

Abstract
The number of patients with osteoporosis or type 2 diabetes mellitus (T2DM) is increasing in aging and westernized societies. Both disorders predispose elderly people to disabling conditions by causing fractures and vascular complications, respectively. Recent animal studies have shown that administration of osteocalcin, which is specifically secreted from osteoblasts, can increase insulin secretion and ameliorate hyperglycemia, obesity, and high triglyceride levels in mice fed a high-fat diet. Moreover, several studies have shown that antagonism of Wnt signaling by oxidative stress contributes to the development of osteoporosis, as well as insulin resistance and hyperlipidemia. Thus, bone metabolism and glucose/fat metabolism seem to be etiologically related to each other. Meta-analyses of multiple clinical studies in humans have shown that hip fracture risk of T2DM patients is increased by 1.4-1.7-fold, although bone mineral density (BMD) is not diminished. Vertebral fracture risk of T2DM patients is also increased, and BMD is not sensitive enough to assess the risk. These findings suggest that bone fragility in T2DM, which is not reflected by BMD, depends on bone quality deterioration rather than bone mass reduction. Thus, surrogate markers are needed to replace the insensitivity of BMD in assessing fracture risks of T2DM patients. Pentosidine, the endogenous secretory receptor for advanced glycation endproducts, and insulin-like growth factor-I seem to be such candidates, although further studies are required to clarify whether or not these markers could predict the occurrence of new fractures of T2DM patients in a prospective fashion.
Keywords: Osteoporosis, Type 2 diabetes mellitus, Fracture risk, Osteocalcin, Wnt signaling
BONE METABOLISM AND GLUCOSE/FAT METABOLISM ARE ASSOCIATED WITH EACH OTHER THROUGH THE ACTION OF OSTEOCALCIN AND WNT SIGNALING
Although osteoporosis and type 2 diabetes mellitus (T2DM) are traditionally viewed as separate disease entities that increase in prevalence with aging, accumulating evidence indicates that there are similar pathophysiological mechanisms underlying them.
Osteocalcin (OC), one of the osteoblast-specific secreted proteins, has several hormonal features and is secreted in the general circulation from osteoblastic cells[1,2]. Recent animal studies have shown that uncarboxylated OC (ucOC) action is related to bone metabolism and glucose metabolism and fat mass[3,4]. Lee et al[3] have shown that OC functions as a hormone that improves glucose metabolism and reduces fat mass, because OC-deficient mice aggravate these processes. Moreover, Ferron et al[4] have shown that recombinant ucOC administration regulates gene expression in β cells and adipocytes (including adiponectin expression), and prevents the development of metabolic diseases, obesity, and T2DM in wild-type mice fed a high-fat diet. Several clinical studies including our own[5-8] have recently shown that serum OC level is associated with glucose and total adiponectin levels, fat mass, as well as atherosclerosis parameters in humans. We have recently shown that serum OC level is negatively correlated with plasma glucose level and atherosclerosis parameters in T2DM patients[5]. Kindblom et al[6] have shown that OC level is inversely related to plasma glucose level and fat mass in elderly non-DM persons. Fernández-Real et al[7] have shown that serum OC level is positively associated with insulin sensitivity in non-DM subjects. Pittas et al[8] have shown that serum OC concentration is inversely associated with fasting plasma glucose (FPG), fasting insulin, homeostasis model assessment for insulin resistance, high-sensitivity C-reactive protein, interleukin-6, body mass index (BMI), and body fat in cross-sectional analyses. They also have found that OC levels are associated with changes in FPG in prospective analyses. We also have found that ucOC is negatively associated with plasma glucose level and fat mass, and positively with adiponectin in T2DM patients[9]. These experimental and clinical findings suggest that bone metabolism and glucose/fat metabolism are etiologically related to each other through the action of ucOC or OC (Figure 1).
Figure 1.

When osteocalcin was administered to obese mice, it increased insulin secretion, and decreased blood glucose level, fat mass, and triglyceride level. In humans, serum carboxylated and uncarboxylated osteocalcin levels were positively correlated with insulin sensitivity and adiponectin level, whereas they were negatively correlated with blood glucose level, fat mass, and atherosclerosis index. Thus, osteoporosis and diabetes are pathophysiologically related to each other through osteocalcin (OC) action in mice and humans.
Wnt signaling is also thought to be a common pathogenetic feature of osteoporosis and DM. Mani et al[10] have shown that a single missense mutation in low-density lipoprotein receptor-related protein 6, the co-receptor for the Wnt-signaling pathway, is genetically linked to osteoporosis as well as DM, hyperlipidemia, and coronary artery disease. In addition, several studies have documented that T-cell-specific transcription factor (TCF)-4, the partner of β-catenin in the canonical Wnt-signaling pathway, is the strongest T2DM susceptibility gene[11-14]. Manolagas and other researchers have suggested that antagonism of Wnt signaling by oxidative stress diverts β-catenin from TCF- to Forkhead box O (FoxO)-mediated transcription, and contributes to the development of osteoporosis as well as insulin resistance and hyperlipidemia[15-18]. Activation of FoxO by reactive oxygen species in early mesenchymal progenitors also leads to decreased osteoblastogenesis by diverting β-catenin away from Wnt signaling[19], the mechanism of which might be implicated in DM-related bone fragility that is described in the following section.
FRACTURE RISK IN T2DM IS NOT REFLECTED BY BONE MINERAL DENSITY
Many clinical studies have also investigated the association between DM and osteoporosis, given that these disorders affect a large proportion of the elderly population. Although bone mineral density (BMD) is considered as a gold standard for evaluating fracture risk in non-DM osteoporosis, accumulating evidence has shown that patients with T2DM have a high fracture rate in spite of the absence of BMD reduction. A recent meta-analysis has shown that T2DM patients have higher hip BMD than non-DM controls (z-score: 0.27), despite an increased risk of hip fracture (1.4-fold)[20], which suggests that BMD values do not reflect bone fragility in T2DM. Another meta-analysis also has shown that hip fracture risk of T2DM patients was increased by 1.7-fold[21].
In contrast, little is known about the risk of vertebral fracture (VF) and its association with BMD. We examined Japanese T2DM patients and non-DM controls about this issue[22,23]. We have found that the presence of T2DM is an independent risk factor for prevalent VFs in women (OR = 1.9) as well as men (OR = 4.7) after adjustment for age, BMI and lumbar BMD by logistic regression analysis. BMD at any site, however, is not significantly associated with the presence of prevalent VFs in T2DM patients, in contrast to the significant association in controls. Comparison of T2DM patients with and without VFs showed no significant differences in BMD values, bone markers, or diabetes status. Receiver operating characteristic analysis has shown that the absolute lumbar, femoral neck, and radial BMD values for detecting prevalent VFs were higher in T2DM patients than controls, whereas their sensitivity and specificity were lower. Figure 2 shows the distribution of lumbar BMD as a function of age in T2DM women. In the control subjects, those with VFs (black dots) were clearly grouped in the region with higher age and lower lumbar BMD. In contrast, T2DM subjects with VFs were scattered widely and there was no association with age or lumbar BMD. Thus, T2DM patients might have an increased risk of VFs independent of BMD or diabetic complication status, which suggests that bone quality, but not bone mass, define bone fragility and cause hip and vertebral fractures in T2DM.
Figure 2.
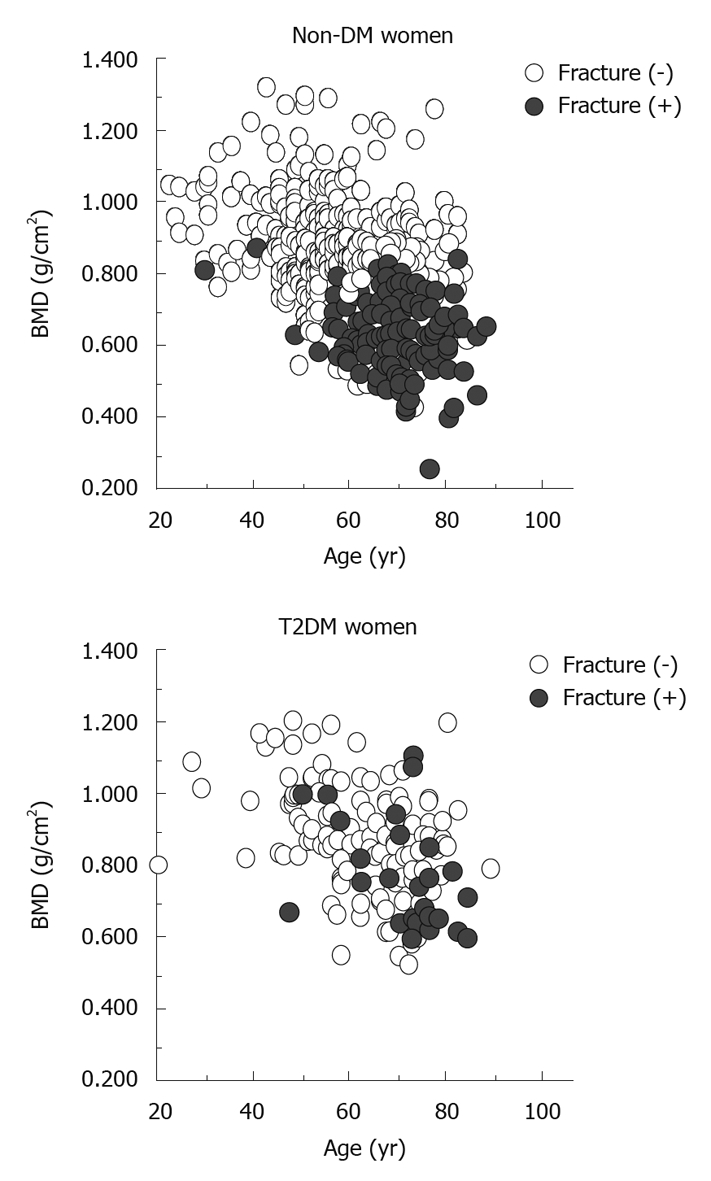
Distribution of bone mineral density in subjects with and without vertebral fractures (black dots and open circles, respectively) as a function of age. BMD: Bone mineral density; T2DM: Type 2 diabetes mellitus.
SURROGATE MARKERS FOR ASSESSING FRACTURE RISK IN T2DM
BMD is not sensitive enough to assess the risk of osteoporotic fractures in T2DM, therefore, the etiology of DM-related bone fragility and diagnostic markers other than BMD need to be explored.
Pentosidine
Formation of advanced glycation end products (AGEs) results from sequential non-enzymatic chemical glycoxidation of protein amino groups[24], collectively called the Maillard reaction. AGEs accumulate in various tissues including kidney, brain, and coronary artery atherosclerotic plaques during normal aging, whereas hyperglycemia results in an accelerated rate of AGE formation, which suggests that AGEs have a pivotal role in the development of complications in DM patients[25,26]. In addition, previous studies have revealed that AGEs accumulate in bone tissue as well[27,28], and that receptor for AGE (RAGE) is expressed in human bone-derived cells[29], which suggests that AGEs are associated with DM-related bone fragility.
Several experimental studies have shown that AGEs have a negative impact on bone. AGEs inhibit the synthesis of type 1 collagen and OC, as well as mature bone nodule formation in osteoblasts[30-32]. We have previously demonstrated that the combination of high glucose and AGEs additionally or synergistically inhibits the mineralization of osteoblastic cells, through glucose-induced increase in expression of RAGE[33]. These findings suggest that AGE accumulation in bone causes osteoblastic dysfunction. AGEs are also known to increase osteoclast activity. Previous in vitro and in vivo experiments[34] have indicated that the number of resorption pits is increased when osteoclasts are cultured on AGE-modified dentin slices, and that AGE-bone particles are resorbed to a much greater extent than non-AGE bone particles when implanted subcutaneously in rats. In addition, RAGE knockout mice display a decreased number of osteoclasts as well as a significantly higher bone mass compared to wild-type mice[35]. AGEs accumulation inhibits the differentiation and mineralization of osteoblasts, while it enhances the activity of osteoclasts, which possibly leads to uncoupling of bone turnover and resultant bone fragility.
AGE accumulation in bone is also negatively associated with material properties[27,28,36]. Collagen crosslinks are known to play crucial roles in the determination of bone strength[37]. AGE-type crosslinks, which are formed spontaneously by non-enzymatic glycation and oxidation reactions, are thought to be associated with brittleness of collagen fibers[38,39], whereas physiological (enzymatic) crosslinks strengthen links of collagen fibers, and lead to the enhancement of bone strength[28,40]. Spontaneously diabetic WBN/Kob rats have been reported to display a decrease in enzymatic crosslinks and an increase in AGE-type crosslinks despite the lack of BMD reduction, which results in deterioration of bone strength[41].
Among the few AGEs characterized to date, pentosidine is one of the well-known AGEs and is chemically well defined[42-44]. Because the formation of pentosidine requires both glycation and oxidation, serum pentosidine levels are considered to be a useful marker for glycoxidation. Several studies have revealed that pentosidine content in cortical or trabecular bone from vertebra or femur is negatively associated with mechanical properties[27,28,36], and that pentosidine content of cortical and trabecular bone derived from patients with femoral neck fracture is higher than that of age-matched controls[45,46]. However, assessment of pentosidine content in bone is not easily done in clinical situations, because invasive procedures like bone biopsy are necessary for preparing specimens. A recent study has revealed that content of pentosidine in plasma shows a significant linear correlation with that in cortical bone[47], which suggests that serum pentosidine level could be used as a surrogate marker for its content in bone and could evaluate bone strength. We have previously shown that serum pentosidine levels are associated with prevalent VFs in postmenopausal women with T2DM (OR = 2.50 per SD increase, Table 1)[48]. This association is independent of BMD, which suggests that it might reflect bone quality rather than BMD. In addition, an observational cohort study has shown that urine pentosidine levels are associated with increased clinical fracture incidence in those with DM (relative hazard 1.42 per SD increase in log pentosidine)[49]. Therefore, serum and urine pentosidine levels might be useful for assessing fracture risk in T2DM women, for which BMD or traditional bone markers are insensitive.
Table 1.
Odds ratio of surrogate markers for the presence of prevalent vertebral fractures
|
Presence of vertebral fractures |
||
| OR (95% CI) | P | |
| Pentosidine (male) | 0.79 (0.41-1.52) | 0.47 |
| Pentosidine (female) | 2.50 (1.09-5.73) | 0.03 |
| esRAGE (male) | 0.46 (0.25-0.84) | 0.012 |
| esRAGE (female) | 0.32 (0.16-0.67) | 0.002 |
| IGF-I (female) | 0.44 (0.23-0.81) | 0.009 |
Multivariate logistic regression analysis was performed with the presence of vertebral fractures as a dependent variable and each of levels of serum insulin-like growth factor (IGF)-I, C-peptide, osteocalcin, and uNTX adjusted for age, duration of diabetes, body weight, height, creatinine, albumin, and HbA1c as independent variables. esRAGE: Endogenous secretory receptor for advanced glycation end product; OR: Odds ratio; CI: Confidence interval.
Endogenous secretory RAGE
RAGE belongs to the immunoglobulin superfamily of cell-surface receptors and is capable of interacting with multiple ligands, including AGEs[50]. When transgenic mice that overexpressed human RAGE in vascular cells were crossbred with a transgenic line that developed insulin-dependent DM shortly after birth, more progressive histological changes of DM nephropathy were observed compared to controls[51], which confirms that RAGE is associated with the development of DM complications. Endogenous secretory RAGE (esRAGE) is a splice variant of one of the naturally occurring secretory forms, and is known to carry all of the extracellular domains but lacks the transmembrane and cytoplasmic domains[52]. esRAGE in the extracellular space is thought to act as a decoy receptor that binds AGEs and reduces the activity of intercellular signal pathways via RAGE[52]. Indeed, administration of a genetically engineered murine soluble RAGE suppresses the development of diabetic atherosclerosis in a dose-dependent manner in streptozotocin-induced apoE-null DM mice[53]. These experimental findings suggest that enhanced RAGE activity is clinically linked to reduced bone strength in DM patients. Given the neutralizing nature of esRAGE, it is possible that the ratio of serum esRAGE to AGE levels could be linked to clinical bone problems, such as fractures, more prominently than either parameter alone.
We have found that the esRAGE/pentosidine ratio in T2DM patients with VFs is significantly lower than in those without VFs. Multivariate logistic regression analysis adjusted for age, height, weight, hemoglobin A1c, serum creatinine, DM duration, therapeutic agents, DM complications, osteoporotic risk factors, and lumbar BMD identified the serum esRAGE level and esRAGE/pentosidine ratio as factors associated with the presence of VFs, independent of BMD in men (OR = 0.46 and 0.34, respectively) and in women (OR = 0.32 and 0.14, respectively) (Table 1)[54]. These results show that serum esRAGE level and esRAGE/pentosidine ratio are more useful than BMD for assessing the risk of VFs in T2DM patients.
Insulin-like growth factor-I
Bone remodeling is regulated by systemic hormones and locally produced factors, which both act in concert to maintain bone mass[55-57]. Insulin-like growth factors (IGFs) are synthesized in osteoblasts and are among the most important regulators of bone cell function due to their anabolic effects on the skeleton[58]. The key role of the IGF system in the local regulation of bone formation is demonstrated by the finding that about 50% of basal bone cell proliferation can be blocked by inhibiting the actions of IGFs that are endogenously produced by bone cells in serum-free culture[59]. In osteoblast-specific knockout mice for IGF-I receptor, a significant reduction in trabecular bone mass and deficient mineralization have been observed[60]. In contrast, circulating IGF-I, which is mainly produced in the liver via regulation by growth hormone and diet, acts in an endocrine manner, which also activates bone remodeling and exerts anabolic effects on bone tissues[61-63]. Liver-specific IGF-I gene null mice show a marked reduction in bone volume, periosteal circumference, and medial lateral width, which suggests that circulating levels of IGF-I also directly regulate bone growth and density[64]. Indeed, our clinical studies have shown that serum IGF-I levels are positively associated with BMD and inversely with the risk of VFs in postmenopausal non-DM women[65,66]. These findings suggest that serum IGF-I levels could be clinically useful for assessing bone mass and the risk of VFs in the non-DM population.
IGFs are also thought to be linked to the pathogenesis of DM-related complications[67]. Impaired production of IGFs could also cause bone complication in DM by diminishing bone cell function[58]. An in vivo study has demonstrated that IGF-I levels in serum and cortical bone are significantly reduced in spontaneously diabetic Goto-Kakizaki rats, which display a significant decrease in BMD in the long bone metaphyses and vertebrae[68]. In contrast, several in vitro studies have shown that the stimulatory actions of IGF-I on osteoblasts are blunted by high glucose concentrations or AGEs. High glucose concentrations significantly impair the proliferative and functional responses of osteoblastic MG-63 cells to IGF-I[69]. AGEs also significantly decrease IGF-I secretion in osteoblastic MC3T3-E1 cells[70]. Thus, high glucose concentrations or AGEs might cause resistance of osteoblasts to IGF-I actions in the local environment.
In T2DM patients, however, the relationship between serum IGF-I levels and bone metabolism has been little documented. We have indicated that serum IGF-I levels are significantly and inversely associated with the presence of VFs in postmenopausal T2DM women (OR = 0.44 per SD increase) in a fashion independent of age, body stature, DM status, renal function, insulin secretion, or lumbar BMD (Table 1)[71]. Accordingly, circulating IGF-I might have a protective effect on VFs, and this effect might be related to bone quality but not to BMD in postmenopausal T2DM women. Thus, serum IGF-I levels as well as esRAGE and pentosidine might be useful for assessing the risk of VFs in T2DM women.
FRACTURE RISK ASSESSMENT OF T2DM PATIENTS IN CLINICAL PRACTICE
As a result of the ineffectiveness of BMD in assessing fracture risks in T2DM, the major clinical problems are how to assess the risks and when to start therapy for preventing fractures in daily practice. Although the above-mentioned markers are potential candidates for such purposes, it is unclear whether or not they could predict the occurrence of new fractures in T2DM patients in a prospective fashion.
In contrast, the presence of prevalent VFs could be used for the assessment of bone quality in individual patients, because a large study on the incidence of VFs in postmenopausal osteoporosis has shown that patients with previous VFs were more likely to suffer from new VFs[72,73] and hip fractures[72] independent of BMD, than those without VFs, during a study period of several years. A patient history of non-VFs is also an established risk factor for additional fractures[74]. We found that 38% of T2DM men and 31% of T2DM women had prevalent VFs by X-ray films, and that 16% each of T2DM men and women had a history of previous non-VFs[23]. Thus, if T2DM patients undergo spinal X-ray examination or are questioned about their fracture histories, it is likely that about half of them will be identified as those who have bone fragility and need osteoporosis treatment for fracture prevention. These procedures are simple and are recommended for all physicians who are engaged in T2DM treatment.
Recently, the fracture risk assessment (FRAX) algorithm has been developed by the WHO, which could assess the fracture risk of an individual even if BMD is not measured[74,75]. This algorithm integrates the influence of several well-validated risk factors for fractures that are independent of BMD, therefore, it might be useful for the case-finding strategy that identifies diabetic patients at high risk for fracture.
CONCLUSION
The fact that BMD is not useful for assessing fracture risks in T2DM seems problematic, because T2DM populations are increasing in every country. T2DM patients may drop out from fracture prevention if doctors diagnose osteoporosis based on BMD values alone. Practitioners should be aware of the importance of detecting pre-existing VFs and fracture histories by spinal X-ray examination and interview, respectively. These procedures could broaden the spectrum of osteoporosis treatments into the T2DM population. Simultaneously, further studies are needed to clarify whether surrogate biochemical markers such as pentosidine, esRAGE, and IGF-I, as well as the WHO FRAX algorithm, could be useful for predicting prospectively the occurrence of new fractures in T2DM patients, with sensitivity and specificity comparable to those of BMD in non-DM osteoporosis.
Footnotes
Peer reviewers: Andrea Giusti, MD, Department of Gerontology and Musculoskeletal Sciences, Galliera Hospital, Via Montallegro 32a/5, Genova 16100, Italy; Vijay K Goel, PhD, Distinguished University Professor, Endowed Chair and McMaster-Gardner Professor of Orthopaedic Bioengineering, Co-Director, Engineering Center for Orthopaedic Research Excellence, Department of Bioengineering and Orthopaedic Surgery, Colleges of Engineering and Medicine, University of Toledo, 5046 NI, MS 303, Toledo, OH 43606, United States
S- Editor Cheng JX L- Editor Kerr C E- Editor Lin YP
References
- 1.Hauschka PV, Lian JB, Cole DE, Gundberg CM. Osteocalcin and matrix Gla protein: vitamin K-dependent proteins in bone. Physiol Rev. 1989;69:990–1047. doi: 10.1152/physrev.1989.69.3.990. [DOI] [PubMed] [Google Scholar]
- 2.Price PA. Gla-containing proteins of bone. Connect Tissue Res. 1989;21:51–57; discussion 57-60. doi: 10.3109/03008208909049995. [DOI] [PubMed] [Google Scholar]
- 3.Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456–469. doi: 10.1016/j.cell.2007.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferron M, Hinoi E, Karsenty G, Ducy P. Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci USA. 2008;105:5266–5270. doi: 10.1073/pnas.0711119105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanazawa I, Yamaguchi T, Yamamoto M, Yamauchi M, Kurioka S, Yano S, Sugimoto T. Serum osteocalcin level is associated with glucose metabolism and atherosclerosis parameters in type 2 diabetes mellitus. J Clin Endocrinol Metab. 2009;94:45–49. doi: 10.1210/jc.2008-1455. [DOI] [PubMed] [Google Scholar]
- 6.Kindblom JM, Ohlsson C, Ljunggren O, Karlsson MK, Tivesten A, Smith U, Mellström D. Plasma osteocalcin is inversely related to fat mass and plasma glucose in elderly Swedish men. J Bone Miner Res. 2009;24:785–791. doi: 10.1359/jbmr.081234. [DOI] [PubMed] [Google Scholar]
- 7.Fernández-Real JM, Izquierdo M, Ortega F, Gorostiaga E, Gómez-Ambrosi J, Moreno-Navarrete JM, Frühbeck G, Martínez C, Idoate F, Salvador J, et al. The relationship of serum osteocalcin concentration to insulin secretion, sensitivity, and disposal with hypocaloric diet and resistance training. J Clin Endocrinol Metab. 2009;94:237–245. doi: 10.1210/jc.2008-0270. [DOI] [PubMed] [Google Scholar]
- 8.Pittas AG, Harris SS, Eliades M, Stark P, Dawson-Hughes B. Association between serum osteocalcin and markers of metabolic phenotype. J Clin Endocrinol Metab. 2009;94:827–832. doi: 10.1210/jc.2008-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanazawa I, Yamaguchi T, Yamauchi M, Yamamoto M, Kurioka S, Yano S, Sugimoto T. Serum undercarboxylated osteocalcin was inversely associated with plasma glucose level and fat mass in type 2 diabetes mellitus. Osteoporos Int. 2010:Epub ahead of print. doi: 10.1007/s00198-010-1184-7. [DOI] [PubMed] [Google Scholar]
- 10.Mani A, Radhakrishnan J, Wang H, Mani A, Mani MA, Nelson-Williams C, Carew KS, Mane S, Najmabadi H, Wu D, et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science. 2007;315:1278–1282. doi: 10.1126/science.1136370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith U. TCF7L2 and type 2 diabetes--we WNT to know. Diabetologia. 2007;50:5–7. doi: 10.1007/s00125-006-0521-z. [DOI] [PubMed] [Google Scholar]
- 12.Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–323. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 13.Owen KR, McCarthy MI. Genetics of type 2 diabetes. Curr Opin Genet Dev. 2007;17:239–244. doi: 10.1016/j.gde.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 14.Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manolagas SC, Almeida M. Gone with the Wnts: beta-catenin, T-cell factor, forkhead box O, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol Endocrinol. 2007;21:2605–2614. doi: 10.1210/me.2007-0259. [DOI] [PubMed] [Google Scholar]
- 16.Hoogeboom D, Burgering BM. Should I stay or should I go: beta-catenin decides under stress. Biochim Biophys Acta. 2009;1796:63–74. doi: 10.1016/j.bbcan.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 17.DeCarolis NA, Wharton KA Jr, Eisch AJ. Which way does the Wnt blow? Exploring the duality of canonical Wnt signaling on cellular aging. Bioessays. 2008;30:102–106. doi: 10.1002/bies.20709. [DOI] [PubMed] [Google Scholar]
- 18.Jin T. The WNT signalling pathway and diabetes mellitus. Diabetologia. 2008;51:1771–1780. doi: 10.1007/s00125-008-1084-y. [DOI] [PubMed] [Google Scholar]
- 19.Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010;31:266–300. doi: 10.1210/er.2009-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes--a meta-analysis. Osteoporos Int. 2007;18:427–444. doi: 10.1007/s00198-006-0253-4. [DOI] [PubMed] [Google Scholar]
- 21.Janghorbani M, Van Dam RM, Willett WC, Hu FB. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am J Epidemiol. 2007;166:495–505. doi: 10.1093/aje/kwm106. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto M, Yamaguchi T, Yamauchi M, Kaji H, Sugimoto T. Bone mineral density is not sensitive enough to assess the risk of vertebral fractures in type 2 diabetic women. Calcif Tissue Int. 2007;80:353–358. doi: 10.1007/s00223-007-9003-7. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto M, Yamaguchi T, Yamauchi M, Kaji H, Sugimoto T. Diabetic patients have an increased risk of vertebral fractures independent of BMD or diabetic complications. J Bone Miner Res. 2009;24:702–709. doi: 10.1359/jbmr.081207. [DOI] [PubMed] [Google Scholar]
- 24.Brownlee M. Advanced protein glycosylation in diabetes and aging. Annu Rev Med. 1995;46:223–234. doi: 10.1146/annurev.med.46.1.223. [DOI] [PubMed] [Google Scholar]
- 25.Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988;318:1315–1321. doi: 10.1056/NEJM198805193182007. [DOI] [PubMed] [Google Scholar]
- 26.Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854. [DOI] [PubMed] [Google Scholar]
- 27.Hernandez CJ, Tang SY, Baumbach BM, Hwu PB, Sakkee AN, van der Ham F, DeGroot J, Bank RA, Keaveny TM. Trabecular microfracture and the influence of pyridinium and non-enzymatic glycation-mediated collagen cross-links. Bone. 2005;37:825–832. doi: 10.1016/j.bone.2005.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Shen X, Li X, Agrawal CM. Age-related changes in the collagen network and toughness of bone. Bone. 2002;31:1–7. doi: 10.1016/s8756-3282(01)00697-4. [DOI] [PubMed] [Google Scholar]
- 29.Takagi M, Kasayama S, Yamamoto T, Motomura T, Hashimoto K, Yamamoto H, Sato B, Okada S, Kishimoto T. Advanced glycation endproducts stimulate interleukin-6 production by human bone-derived cells. J Bone Miner Res. 1997;12:439–446. doi: 10.1359/jbmr.1997.12.3.439. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto T, Ozono K, Miyauchi A, Kasayama S, Kojima Y, Shima M, Okada S. Role of advanced glycation end products in adynamic bone disease in patients with diabetic nephropathy. Am J Kidney Dis. 2001;38:S161–S164. doi: 10.1053/ajkd.2001.27428. [DOI] [PubMed] [Google Scholar]
- 31.Katayama Y, Akatsu T, Yamamoto M, Kugai N, Nagata N. Role of nonenzymatic glycosylation of type I collagen in diabetic osteopenia. J Bone Miner Res. 1996;11:931–937. doi: 10.1002/jbmr.5650110709. [DOI] [PubMed] [Google Scholar]
- 32.Sanguineti R, Storace D, Monacelli F, Federici A, Odetti P. Pentosidine effects on human osteoblasts in vitro. Ann N Y Acad Sci. 2008;1126:166–172. doi: 10.1196/annals.1433.044. [DOI] [PubMed] [Google Scholar]
- 33.Ogawa N, Yamaguchi T, Yano S, Yamauchi M, Yamamoto M, Sugimoto T. The combination of high glucose and advanced glycation end-products (AGEs) inhibits the mineralization of osteoblastic MC3T3-E1 cells through glucose-induced increase in the receptor for AGEs. Horm Metab Res. 2007;39:871–875. doi: 10.1055/s-2007-991157. [DOI] [PubMed] [Google Scholar]
- 34.Miyata T, Notoya K, Yoshida K, Horie K, Maeda K, Kurokawa K, Taketomi S. Advanced glycation end products enhance osteoclast-induced bone resorption in cultured mouse unfractionated bone cells and in rats implanted subcutaneously with devitalized bone particles. J Am Soc Nephrol. 1997;8:260–270. doi: 10.1681/ASN.V82260. [DOI] [PubMed] [Google Scholar]
- 35.Ding KH, Wang ZZ, Hamrick MW, Deng ZB, Zhou L, Kang B, Yan SL, She JX, Stern DM, Isales CM, et al. Disordered osteoclast formation in RAGE-deficient mouse establishes an essential role for RAGE in diabetes related bone loss. Biochem Biophys Res Commun. 2006;340:1091–1097. doi: 10.1016/j.bbrc.2005.12.107. [DOI] [PubMed] [Google Scholar]
- 36.Viguet-Carrin S, Roux JP, Arlot ME, Merabet Z, Leeming DJ, Byrjalsen I, Delmas PD, Bouxsein ML. Contribution of the advanced glycation end product pentosidine and of maturation of type I collagen to compressive biomechanical properties of human lumbar vertebrae. Bone. 2006;39:1073–1079. doi: 10.1016/j.bone.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 37.Saito M, Marumo K. Collagen cross-links as a determinant of bone quality: a possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporos Int. 2010;21:195–214. doi: 10.1007/s00198-009-1066-z. [DOI] [PubMed] [Google Scholar]
- 38.Garnero P, Borel O, Gineyts E, Duboeuf F, Solberg H, Bouxsein ML, Christiansen C, Delmas PD. Extracellular post-translational modifications of collagen are major determinants of biomechanical properties of fetal bovine cortical bone. Bone. 2006;38:300–309. doi: 10.1016/j.bone.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 39.Vashishth D. The role of the collagen matrix in skeletal fragility. Curr Osteoporos Rep. 2007;5:62–66. doi: 10.1007/s11914-007-0004-2. [DOI] [PubMed] [Google Scholar]
- 40.Banse X, Sims TJ, Bailey AJ. Mechanical properties of adult vertebral cancellous bone: correlation with collagen intermolecular cross-links. J Bone Miner Res. 2002;17:1621–1628. doi: 10.1359/jbmr.2002.17.9.1621. [DOI] [PubMed] [Google Scholar]
- 41.Saito M, Fujii K, Mori Y, Marumo K. Role of collagen enzymatic and glycation induced cross-links as a determinant of bone quality in spontaneously diabetic WBN/Kob rats. Osteoporos Int. 2006;17:1514–1523. doi: 10.1007/s00198-006-0155-5. [DOI] [PubMed] [Google Scholar]
- 42.Odetti P, Fogarty J, Sell DR, Monnier VM. Chromatographic quantitation of plasma and erythrocyte pentosidine in diabetic and uremic subjects. Diabetes. 1992;41:153–159. doi: 10.2337/diab.41.2.153. [DOI] [PubMed] [Google Scholar]
- 43.Monnier VM, Sell DR, Nagaraj RH, Miyata S, Grandhee S, Odetti P, Ibrahim SA. Maillard reaction-mediated molecular damage to extracellular matrix and other tissue proteins in diabetes, aging, and uremia. Diabetes. 1992;41 Suppl 2:36–41. doi: 10.2337/diab.41.2.s36. [DOI] [PubMed] [Google Scholar]
- 44.Grandhee SK, Monnier VM. Mechanism of formation of the Maillard protein cross-link pentosidine. Glucose, fructose, and ascorbate as pentosidine precursors. J Biol Chem. 1991;266:11649–11653. [PubMed] [Google Scholar]
- 45.Saito M, Fujii K, Soshi S, Tanaka T. Reductions in degree of mineralization and enzymatic collagen cross-links and increases in glycation-induced pentosidine in the femoral neck cortex in cases of femoral neck fracture. Osteoporos Int. 2006;17:986–995. doi: 10.1007/s00198-006-0087-0. [DOI] [PubMed] [Google Scholar]
- 46.Saito M, Fujii K, Marumo K. Degree of mineralization-related collagen crosslinking in the femoral neck cancellous bone in cases of hip fracture and controls. Calcif Tissue Int. 2006;79:160–168. doi: 10.1007/s00223-006-0035-1. [DOI] [PubMed] [Google Scholar]
- 47.Odetti P, Rossi S, Monacelli F, Poggi A, Cirnigliaro M, Federici M, Federici A. Advanced glycation end products and bone loss during aging. Ann N Y Acad Sci. 2005;1043:710–717. doi: 10.1196/annals.1333.082. [DOI] [PubMed] [Google Scholar]
- 48.Yamamoto M, Yamaguchi T, Yamauchi M, Yano S, Sugimoto T. Serum pentosidine levels are positively associated with the presence of vertebral fractures in postmenopausal women with type 2 diabetes. J Clin Endocrinol Metab. 2008;93:1013–1019. doi: 10.1210/jc.2007-1270. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz AV, Garnero P, Hillier TA, Sellmeyer DE, Strotmeyer ES, Feingold KR, Resnick HE, Tylavsky FA, Black DM, Cummings SR, et al. Pentosidine and increased fracture risk in older adults with type 2 diabetes. J Clin Endocrinol Metab. 2009;94:2380–2386. doi: 10.1210/jc.2008-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du Yan S, Hofmann M, Yan SF, Pischetsrieder M, Stern D, et al. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem. 1999;274:31740–31749. doi: 10.1074/jbc.274.44.31740. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto Y, Kato I, Doi T, Yonekura H, Ohashi S, Takeuchi M, Watanabe T, Yamagishi S, Sakurai S, Takasawa S, et al. Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J Clin Invest. 2001;108:261–268. doi: 10.1172/JCI11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, Yasui K, Takeuchi M, Makita Z, Takasawa S, et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J. 2003;370:1097–1109. doi: 10.1042/BJ20021371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ Jr, Chow WS, Stern D, Schmidt AM. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 54.Yamamoto M, Yamaguchi T, Yamauchi M, Sugimoto T. Low serum level of the endogenous secretory receptor for advanced glycation end products (esRAGE) is a risk factor for prevalent vertebral fractures independent of bone mineral density in patients with type 2 diabetes. Diabetes Care. 2009;32:2263–2268. doi: 10.2337/dc09-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Canalis E. The hormonal and local regulation of bone formation. Endocr Rev. 1983;4:62–77. doi: 10.1210/edrv-4-1-62. [DOI] [PubMed] [Google Scholar]
- 56.Mohan S, Baylink DJ. Bone growth factors. Clin Orthop Relat Res. 1991:30–48. [PubMed] [Google Scholar]
- 57.Ueland T. Bone metabolism in relation to alterations in systemic growth hormone. Growth Horm IGF Res. 2004;14:404–417. doi: 10.1016/j.ghir.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 58.McCarthy TL, Centrella M, Canalis E. Insulin-like growth factor (IGF) and bone. Connect Tissue Res. 1989;20:277–282. doi: 10.3109/03008208909023897. [DOI] [PubMed] [Google Scholar]
- 59.Mohan S. Insulin-like growth factor binding proteins in bone cell regulation. Growth Regul. 1993;3:67–70. [PubMed] [Google Scholar]
- 60.Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, Malluche H, Zhao G, Rosen CJ, Efstratiadis A, et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005–44012. doi: 10.1074/jbc.M208265200. [DOI] [PubMed] [Google Scholar]
- 61.Johansson AG, Lindh E, Ljunghall S. Insulin-like growth factor I stimulates bone turnover in osteoporosis. Lancet. 1992;339:1619. doi: 10.1016/0140-6736(92)91889-g. [DOI] [PubMed] [Google Scholar]
- 62.Schwander JC, Hauri C, Zapf J, Froesch ER. Synthesis and secretion of insulin-like growth factor and its binding protein by the perfused rat liver: dependence on growth hormone status. Endocrinology. 1983;113:297–305. doi: 10.1210/endo-113-1-297. [DOI] [PubMed] [Google Scholar]
- 63.Spencer EM, Liu CC, Si EC, Howard GA. In vivo actions of insulin-like growth factor-I (IGF-I) on bone formation and resorption in rats. Bone. 1991;12:21–26. doi: 10.1016/8756-3282(91)90050-s. [DOI] [PubMed] [Google Scholar]
- 64.Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL, Ooi GT, Setser J, Frystyk J, Boisclair YR, et al. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest. 2002;110:771–781. doi: 10.1172/JCI15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sugimoto T, Nishiyama K, Kuribayashi F, Chihara K. Serum levels of insulin-like growth factor (IGF) I, IGF-binding protein (IGFBP)-2, and IGFBP-3 in osteoporotic patients with and without spinal fractures. J Bone Miner Res. 1997;12:1272–1279. doi: 10.1359/jbmr.1997.12.8.1272. [DOI] [PubMed] [Google Scholar]
- 66.Yamaguchi T, Kanatani M, Yamauchi M, Kaji H, Sugishita T, Baylink DJ, Mohan S, Chihara K, Sugimoto T. Serum levels of insulin-like growth factor (IGF); IGF-binding proteins-3, -4, and -5; and their relationships to bone mineral density and the risk of vertebral fractures in postmenopausal women. Calcif Tissue Int. 2006;78:18–24. doi: 10.1007/s00223-005-0163-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thrailkill KM. Insulin-like growth factor-I in diabetes mellitus: its physiology, metabolic effects, and potential clinical utility. Diabetes Technol Ther. 2002;2:69–80. doi: 10.1089/152091599316775. [DOI] [PubMed] [Google Scholar]
- 68.Ahmad T, Ugarph-Morawski A, Lewitt MS, Li J, Sääf M, Brismar K. Diabetic osteopathy and the IGF system in the Goto-Kakizaki rat. Growth Horm IGF Res. 2008;18:404–411. doi: 10.1016/j.ghir.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 69.Terada M, Inaba M, Yano Y, Hasuma T, Nishizawa Y, Morii H, Otani S. Growth-inhibitory effect of a high glucose concentration on osteoblast-like cells. Bone. 1998;22:17–23. doi: 10.1016/s8756-3282(97)00220-2. [DOI] [PubMed] [Google Scholar]
- 70.McCarthy AD, Etcheverry SB, Cortizo AM. Effect of advanced glycation endproducts on the secretion of insulin-like growth factor-I and its binding proteins: role in osteoblast development. Acta Diabetol. 2001;38:113–122. doi: 10.1007/s005920170007. [DOI] [PubMed] [Google Scholar]
- 71.Kanazawa I, Yamaguchi T, Yamamoto M, Yamauchi M, Yano S, Sugimoto T. Serum insulin-like growth factor-I level is associated with the presence of vertebral fractures in postmenopausal women with type 2 diabetes mellitus. Osteoporos Int. 2007;18:1675–1681. doi: 10.1007/s00198-007-0430-0. [DOI] [PubMed] [Google Scholar]
- 72.Black DM, Arden NK, Palermo L, Pearson J, Cummings SR. Prevalent vertebral deformities predict hip fractures and new vertebral deformities but not wrist fractures. Study of Osteoporotic Fractures Research Group. J Bone Miner Res. 1999;14:821–828. doi: 10.1359/jbmr.1999.14.5.821. [DOI] [PubMed] [Google Scholar]
- 73.Liberman UA, Weiss SR, Bröll J, Minne HW, Quan H, Bell NH, Rodriguez-Portales J, Downs RW Jr, Dequeker J, Favus M. Effect of oral alendronate on bone mineral density and the incidence of fractures in postmenopausal osteoporosis. The Alendronate Phase III Osteoporosis Treatment Study Group. N Engl J Med. 1995;333:1437–1443. doi: 10.1056/NEJM199511303332201. [DOI] [PubMed] [Google Scholar]
- 74.Kanis JA, Borgstrom F, De Laet C, Johansson H, Johnell O, Jonsson B, Oden A, Zethraeus N, Pfleger B, Khaltaev N. Assessment of fracture risk. Osteoporos Int. 2005;16:581–589. doi: 10.1007/s00198-004-1780-5. [DOI] [PubMed] [Google Scholar]
- 75.Kanis JA, Oden A, Johansson H, Borgström F, Ström O, McCloskey E. FRAX and its applications to clinical practice. Bone. 2009;44:734–743. doi: 10.1016/j.bone.2009.01.373. [DOI] [PubMed] [Google Scholar]