis [69], but not in the definitive vessel assembly, which is closely dependent on VEGFR-2 [70]. On the other hand, FGF-2/FGFRs system is involved in vasculogenesis [71].
In active MM, plasma cells secrete VEGF and FGF-2 [72,61] and induce macrophage to secrete their own VEGF and FGF-2 [3].
In healthy subjects, cells of monocyte lineage (other mesodermal-derived cells) can generate endothelial cell progenitors [73] or act as pluripotent stem cells [74]. They can develop an endothelial cell phenotype, especially when stimulated by VEGF and/or bFGF [74,75], and produce a functional capillary-like mesh [76] permeable by blood cells [77], hence recapitulating embryo vasculogenesis [55].
Bone marrow monocytes and macrophages of MM patients can be induced to assume a number of endothelial cell properties and form capillary-like structures in vitro through vasculogenesis. Moreover, macrophages contribute to build neovessels in MM through vasculogenic mimicry, and in MGUS they are prone to a vascular switch that marches in step with the progression toward MM [69]. In fact, MM bone marrow macrophages exposed to VEGF and bFGF develop a number of phenotypic properties similar to those of paired bone marrow endothelial cells, and form capillary-like structures morphologically mimicking those produced by MM endothelial cells. At the ultrastructural level, MM macrophages exhibit numerous cytoplasmic extroversions arranged in tube-like structures [69]. All these features are lacking or minimal in macrophages of patients with MGUS or with benign anemia which, however, will become phenotypically and functionally similar to those of MM under angiogenic stimulation [69]. Bone marrow biopsies of MM, but not of MGUS, harbour ‘mosaic’ vessels since these are formed by MM endothelial cells, endothelial cell-like macrophages and macrophages themselves [69].
Mast cells
Mast cells density is strictly correlated with the extent of pathological angiogenesis, occurring in chronic inflammation and tumors [78,79]. Mast cells accumulation has been associated with enhanced growth and invasion of several solid and haematological malignancies [63] and they also act as a host response to neoplasia and display tumoricidal activity in experimental settings [80]. Mast cells are recruited via several mediators produced by tumor cells, such as c-kit receptor or Stem Cell Factor (SCF) [79, 81], FGF-2, VEGF and platelet derived growth factor (PDGF). Mast cells contain several angiogenic factors including tryptase, chymase, heparin and histamine [82, 83], TGF-β, TNF-α [84], IL-8 [85], FGF-2 [86] and VEGF [87]. Heparin may induce endothelial cell proliferation and migration [80]; histamine has angiogenic effect through both H1 and H2 receptors [82] and also contribute to the hyperpermeability of new formed microvessels during tumor angiogenesis, increasing leakage of plasma proteins and hence deposition of fibrin [80]. Degradation products of fibrin, in turn, are angiogenic in vivo [88]. Moreover, in vitro experiments demonstrated that histamine induces VEGF production in the granulation tissue [89]. Tryptase is the predominant protease in mast cells and it is a potent mitogen for fibroblasts, smooth muscle cells, and epithelial cells [90,91] and could play an important role in neovascularization favouring the formation of capillary structures via a direct action on endothelial cells [92] or by activating latent metalloproteinases and plasminogen activator [93]. It has been demonstrated that the new-vessels wall appear lined also by typical tryptase-positive mast cells, which are connected by a junctional system with the endothelial cells. Because mast cells keep their lineage marker, they can be regarded as cells that do not transdifferentiate into endothelial cells. This behaviour of mast cells can thus be regarded as an example of vasculogenesis mimicry [94].
It has been also demonstrated that bone marrow angiogenesis, evaluated as microvessel area, and mast cells counts are highly correlated in patients with MM [63]. Both parameters increase simultaneously in active MM [93]. Moreover it has been demonstrated a significant correlation between vessel count and the number of both mast cells and VEGF-expressing cells revealing that mast cells express VEGF mRNA [95].
Targeting antiangiogenesis
The actual therapeutic strategies of MM consists of conventional chemotherapy in combination with biologically based therapies in various settings, targeting not only the MM plasma cells but also its microenvironment and new therapeutic targets are currently available [77].
Proteasome inhibitor bortezomib (Velcade, formerly PS-341), a boronic acid dipeptide, is a potent, highly selective, and reversible proteasome inhibitor that targets 26S proteasome complex and inhibits its function [96]. The 26S proteasome is an ATP-dependent multicatalytic protease mediating intracellular protein degradation [97]. Proteasomal degradation of mis-folded or damaged proteins proceeds by recognition of polyubiquitinated proteins by the 19S regulatory subunit of the 26S protease and subsequently hydrolysis to small polypeptides [97]. Besides eliminating damaged/misfolded proteins, the proteasome also regulates key cellular processes, including modulation of transcription factors, such as NF-κB, cell cycle progression, inflammation, immune surveillance, growth arrest, and apoptosis [98].
Bortezomib has inhibitory effects on the NF-κB activity in MM cells. NF-κB is a major transcriptional factor which mediates the expression of many proteins including cytokines, chemokines, cell adhesion molecules, as well as those involved in anti-apoptosis and cellular growth control [98]. Its activity is regulated by association with IκB family proteins [99]. Varius stimuli, including cytokines, such as TNFα and IL-1β, trigger phosphorylation of IκB protein by IκB kinase [100]. Phosphorylated IκB is subsequently polyubiquitinated by specific enzymes and de-gradated by the 26S proteasome [100], which allows p50/p65 NF-κB nuclear translocation and binding to consensus motifits in the promoter region of target genes [98]. Expression of adhesion molecules, such as ICAM-1 and VCAM-1, on both MM cells and bone marrow stromal cells are also regulated by NF-κB [22, 101]. Thus inhibition of NF-κB by bortezomib down-regulates these adhesion molecules, thereby enhancing susceptibility of MM cells to therapeutic agents in the context of the bone marrow milieu [98]. Another important aspect is that induction of IL-6 transcription and secretion by bone marrow stromal cells is mediated via NF-κB activation which, in turn, increases secretion of other cytokines, such as VEGF, from MM plasma cells [18]. Furthermore, MM cell adherence to the bone marrow stromal cells triggers IL-6 secretion via NF-κB activation, associated with an increased MM cell growth [98] and leads to a reduction of VEGF secretion. Bortezomib significantly blocks both constitutive and MM cell adhesion induced by IL-6 secretion from bone marrow stromal cells [98]. Bortezomib is also directly cytotoxic, triggering stress response and apoptotic signalling via multiple pathways [98]. As the result of inhibition of proteasome activity, it causes the accumulation of misfolded polyubiquinated proteins, resulting in endoplasmic reticulum stress which triggers caspase-4 and downstream signalling [102]. Bortezomib also induces ROS which play a critical role in the initiation of the apoptotic cascades by disruption of membrane potential and the release of cytochrome c from mitochondria, followed by caspase-9 activation [103]. Proteasome inhibitors have a potent activity against mitotic endothelial cells, so they target aberrant blood vessel development associated with tumor growth, in fact, bortezomib inhibits the proliferation of MM endothelial cells associated with downregulation of VEGF, IL-6, IGF-I, Ang-1 and Ang-2 [98]. Moreover, bortezomib inhibits DNA repair activity by cleavage of DNA dependent protein kinase catalytic subunit (DNA-PKcs), thereby restoring sensitivity to DNA-damaging chemotherapeutic agents, such as doxorubicin and melphalan [104]. Bortezomib also down-regulates caveolin-1 tyrosine phosphorylation, which is required for VEGF-mediated MM cell migration, and also blocks the caveolin-1 phosphorylation induced by VEGF (transcriptional target of NF-κB) in endothelial cells, thereby inhibiting ERK-dependent cell proliferation. It inhibits the transcription of important adhesion molecules such as ICAM-1, VCAM1 and E-selectin [105].
Thalidomide has a direct tumoricidal activity, an antiangiogenic effect and modulates TNF-α signalling through direct and/or indirect effects on the tumour microenvironment [106], reduces FGF-2 [107], VEGF and IL-6 secretion in bone marrow stromal cells and by MM cells [108]. It also stimulates the activation and expansion of T cells and augments NK-cell -mediated cytotoxicity through its direct effect on T cells with a consequent increase in IL-2 and interferon gamma (IFN-γ) secretion [109], and interferes with NF-κB activity by blocking its ability to bind to DNA or suppresses IκB kinase activity, thus abrogating normal inflammatory cytokine production [110]. Thalidomide also disrupts the host marrow-MM cell interactions by selective modulation of the density of cell surface adhesion molecules [111]. Treatment with thalidomide is associated with sedation, fatigue, constipation, rash, deep-vein thrombosis, and peripheral neuropathy [112]. Lenalidomide, a derivative of thalidomide, is less toxic and more potent than the parent drug [113]. In patients with relapsed or refractory MM, lenalidomide can overcome resistance not only to conventional chemotherapy but also to thalidomide [114,115].
The bisphosphonates are other compounds that, although originally used to reduce bone loss in MM due to an anti-osteoclast activity, have also been shown to have a direct effect on MM cells [116]. In fact, zoledronic acid has a direct cytotoxic activity on tumor cells and suppresses angiogenesis [117, 118], inhibits FGF-2 - and VEGF-dependent proliferation of endothelial cells and inhibits VEGFR-2 in an autocrine loop [116]. Neridronate exerts its antiangiogenic activity through both a direct effect on endothelial cell proliferative activity and inhibitory effect on the responsivity of the endothelial cells to the proliferative stimuli mediated by angiogenic cytokines [119]. The use of bisphosphonates can cause the osteonecrosis of the jaw (ONJ), a long-lasting disorder that occurs mainly in breast cancer and MM patients treated with intravenous bisphosphonates [120].
Conclusions
The bone marrow microenvironment plays a crucial role in the pathophysiology of MM. It is involved in the crosstalk between plasma cells and bone marrow stromal cells, which increases the survival, proliferation and migration of tumor cells themselves, and represents the substrate for angiogenesis which favours the disease progression. Due to interaction with active microenvironment, MM plasma cells also acquire drug resistance giving less opportunity to therapy response. Many research studies have tried to better understand the biological mechanisms and the genetic basis of all the interactions between MM cells and bone marrow stromal cells. VEGF, FGF-2, IL-6, macrophages, mast cells, and many others cells and molecules, play the most important role in this process. Furthermore, several studies have focused their investigation on novel drugs targeting the MM plasma cells and the microenvironment cells. Good results have been already obtained, but MM still remains an incurable malignancy, indicating that the role of bone marrow microenvironment is important in MM progression, but its role is still not completely clear. The future goal for MM therapy may be the simultaneous block of plasma cell proliferation and survival, plasma cells/ bone marrow stromal cells interaction, and bone marrow stromal cells activity by the combination of biological target drugs.
Acknowledgments
The authors would like to thank Prof. C. Perillo for editing the English version of this manuscript.
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC), Investigator Grant, and Special Program Molecular Clinical Oncology 5 per mille n. 9965, Milan, and the Ministry of Health, Progetto Oncologia 2006, Humanitas Mirasole S.p.A., Rome, Italy.
Disclosure of potential conflict of interest
The authors declare no competing financial interests. The authors are fully responsible for the content and editorial decisions for the manuscript.
References
- 1.Kastrinakis NG, Gorgoulis VG, Foukas PG, Dimopoulos MA, Kittas C. Molecular aspects of multiple myeloma. Ann Oncol. 2000;11:1217–1228. doi: 10.1023/a:1008331714186. [DOI] [PubMed] [Google Scholar]
- 2.Dimopoulos MA, Kastritis E, Anagnostopoulos A. Hematological malignancies: myeloma. Ann Oncol Suppl. 2006;10:137–143. doi: 10.1093/annonc/mdl251. [DOI] [PubMed] [Google Scholar]
- 3.Ribatti D, Nico B, Vacca A. Importance of the bone marrow microenvironment in inducing the angiogenic response in multiple myeloma. Oncogene. 2006;25:4257–4266. doi: 10.1038/sj.onc.1209456. [DOI] [PubMed] [Google Scholar]
- 4.Vacca A, Ribatti D. Bone marrow angiogenesis in multiple myeloma. Leukemia. 2006;20:193–199. doi: 10.1038/sj.leu.2404067. [DOI] [PubMed] [Google Scholar]
- 5.The International Myeloma Working Group. Criteria for the classification of monoclonal gammo-pathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121:749–757. [PubMed] [Google Scholar]
- 6.Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23:3–9. doi: 10.1038/leu.2008.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stasi R, Amadori S. The role of angiogenesis in hematologic Malignancies. J of Hematotherapy & stem cell research. 2002;11:49–68. doi: 10.1089/152581602753448531. [DOI] [PubMed] [Google Scholar]
- 8.Vacca A, Ribatti D, Roncali L, Ranieri G, Serio G, Silvestris F, Dammacco F. Bone marrow angiogenesis and progression in multiple myeloma. Br J Haematol. 1994;87:503–508. doi: 10.1111/j.1365-2141.1994.tb08304.x. [DOI] [PubMed] [Google Scholar]
- 9.Risau W, Sariola H, Zerwes HG, Sasse J, Ekblom P, Kemler R, Doetschman T. Vasculogenesis and angiogenesis in embryonic-stem-cell-derived embryoid bodies. Development. 1988;102:471–478. doi: 10.1242/dev.102.3.471. [DOI] [PubMed] [Google Scholar]
- 10.Iruela-Arispe ML, Dvorak HF. Angiogenesis: a dynamic balance of stimulators and inhibitors. Thromb Haemost. 1997;78:672–677. [PubMed] [Google Scholar]
- 11.Folkman J, Browder T, Palmblad J. Angiogenesis research: guidelines for translation to clinical application. Thromb Haemost. 2001;86:23–33. [PubMed] [Google Scholar]
- 12.Döme B, Hendrix MJ, Paku S, Tóvári J, Tímár J. Alternative vascularisation mechanisms in cancer: pathology and therapeutic implications. Am J Pathol. 2007;170:1–15. doi: 10.2353/ajpath.2007.060302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vacca A, Ribatti D, Roccaro AM, Frigeri A, Dammacco F. Bone marrow angiogenesis in patients with active multiple myeloma. Semin Oncol. 2001;28:543–550. doi: 10.1016/s0093-7754(01)90022-3. [DOI] [PubMed] [Google Scholar]
- 14.Hynes RO. A reevaluation of integrins as regulators of angiogenesis. Nat Med. 2002;8:918–921. doi: 10.1038/nm0902-918. [DOI] [PubMed] [Google Scholar]
- 15.Asosingh K, De Raeve H, Menu E, Van Riet I, Van Marck E, Van Camp B, Vanderkerken K. Angiogenic switch during 5T2MM murine myeloma tumorigenesis: role of CD45 heterogeneity. Blood. 2004;103:3131–3177. doi: 10.1182/blood-2003-08-2946. [DOI] [PubMed] [Google Scholar]
- 16.Vacca A, Ria R, Ribatti D, Semeraro F, Djonov V, Di Raimondo F, Dammacco F. A paracrine loop in the vascular endothelial growth factor pathway triggers tumor angiogenesis and growth in multiple myeloma. Haematologica. 2003;88:176–185. [PubMed] [Google Scholar]
- 17.Podar K, Tai YT, Davies FE, Lentzsch S, Sattler M, Hideshima T, Lin BK, Gupta D, Shima Y, Chauhan D, Mitsiades C, Raje N, Richardson P, Anderson KC. Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood. 2001;98:428–435. doi: 10.1182/blood.v98.2.428. [DOI] [PubMed] [Google Scholar]
- 18.Dankbar B, Padró T, Leo R, Feldmann B, Kropff M, Mesters RM, Serve H, Berdel WE, Kienast J. Vascular endothelial growth factor and inter-leukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood. 2000;95:2630–2636. [PubMed] [Google Scholar]
- 19.Hideshima T, Podar K, Chauhan D, Anderson KC. Cytokines and signal transduction. Best Pract Res Clin Haematol. 2005;18:509–524. doi: 10.1016/j.beha.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 21.Hideshima T, Nakamura N, Chauhan D, Anderson KC. Biologic sequelae of interleukin-6 induced PI3-K/Akt signaling in multiple myeloma. Oncogene. 2001;20:5991–6000. doi: 10.1038/sj.onc.1204833. [DOI] [PubMed] [Google Scholar]
- 22.Hideshima T, Chauhan D, Schlossman R, Richardson P, Anderson KC. The role of tumor necrosis factor alpha in the pathophysiology of human multiple myeloma: therapeutic applications. Oncogene. 2001;20:4519–4527. doi: 10.1038/sj.onc.1204623. [DOI] [PubMed] [Google Scholar]
- 23.Chauhan D, Uchiyama H, Akbarali Y, Urashima M, Yamamoto K, Libermann TA, Anderson KC. Multiple myeloma cell adhesion-induced inter-leukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood. 1996;87:1104–1112. [PubMed] [Google Scholar]
- 24.Hideshima T, Chauhan D, Hayashi T, Podar K, Akiyama M, Gupta D, Richardson P, Munshi N, Anderson KC. The biological sequelae of stromal cell-derived factor-1alpha in multiple myeloma. Mol Cancer Ther. 2002;1:539–544. [PubMed] [Google Scholar]
- 25.Tai YT, Podar K, Gupta D, Lin B, Young G, Akiyama M, Anderson KC. CD40 activation induces p53-dependent vascular endothelial growth factor secretion in human multiple myeloma cells. Blood. 2002;99:1419–27. doi: 10.1182/blood.v99.4.1419. [DOI] [PubMed] [Google Scholar]
- 26.Vacca A, Scavelli C, Serini G, Di Pietro G, Cirulli T, Merchionne F, Ribatti D, Bussolino F, Guidolin D, Piaggio G, Bacigalupo A, Dammacco F. Loss of inhibitory semaphorin 3A (SEMA3A) autocrine loops in bone marrow endothelial cells of patients with multiple myeloma. Blood. 2006;108:1661–1667. doi: 10.1182/blood-2006-04-014563. [DOI] [PubMed] [Google Scholar]
- 27.Ribatti D, Vacca A, Rusnati M, Presta M. The discovery of basic fibroblastic growth factor/ fibroblast growth factor-2 and its role in haema-tological malignancies. Cytokine Growth Factor. Rev2007;18:327–334. doi: 10.1016/j.cytogfr.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 28.Mitsiades CS, Mitsiades NS, Munshi NC, Richardson PG, Anderson KC. The role of the bone marrow microenvironment in the pathophysiology and therapeutic management of multiple myeloma: interplay of growth factors, their receptors and stromal interactions. Eur J Cancer. 2006;42:1564–1573. doi: 10.1016/j.ejca.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 29.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 30.Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M, Iuliano R, Palumbo T, Pichiorri F, Roldo C, Garzon R, Sevignani C, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 31.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 32.Calin GA, Croce CM. MicroRNAs and chromosomal abnormalities in cancer cells. Oncogene. 2006;25:6202–6210. doi: 10.1038/sj.onc.1209910. [DOI] [PubMed] [Google Scholar]
- 33.Löffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermüller J, Kretzschmar AK, Burger R, Gramatzki M, Blumert C, Bauer K, Cvijic H, Ullmann AK, Stadler PF, Horn F. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood. 2007;110:1330–1333. doi: 10.1182/blood-2007-03-081133. [DOI] [PubMed] [Google Scholar]
- 34.Pichiorri F, Suh SS, Ladetto M, Kuehl M, Palumbo T, Drandi D, Taccioli C, Zanesi N, Alder H, Hagan JP, Munker R, Volinia S, Boccadoro M, Garzon R, Palumbo A, Aqeilan RI, Croce CM. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc Natl Acad Sci U.S.A. 2008;105:12885–12890. doi: 10.1073/pnas.0806202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ronchetti D, Lionetti M, Mosca L, Agnelli L, Andronache A, Fabris S, Deliliers GL, Neri A. An integrative genomic approach reveals coordinated expression of intronic miR-335, miR-342, and miR-561 with deregulated host genes in multiple myeloma. BMC Med Genomics. 2008;1:37. doi: 10.1186/1755-8794-1-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roccaro AM, Sacco A, Thompson B, Leleu X, Azab AK, Azab F, Runnels J, Jia X, Ngo HT, Melhem MR, Lin CP, Ribatti D, Rollins BJ, Witzig TE, Anderson KC, Ghobrial IM. MicroRNAs 15a and 16 regulate tumor proliferation in multiple myeloma. Blood. 2009;113:6669–6680. doi: 10.1182/blood-2009-01-198408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 38.Shalaby F, Ho J, Stanford WL, Fischer KD, Schuh AC, Schwartz L, Bernstein A, Rossant J. A requirement for Flk-1 in primitive and definitive hematopoiesis and vasculogenesis. Cell. 1999;89:981–990. doi: 10.1016/s0092-8674(00)80283-4. [DOI] [PubMed] [Google Scholar]
- 39.Gendron RL, Tsai FY, Paradis H, Arceci RJ. Induction of embryonic vasculogenesis by bFGF and LIF in vitro and in vivo. Dev Biol. 1996;177:332–346. doi: 10.1006/dbio.1996.0167. [DOI] [PubMed] [Google Scholar]
- 40.Vacca A, Ria R, Semeraro F, Merchionne F, Coluccia M, Boccarelli A, Scavelli C, Nico B, Gernone A, Battelli F, Tabilio A, Guidolin D, Petrucci MT, Ribatti D, Dammacco F. Endothelial cells in the bone marrow of patients with multiple myeloma. Blood. 2003;102:3340–3348. doi: 10.1182/blood-2003-04-1338. [DOI] [PubMed] [Google Scholar]
- 41.Holmgren L, O'Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1:149–153. doi: 10.1038/nm0295-149. [DOI] [PubMed] [Google Scholar]
- 42.Dejana E. Endothelial adherens junctions: implications in the control of vascular permeability and angiogenesis. J Clin Invest. 1996;98:1949–1953. doi: 10.1172/JCI118997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ausprunk DH, Falterman K, Folkman J. The sequence of events in the regression of corneal capillaries. Lab Invest. 1978;38:284–294. [PubMed] [Google Scholar]
- 44.Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156:1363–1380. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Folberg R, Hendrix MJ, Maniotis AJ. Vasculogenic mimicry and tumor angiogenesis. Am J Pathol. 2000;156:361–381. doi: 10.1016/S0002-9440(10)64739-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. 1997;3:177–182. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- 47.Ria R, Piccoli C, Cirulli T, Falzetti F, Mangialardi G, Guidolin D, Tabilio A, Di Renzo N, Guarini A, Ribatti D, Dammacco F, Vacca A. Endothelial differentiation of hematopoietic stem and progenitor cells from patients with multiple myeloma. Clin Cancer Res. 2008;14:1678–1685. doi: 10.1158/1078-0432.CCR-07-4071. [DOI] [PubMed] [Google Scholar]
- 48.Carlos T, Kovach N, Schwartz B, Rosa M, Newman B, Wayner E, Benjamin C, Osborn L, Lobb R, Harlan J. Human monocytes bind to two cyto-kine-induced adhesive ligands on cultured human endothelial cells: endothelial-leukocyte adhesion molecule-1 and vascular cell adhesion molecule-1. Blood. 1991;7:2266–2271. [PubMed] [Google Scholar]
- 49.Vacca A, Frigeri A, Ribatti D, Nicchia GP, Nico B, Ria R, Svelto M, Dammacco F. Microvessel overexpression of aquaporin 1 parallels bone marrow angiogenesis in patients with active multiple myeloma. Br J Haematol. 2001;113:415–421. doi: 10.1046/j.1365-2141.2001.02738.x. [DOI] [PubMed] [Google Scholar]
- 50.Vacca A, Ria R, Ribatti D, Semeraro F, Djonov V, Di Raimondo F, Dammacco F. A paracrine loop in the vascular endothelial growth factor path way triggers tumor angiogenesis and growth in multiple myeloma. Haematologica. 2003;88:176–185. [PubMed] [Google Scholar]
- 51.Ribatti D, Nico B, Morbidelli L, Donnini S, Ziche M, Vacca A, Roncali L, Presta M. Cell-mediated delivery of fibroblast growth factor-2 and vascular endothelial growth factor onto the chick chorioallantoic membrane: endothelial fenestration and angiogenesis. J Vasc Res. 2001;38:389–397. doi: 10.1159/000051070. [DOI] [PubMed] [Google Scholar]
- 52.Ria R, Vacca A, Russo F, Cirulli T, Massaia M, Tosi P, Cavo M, Guidolin D, Ribatti D, Dammacco F. A VEGF-dependent autocrine loop mediates proliferation and capillarogenesis in bone marrow endothelial cells of patients with multiple myeloma. Thromb Haemost. 2004;92:1438–1445. doi: 10.1160/TH04-06-0334. [DOI] [PubMed] [Google Scholar]
- 53.Pellegrino A, Ria R, Di Pietro G, Cirulli T, Surico G, Pennisi A, Morabito F, Ribatti D, Vacca A. Bone marrow endothelial cells in multiple myeloma secrete CXC-chemokines that mediate interactions with plasma cells. Br J Haematol. 2005;129:248–256. doi: 10.1111/j.1365-2141.2005.05443.x. [DOI] [PubMed] [Google Scholar]
- 54.Vande Broek I, Asosingh K, Vanderkerken K, Straetmans N, Van Camp B, Van Riet I. chemokine receptor CCR2 is expressed by human multiple myeloma cells and mediates migration to bone marrow stromal cell-produced monocyte chemotactic proteins MCP-1, -2, -3. Br J Cancer. 2003;88:855–862. doi: 10.1038/sj.bjc.6600833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73–91. doi: 10.1146/annurev.cb.11.110195.000445. [DOI] [PubMed] [Google Scholar]
- 56.McDonald DM, Foss AJ. Endothelial cells of tumor vessels: abnormal but not absent. Cancer Metastasis Rev. 2001;19:109–120. doi: 10.1023/a:1026529222845. [DOI] [PubMed] [Google Scholar]
- 57.Ribatti D. Postnatal vasculogenesis. Mech Dev. 2001;100:157–163. doi: 10.1016/s0925-4773(00)00522-0. [DOI] [PubMed] [Google Scholar]
- 58.Gehling UM, Ergün S, Schumacher U, Wagener C, Pantel K, Otte M, Schuch G, Schafhausen P, Mende T, Kilic N, Kluge K, Schäfer B, Hossfeld DK, Fiedler W. In vitro differentiation of endothelial cells from AC133-positive progenitor cells. Blood. 2000;95:3106–3112. [PubMed] [Google Scholar]
- 59.Asahara T. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 60.Ziegler BL. KDR receptor: a key marker defining hematopoietic stem cells. Science. 1999;285:1553–1558. doi: 10.1126/science.285.5433.1553. [DOI] [PubMed] [Google Scholar]
- 61.Vacca A, Ribatti D, Presta M, Minischetti M, Iurlaro M, Ria R, Albini A, Bussolino F, Dammacco F. Bone marrow neovascularization, plasma cell angiogenic potential and matrix metalloproteinase-2 secretion parallel progression of human multiple myeloma. Blood. 1999;93:3064–3073. [PubMed] [Google Scholar]
- 62.Zhang H, Vakil V, Braunstein M, Smith EL, Maroney J, Chen L, Dai K, Berenson JR, Hussain MM, Klueppelberg U, Norin AJ, Akman HO, Ozçelik T, Batuman OA. Circulating endothelial progenitor cells in multiple myeloma: implications and significance. Blood. 2005;105:3286–3294. doi: 10.1182/blood-2004-06-2101. [DOI] [PubMed] [Google Scholar]
- 63.Ribatti D, Crivellato E, Roccaro AM, Ria R, Vacca A. Mast cell contribution to angiogenesis related to tumour progression. Clin Exp Allergy. 2004;34:1660–1664. doi: 10.1111/j.1365-2222.2004.02104.x. [DOI] [PubMed] [Google Scholar]
- 64.Leek RD, Landers RJ, Harris AL, Lewis CE. Necrosis correlates with high vascular density and focal macrophages infiltration in invasive carcinoma of the breast. Br J Cancer. 1999;79:991–995. doi: 10.1038/sj.bjc.6690158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bingle L, Brown NJ, Lewis CE. The role of tumor associated macrophages in tumor progression: implications for new anticancer therapies. J Pathol. 2002;196:254–65. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 66.Jenkins DC, Charles IG, Thomsen LL, Moss DW, Holmes LS, Baylis SA, Rhodes P, Westmore K, Emson PC, Moncada S. Role of nitric oxide in tumor growth. Proc Natl Acad Sci USA. 1995;92:4392–6. doi: 10.1073/pnas.92.10.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moldovan NI. Functional adaptation, the key to plasticity of cardiovascular ‘stem’ cells? Stem Cells Dev. 2005;14:111–121. doi: 10.1089/scd.2005.14.111. [DOI] [PubMed] [Google Scholar]
- 68.Gruber BL, Marchese MJ, Kew R. Angiogenic factors stimulate mast cell migration. Blood. 1995;86:2488–2493. [PubMed] [Google Scholar]
- 69.Scavelli C, Nico B, Cirulli T, Ria R, Di Pietro G, Mangieri D, Bacigalupo A, Mangialardi G, Coluccia AM, Caravita T, Molica S, Ribatti D, Dammacco F, Vacca A. Vasculogenic mimicry by bone marrow macrophages in patients with multiple myeloma. Oncogene. 2008;27:663–674. doi: 10.1038/sj.onc.1210691. [DOI] [PubMed] [Google Scholar]
- 70.Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marmé D. Migration of human mono-cytes in response to vescular endothelial growth factor (VEGF) is mediated via the VEGF receptor Flt-1. Blood. 1996;87:3336–3343. [PubMed] [Google Scholar]
- 71.Shalaby F, Ho J, Stanford WL, Fischer KD, Schuh AC, Schwartz L, Bernstein A, Rossant J. A requirement for Flk-1 in primitive and definitive hematopoiesis and vasculogenesis. Cell. 1999;89:981–990. doi: 10.1016/s0092-8674(00)80283-4. [DOI] [PubMed] [Google Scholar]
- 72.Bellamy WT, Richter L, Frutiger Y, Grogan TM. Expression of vascular endothelial growth factor and its receptors in haematological malignancies. Cancer Res. 1999;59:728–733. [PubMed] [Google Scholar]
- 73.Peichev M, Naiyer AJ, Pereira D, Zhu Z, Lane WJ, Williams M, Oz MC, Hicklin DJ, Witte L, Moore MA, Rafii S. Expression of VEGFR-2 and AC133 by circulating human CD34+ cells identifies a population of functional endothelial precursors. Blood. 2000;95:952–958. [PubMed] [Google Scholar]
- 74.Rehman J, Li J, Orschell CM, March KL. Peripheral blood ‘endothelial progenitor cells’ are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation. 2003;107:1164–1169. doi: 10.1161/01.cir.0000058702.69484.a0. [DOI] [PubMed] [Google Scholar]
- 75.Zhao Y, Glesne D, Huberman E. A human peripheral blood monocyte-derived subset acts as pluripotent stem cells. Proc Natl Acad Sci USA. 2003;100:2426–2431. doi: 10.1073/pnas.0536882100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fernandez Pujol B, Lucibello FC, Gehling UM, Lindemann K, Weidner N, Zuzarte ML, Adamkiewicz J, Elsässer HP, Müller R, Havemann K. Endothelial-like cells derived from human CD14 positive monocytes. Differentiation. 2000;65:287–300. doi: 10.1046/j.1432-0436.2000.6550287.x. [DOI] [PubMed] [Google Scholar]
- 77.Sirohi B, Powles R, Mehta J, Treleaven J, Raje N, Kulkarni S, Rudin C, Bhagwati N, Horton C, Saso R, Singhal S, Parikh R. The implication of compromised renal function at presentation in myeloma: similar outcome in patients who receive high-dose therapy: a singlecenter study of 251 previously untreated patients. Med Oncol. 2001;18:39–50. doi: 10.1385/mo:18:1:39. [DOI] [PubMed] [Google Scholar]
- 78.Meininger CJ, Zetter BR. Mast cells and angiogenesis. Semin Cancer Biol. 1995;3:73–79. [PubMed] [Google Scholar]
- 79.Norrby K, Woolley D. Role of mast cells in mito-genesis and angiogenesis in normal tissue and tumour tissue. Advanc Biosci. 1993;89:71–115. [Google Scholar]
- 80.Ribatti D. The crucial role of vascular permeability factor/vascular endothelial growth factor in angiogenesis: a historical review. Br J Haematol. 2005;128:303–309. doi: 10.1111/j.1365-2141.2004.05291.x. [DOI] [PubMed] [Google Scholar]
- 81.Poole TJ, Zetter BR. Mast cell chemotaxis to tumor derived factors. Cancer Res. 1983;43:5857–5862. [PubMed] [Google Scholar]
- 82.Sörbo J, Jakobsson A, Norrby K. Mast cell histamine is angiogenic through receptors for hista-mine 1 and histamine 2. Int J Exp Pathol. 1994;75:43–50. [PMC free article] [PubMed] [Google Scholar]
- 83.Blair RJ, Meng H, Marchese MJ, Ren S, Schwartz LB, Tonnesen MG, Gruber BL. Human mast cells stimulate vascular tube formation. Tryptase is a novel, potent angiogenic factor. J Clin Invest. 1997;99:2691–2700. doi: 10.1172/JCI119458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Möller A, Lippert U, Lessmann D, Kolde G, Hamann K, Welker P, Schadendorf D, Rosenbach T, Luger T, Czarnetzki BM. Human mast cells produce IL-8. J Immunol. 1998;151:3261–3266. [PubMed] [Google Scholar]
- 85.Grützkau A, Krüger-Krasagakes S, Kögel H, Möller A, Lippert U, Henz BM. Detection of intracellular interleukin-8 in human mast cells: flow cytometry as a guide for immunoelectron microscopy. J Histochem Cytochem. 1997;45:935–945. doi: 10.1177/002215549704500703. [DOI] [PubMed] [Google Scholar]
- 86.Qu Z, Kayton RJ, Ahmadi P, Liebler JM, Powers MR, Planck SR, Rosenbaum JT. Ultrastructural immunolocalization of basic fibroblast growth factor in mast cell secretory granules: morphological evidence for bFGF release through de-granulation. J Histochem Cytochem. 1998;46:1119–1128. doi: 10.1177/002215549804601004. [DOI] [PubMed] [Google Scholar]
- 87.Grützkau A, Krüger-Krasagakes S, Baumeister H, Schwarz C, Kögel H, Welker P, Lippert U, Henz BM, Möller A. Synthesis, storage, and release of vascular endothelial growth factor/ vascular permeability factor (VEGF/VPF) by human mast cells: implications for the biological significance of VEGF 206. Mol Biol Cell. 1998;9:875–884. doi: 10.1091/mbc.9.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thompson WD, Campbell R, Evans T. Fibrin degradation and angiogenesis: quantitative analysis of the angiogenic response in the chick chorioallantoic membrane. J Pathol. 1995;145:27–37. doi: 10.1002/path.1711450103. [DOI] [PubMed] [Google Scholar]
- 89.Ghosh AK, Hirasawa N, Ohuchi K. Enhancement by histamine of vascular endothelial growth factor production in granulation tissue via H2 receptors. Br J Pharmacol. 2001;134:1419–1428. doi: 10.1038/sj.bjp.0704372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brown JK, Tyler CL, Jones CA, Ruoss SJ, Hartmann T, Caughey GH. Tryptase, the dominant secretory granular protein in human mast cells, is a potent mitogen for cultured dog tracheal smooth muscle cells. Am J Respir Cell Mol Biol. 1995;13:227–236. doi: 10.1165/ajrcmb.13.2.7626290. [DOI] [PubMed] [Google Scholar]
- 91.Cairns JA, Walls AF. Mast cell tryptase is a mitogen for epithelial cells. Stimulation of IL-8 production and intercellular adhesion molecule-1 expression. J Immunol. 1996;156:275–283. [PubMed] [Google Scholar]
- 92.Stack MS, Johnson DA. Human mast cell tryptase activates single-chain urinary-type plasmi-nogen activator (pro-urokinase) J Biol Chem. 1994;269:9416–9419. [PubMed] [Google Scholar]
- 93.Ribatti D, Vacca A, Nico B, Quondamatteo F, Ria R, Minischetti M, Marzullo A, Herken R, Roncali L, Dammacco F. Bone marrow angiogenesis and mast cell density increase simultaneously with progression of human multiple myeloma. Br J Cancer. 1999;79:451–455. doi: 10.1038/sj.bjc.6690070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nico B, Mangieri D, Crivellato E, Vacca A, Ribatti D. Mast Cells contribute to vasculogenic mimicry in multiple myeloma. Stem Cells and Development. 2008;17:19–22. doi: 10.1089/scd.2007.0132. [DOI] [PubMed] [Google Scholar]
- 95.Fukushima N, Satoh T, Sano M, Tokunaga O. Angiogenesis and mast cell in non Hodgkin's lymphoma; a strong correlation in angioim-munoblastic T-cell lymphoma. Leuk Lymphoma. 2001;42:709–720. doi: 10.3109/10428190109099333. [DOI] [PubMed] [Google Scholar]
- 96.Roccaro AM, Hideshima T, Raje N, Kumar S, Ishitsuka K, Yasui H, Shiraishi N, Ribatti D, Nico B, Vacca A, Dammacco F, Richardson PG, Anderson KC. Bortezomib mediates antiangio-genesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 2006;66:184–191. doi: 10.1158/0008-5472.CAN-05-1195. [DOI] [PubMed] [Google Scholar]
- 97.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001;8:739–758. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 98.Roccaro AM, Hideshima T, Richardson PG, Russo D, Ribatti D, Vacca A, Dammacco F, Anderson KC. Bortezomib as an antitumor agent. Curr Pharmac Biothechn. 2006;7:441–448. doi: 10.2174/138920106779116865. [DOI] [PubMed] [Google Scholar]
- 99.Beg AA, Baldwin AS., Jr The I kappa B proteins: multifunctional regulators of Rel/NF-kappa B transcription factors. Genes Dev. 1993;7:2064–2070. doi: 10.1101/gad.7.11.2064. [DOI] [PubMed] [Google Scholar]
- 100.Zandi E, Chen Y, Karin M. Direct phosphorylation of IkappaB by IKKalpha and IKKbeta: discrimination between free and NF-kappaB-bound substrate. Science. 1998;281:1360–1363. doi: 10.1126/science.281.5381.1360. [DOI] [PubMed] [Google Scholar]
- 101.Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V, Adams J, Anderson KC. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277:16639–16647. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 102.Nawrocki ST, Carew JS, Dunner K, Jr, Boise LH, Chiao PJ, Huang P, Abbruzzese JL, McConkey DJ. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005;65:11510–11519. doi: 10.1158/0008-5472.CAN-05-2394. [DOI] [PubMed] [Google Scholar]
- 103.Ling YH, Liebes L, Zou Y, Perez-Soler R. J Biol Chem, Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J Biol Chem. 2003;278:33714–33723. doi: 10.1074/jbc.M302559200. [DOI] [PubMed] [Google Scholar]
- 104.Mitsiades N, Mitsiades CS, Richardson PG, Poulaki V, Tai YT, Chauhan D, Fanourakis G, Gu X, Bailey C, Joseph M, Libermann TA, Schlossman R, Munshi NC, Hideshima T, Anderson KC. The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood. 2003;101:2377–2380. doi: 10.1182/blood-2002-06-1768. [DOI] [PubMed] [Google Scholar]
- 105.Pajonk F, McBride WH. The proteasome in cancer biology and treatment. Radiat Res. 2001;156:447–459. doi: 10.1667/0033-7587(2001)156[0447:tpicba]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 106.Segeren CM, Sonneveld P, van der Holt B, Vellenga E, Croockewit AJ, Verhoef GE, Cornelissen JJ, Schaafsma MR, van Oers MH, Wijermans PW, Fibbe WE, Wittebol S, Schouten HC, van Marwijk Kooy M, Biesma DH, Baars JW, Slater R, Steijaert MM, Buijt I, Lokhorst HM, Dutch-Belgian Hemato-Oncology Cooperative Study Group Overall and event-free survival are not improved by the use of myeloablative therapy following intensified chemotherapy in previously untreated patients with multiple myeloma: a prospective randomized phase 3 study. Blood. 2003;101:2144–2151. doi: 10.1182/blood-2002-03-0889. [DOI] [PubMed] [Google Scholar]
- 107.D'Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91:4082–4085. doi: 10.1073/pnas.91.9.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ribatti D, Vacca A. Novel Therapeutic Approches Targeting Vascular Endothelial Growth Factor and its receptors in haematological malignancies. Curr Cancer Drug Targets. 2005;5:573–578. doi: 10.2174/156800905774932806. [DOI] [PubMed] [Google Scholar]
- 109.Davies FE, Raje N, Hideshima T, Lentzsch S, Young G, Tai YT, Lin B, Podar K, Gupta D, Chauhan D, Treon SP, Richardson PG, Schlossman RL, Morgan GJ, Muller GW, Stirling DI, Anderson KC. Thalidomide and immunomodulatory derivatives augument natural killer cell cytotoxicity in multiple myeloma. Blood. 2001;98:210–216. doi: 10.1182/blood.v98.1.210. [DOI] [PubMed] [Google Scholar]
- 110.Juliusson G, Celsing F, Turesson I, Lenhoff S, Adriansson M, Malm C. Frequent good partial remissions from thalidomide including best response ever in patients with advanced refractory and relapsed myeloma. Br J Haematol. 2000;109:89–96. doi: 10.1046/j.1365-2141.2000.01983.x. [DOI] [PubMed] [Google Scholar]
- 111.Gupta D, Treon SP, Shima Y, Hideshima T, Podar K, Tai YT, Lin B, Lentzsch S, Davies FE, Chauhan D, Schlossman RL, Richardson P, Ralph P, Wu L, Payvandi F, Muller G, Stirling DI, Anderson KC. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factoor secretion: therapeutic application. Leukemia. 2001;15:1950–1961. doi: 10.1038/sj.leu.2402295. [DOI] [PubMed] [Google Scholar]
- 112.Dimopoulos M, Spencer A, Attal M, Prince HM, Harousseau JL, Dmoszynska A, San Miguel J, Hellmann A, Facon T, Foà R, Corso A, Masliak Z, Olesnyckyj M, Yu Z, Patin J, Zeldis JB, Knight RD, Multiple Myeloma (010) Study Investigators Lenalidomide plus Dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357:2123–2132. doi: 10.1056/NEJMoa070594. [DOI] [PubMed] [Google Scholar]
- 113.García-Sanz R, González-Porras JR, Hernández JM, Polo-Zarzuela M, Sureda A, Barrenetxea C, Palomera L, López R, Grande-García C, Alegre A, Vargas-Pabón M, Gutiérrez ON, Rodríguez JA, San Miguel JF. The oral combination of thalidomide, cyclophosphamide and dexamethasone (ThaCyDex) is effective in relapsed/refractory multiple myeloma. Leukemia. 2004;18:856–863. doi: 10.1038/sj.leu.2403322. [DOI] [PubMed] [Google Scholar]
- 114.Richardson PG, Schlossman RL, Weller E, Hideshima T, Mitsiades C, Davies F, LeBlanc R, Catley LP, Doss D, Kelly K, McKenney M, Mechlowicz J, Freeman A, Deocampo R, Rich R, Ryoo JJ, Chauhan D, Balinski K, Zeldis J, Anderson KC. Immunomodulatory drug CC-5013 overcomes drug resistence and is well tolerated in patients with relapsed multiple myeloma. Blood. 2002;100:3063–3067. doi: 10.1182/blood-2002-03-0996. [DOI] [PubMed] [Google Scholar]
- 115.Richardson PG, Blood E, Mitsiades CS, Jagannath S, Zeldenrust SR, Alsina M, Schlossman RL, Rajkumar SV, Desikan KR, Hideshima T, Munshi NC, Kelly-Colson K, Doss D, McKenney ML, Gorelik S, Warren D, Freeman A, Rich R, Wu A, Olesnyckyj M, Wride K, Dalton WS, Zeldis J, Knight R, Weller E, Anderson KC. A randomized phase 2 study of lenalidomide therapy for patients with relapsed or relapsed and refractory multiple myeloma. Blood. 2006;108:3458–3464. doi: 10.1182/blood-2006-04-015909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Scavelli C, Di Pietro G, Cirulli T, Coluccia M, Boccarelli A, Giannini T, Mangialardi G, Bertieri R, Coluccia AM, Ribatti D, Dammacco F, Vacca A. Zoledronic acid affects over-angiogenic phenotype of endothelial cells in patients with multiple myeloma. Mol Cancer Ther. 2007;6:3256–3262. doi: 10.1158/1535-7163.MCT-07-0311. [DOI] [PubMed] [Google Scholar]
- 117.Clezardin P. The antitumor potential of bisphosphonates. Semin Oncol. 2002;29:33–42. doi: 10.1053/sonc.2002.37420. [DOI] [PubMed] [Google Scholar]
- 118.Wood J, Bonjean K, Ruetz S, Bellahcène A, Devy L, Foidart JM, Castronovo V, Green JR. Novel antiangiogenic effects of the bisphosphonates compound zoledronic acid. J Pharmacol Exp Ther. 2002;302:1055–1061. doi: 10.1124/jpet.102.035295. [DOI] [PubMed] [Google Scholar]
- 119.Ribatti D, Nico B, Mangieri D, Maruotti N, Longo V, Vacca A, Cantatore FP. Neridronate inhibits angiogenesis in vitro and in vivo. Clin Rheumatol. 2007;26:1094–1098. doi: 10.1007/s10067-006-0455-3. [DOI] [PubMed] [Google Scholar]
- 120.Hoff AO, Toth BB, Altundag K, Johnson MM, Warneke CL, Hu M, Nooka A, Sayegh G, Guarneri V, Desrouleaux K, Cui J, Adamus A, Gagel RF, Hortobagyi GN. Frequency and Risk Factors Associated With Osteonecrosis of the Jaw in Cancer Patients Treated With Intravenous Bisphosphonates. J Bone Miner Res. 2008;23:826–836. doi: 10.1359/JBMR.080205. [DOI] [PMC free article] [PubMed] [Google Scholar]