

Abstract
Gefitinib, a small molecule inhibitor of the epidermal growth factor receptor tyrosine kinase, has been shown to induce autophagy as well as apoptosis in tumor cells. Yet, how to exploit autophagy and apoptosis to improve therapeutic efficacy of this drug against cancer remains to be explored. We reported here that MK-2206, a potent allosteric Akt inhibitor currently in Phase I trials in patients with solid tumors, could reinforce the cytocidal effect of gefitinib against glioma. We found that co-treatment with gefitinib and MK-2206 increased the cytotoxicity of this growth factor receptor inhibitor in the glioma cells, and the Compusyn synergism/antagonism analysis showed that MK-2206 acted synergistically with gefitinib. The benefit of the combinatorial treatment was also demonstrated in an intracranial glioma mouse model. In the presence of MK-2206, there was a significant increase in apoptosis in glioma cells treated with gefitinib. MK-2206 also augmented the autophagy-inducing effect of gefitinib, as evidenced by increased levels of the autophagy marker, LC3-II. Inhibition of autophagy by silencing of the key autophagy gene, beclin 1 or 3-MA, further increased the cytotoxicity of this combinatorial treatment, suggesting that autophagy induced by these agents plays a cytoprotective role. Notably, at 48 hours following the combinatorial treatment, the level of LC3-II began to decrease but Bim was significantly elevated, suggesting a switch from autophagy to apoptosis. Based on the synergistic effect of MK-2206 on gefitinib observed in this study, the combination of these two drugs may be utilized as a new therapeutic regimen for malignant glioma.
Keywords: MK-2206, gefitinib, apoptosis, autophagy, glioblastoma
Introduction
Activation of epidermal growth factor receptor (EGFR), a cell surface protein belonging to the ErbB receptor tyrosine kinase family, exerts stimulatory effects on a number of oncogenic signaling cascades such as RAS, phosphoinositide 3-kinase (PI3K), and mitogen-activated protein kinase (MAPK) pathways, thereby promoting proliferation, growth and survival of tumor cells (1). Based on the essential role of the EGFR-initiated signaling in tumor development and progression, this receptor tyrosine kinase has been actively pursued as a therapeutic target for cancer treatment (2). Thus far, EGFR inhibitors such as gefitinib, cetuximab, and erlotinib have already been approved as targeted therapies for treating patients with lung cancer, colon cancer, breast cancer, and some other types of malignancies (3).
Malignant gliomas are the most common and aggressive type of primary brain tumors in humans. Despite optimal treatment with surgery, chemotherapy and radiation therapy, the prognosis of this disease remains poor. Thus, more effective therapeutic interventions are urgently needed in order to improve the treatment of this malignancy. Since mutations or aberrant expression of EGFR is frequent in this type of cancer, use of inhibitors of EGFR is believed to be an effective therapeutic intervention for patients with malignant gliomas (4-5). Indeed, EGFR inhibitors such as lapatinib have been reported to exhibit inhibitory effects on proliferation and migration of glioma cells and to activate apoptosis (6). Gefitinib (Iressa, ZD1839), another selective inhibitor of EGFR tyrosine kinase, has also been shown to possess inhibitory effects on both cell viability and invasion in maligant gliomas that have amplification of EGFR (7). In addition, inhibitors of EGFR such as erlotinib and gefitinib have been reported to inhibit proliferation of tumor-initiating cells in human glioma (8). Nevertheless, in the reported clinical trials, gefitinib only demonstrated modest anti-glioma efficacy (9). More recently, it was shown that, although gefitinib can reach the brain tumors in high concentrations and efficiently dephosphorylate EGFR, this drug is not sufficient to repress the activity of the pathway (10). It is likely that single molecular targeted therapy may not be enough to control the complex pathogenesis of glioblastoma, and that targeting multiple signaling molecules of the EGFR-initiated pathways may produce better anti-tumor effects.
The Akt serine/threonine kinase family, which consists of Akt1, Akt2 and Akt3, is a central component of the PI3 kinase pathway, and aberrant activation of Akt is found to be common in malignant glioma (11-12). The activity of Akt is closely regulated by the EGFR signaling, and has inhibitory effects on apoptosis. Recently, inhibiting Akt has been shown to possess autophagy-inducing effect in addition to apoptogenic effect (13), so as do the EGFR inhibitors including gefitinib (14-15). Autophagy is a catabolic process that degrades cytoplasmic components via the lysosomal machinery. Induction of autophagy can promote either cell survival or cell death, depending on different circumstances (16). In light of the critical role of Akt in the EGFR-initiated pathway, we sought to determine whether dual inhibitions of EGFR and Akt by corresponding inhibitors would reinforce the anti-glioma efficacy of EGFR inhibitors, and what roles autophagy would play in determining the anti-tumor efficacy of the combinatorial therapy. It has been reported that the novel small molecule allosteric inhibitor of Akt, MK-2206, which is being tested both in pre-clinical settings and clinical trials as an anticancer agent, can synergistically enhance the antitumor efficacy of some conventional chemotherapeutic drugs and molecular targeted agents in pre-clinical models of lung cancer, ovarian cancer and breast cancer (17-19). In this study, we focused on testing whether combining MK-2206 with gefitinib would improve the anti-glioma activity of this EGFR inhibitor, and how induction of autophagy would affect apoptotic cell death caused by dual inhibitions of EGFR and Akt. We found that MK-2206 synergistically enhanced the cytocidal effects of gefitinib on glioma cells, and the mechanism of this synergism involved the Akt inhibition-mediated modulation of apoptosis and autophagy in tumor cells treated with the targeted therapies. The results of the current study recommend that that combined administration of gefitinib and MK-2206 should be considered as a new therapeutic tactic that warrants clinical investigation for treating patients with malignant brain tumors.
Materials and Methods
Cell lines and culture
The human glioblastoma cell lines, T98G, LN229, and U87MG were purchased from American Type Culture Collection. These glioma cell lines all express EGFR. T98G cells were cultured in Ham's F-10: DMEM (10:1) medium, and LN229, U251 and U87MG cells were cultured in DMEM medium. These media were supplemented with 10% fetal bovine serum, 100 units/mL of penicillin, and 100 μg/mL of streptomycin. Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2/95% air. All cell cultures were monitored routinely and found to be free of contamination by mycoplasma or fungi. All cell lines were discarded after three months and new lines propagated from frozen stocks.
Reagents and antibodies
MK-2206 was a gift from Merck & Co. Inc. Gefitinib was purchased from LC Laboratories. 3-MA, Bafilomycin A1 and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma. Antibodies to LC-3, Akt, Bim, survivin, phospho-mTOR, mTOR, phospho-S6 kinase (Ser371), S6 kinase, phospho-EGFR and EGFR, were purchased from Cell Signaling Technologies. Antibodies to Beclin 1 and α-tubulin were purchased from Santa Cruz. All cell culture media were purchased from Invitrogen. Western blot reagents were obtained from Pierce Biotechnology.
Apoptosis assay
Apoptosis was determined by flow cytometric analysis of Annexin V and 7-AAD staining. Briefly, 100 μl of Guava Nexin reagent (Millipore) was added to 1 × 105 cells (in 100 μl) and the cells were then incubated with the reagent for 20 min at room temperature in the dark. At the end of incubation, the samples were analyzed by a Guava EasyCyte™ Plus FlowCytometry System (Millipore).
Western blot
Cells were lysed in the M-PER mammalian protein extraction reagent (Thermo Scientific) supplemented with a protease inhibitor cocktail (Roche) at room temperature for 5 minutes, followed by centrifugation at 14,000 × g for 10 minutes. Protein concentrations of cell lysates were measured using the Bio-Rad DC assay reagent (Bio-Rad). Proteins (20-40 μg) were resolved by SDS-PAGE and then transferred to PVDF membrane (Bio-Rad). The PVDF membranes were incubated with the respective antibodies in 3% BSA/TBST at 4°C for overnight, followed by incubation with secondary antibody at room temperature for 1 h. Protein signals were detected by ECL method following the manufacturer's protocol.
siRNA transfection
siRNA targeting Beclin 1 was prepared by Dharmacon Research. Scrambled siRNA was used as a control. Transfection of siRNA was performed according to the manufacturer's protocol. Briefly, cells in exponential phase of growth were plated in six-well tissue culture plates at 1× 105cells per well, grown for 24 h, then transfected with siRNA using Oligofectamine and OPTI-MEM I–reduced serum medium. The concentrations of siRNAs were chosen based on dose-response studies.
Cellular viability assay
Cell viability was measured by MTT assay. Briefly, cells were plated at 5×103 cells per well in 96-well tissue culture plates, and subjected to different treatments at 37 °C for 72h in a humidified atmosphere containing 5% CO2/95% air. At the end of treatments, MTT was added to each well and incubated for another 4 h. The formazan product was dissolved in DMSO and read at 570 nm on a Victor3 Multi Label plate reader (PerkinElmer).
Animal experiments
Six-week-old male BALB/c nude mice were used for intra-cerebral implantation of glioma cells. Briefly, human glioma cells LN229 (1 × 105 cells in 15 μl of DMEM medium) were injected into the brains at 4 mm depth under anesthesia with chloralic hydras (4%, 2ml/kg, ip). Three days after tumor cell implantation, mice were randomly divided into four groups (15 mice/group). Treatments were begun on day 4, according to the following regimens: gefitinib, 80 mg/kg, p.o., for 5 consecutive days; MK-2206, 100 mg/kg, p.o., three times per week for 2 weeks; gefitinib + MK-2206, given as same as above; control, 10% DMSO in saline, p.o., given as same as above. Animal body weight and physical signs were monitored daily during the experiments. The mice were housed in a temperature-controlled and light-controlled environment in the animal facility. At day 17 post-inoculation, the mice were euthanized, and the brains were fixed in 10% buffered formalin, embedded in paraffin, and then stained with hematoxillin-eosin (H&E). The animal experiments were approved by the Institutional Animal Care and Use Committee of our university.
Statistical analysis
The differences between treatments were analyzed using a two-sample t-test. The survival curves of the tumor - bearing mice subjected to different treatments were estimated using Kaplan-Meier method and compared by log-rank statistic analysis.
Results
Gefitinib simultaneously induces apoptosis and autophagy in human glioma cells
Apoptosis and autophagy are the two cellular processes likely to alter efficacy of a therapeutic agent. In this study, we observed that treatment of the human glioma cell lines LN229 and T98G with gefitinib, which decreased the phosphorylation of p-EGFR (Fig. 1B), triggered both apoptosis and autophagy in a dose-dependent manner, as evidenced by increases in the number of cells with Annexin V staining (Fig. 1C), an indicator of apoptosis, and in the amount of LC3 II (Fig. 1D), a specific marker of autophagy. We also demonstrated that in the presence of bafilomycin A1 (an inhibitor of lysosomal protease), the accumulation of LC3 II was more abundant, indicating an increase in autophagic flux (Fig. 1D). Stimulatory effect of low concentration (0.5 μM) but inhibitory effect of high concentrations (1 and 5 μM) of gefitinib on EGFR phosphorylation was also observed by others (20). Treatment with gefitinib also caused a dose-dependent decrease in the phosphorylation of Akt (Fig. 1E), which indicates an inhibition of Akt activity. These results imply that activation of apoptosis and autophagy in response to gefitinib treatment could be a part of cellular processes that determine the cytocidal efficacy of this EGFR inhibitor.
Figure 1. Simultaneous induction of apoptosis and autophagy, and inhibition of Akt, by gefitinib in human glioma cells.
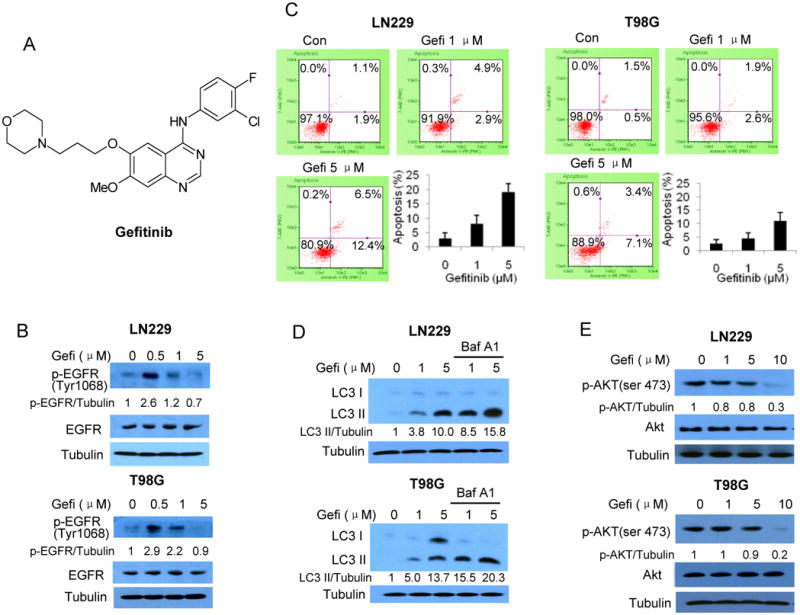
(A) Chemical structure of gefitinib. (B) LN229 and T98G cells were treated with gefitinib for 24 h; the levels of p-EGFR and EGFR were examined by Western blot. Tubulin was used as a loading control. (C) LN229 and T98G cells cultured in medium supplemented with 10% fatal bovine serum were treated with gefitinib for 60 h, and apoptosis was examined by measuring Annexin V staining using flow cytometry. (D) LN229 and T98G cells were treated with gefitinib for 24 h in the absence or presence of Bafilomycin A1, and the levels of LC3 were examined by Western blot. Tubulin was used as a loading control. (E) LN229 and T98G cells were treated with gefitinib for 48 h, and the levels of Akt and p-Akt were measured by Western blot. Tubulin was used as a loading control.
The Akt inhibitor, MK-2206, synergistically increases the cytocidal effect of gefitinib on human glioma cells
As gefitinib showed an inhibitory effect on Akt activity (Fig. 1E), and this action might account, at least in part, for its pro-apoptotic effect, we next tested in LN229, T98G and U87MG human glioma cell lines whether combining an Akt inhibitor with gefitinib would enhance the cytocidal efficacy of this EGFR inhibitor. Fig. 2 shows that the novel allosteric small molecule Akt inhibitor MK-2206, at a non-cytotoxic concentration (0.5 μM), inhibited the activation of Akt (Fig. 2B) and increased the cytocidal activity of gefitinib against the glioma cells (Fig. 2C, D and E). In the presence of MK-2206, the IC50s of gefitinib were decreased ∼2-3 fold (Fig. 2F). Although the IC50s of gefitinib in the presence of MK-2206 were still in the μM ranges, these concentrations are achievable in the brain tumor tissues, as gefitinib has a high ability to penetrate into solid tumor tissues including glioma (21-23), and gefitinib concentration in tumor tissues can be 10 – 40 fold higher than that in blood (21-26). Analysis of the effects of the drug combination using the Compusyn synergism/antagonism analysis software (ComboSyn, Inc., Paramus, NJ) informed that MK-2206 acted synergistically with gefitinib in producing cytocidal activity. The combination indexes (CI) obtained were all below 1.0 (range: 0.16 ∼ 0.85) (Fig. 2G), indicating that synergism varies between strong and moderate (27).
Figure 2. Effects of gefitinib on viability of glioma cells in the absence or presence of MK-2206.

(A) Chemical structure of MK-2206. (B) LN229 and T98G cells were treated with MK-2206 for 24 h, and the levels of p-Akt and Akt were examined by Western blot. Tubulin was used as a loading control. (C, D, and E) LN229, T98G and U87MG human glioma cells cultured in medium supplemented with 10% fetal bovine serum were treated with a series of concentrations of gefitinib for 72h in the absence or presence of 0.5 μM of MK-2206. At the end of treatments, the cell viability was measured by MTT assay. Each bar represents the mean ± SE of three experiments. *p< 0.05, **p<0.01. (F) IC50s of gefitinib in the absence or presence of 0.5 μM of MK-2206. (G) Combination indices for gefitinib and MK-2206 combination therapy, as computed by CompuSyn.
MK-2206 augments both apoptogenic and autophagenic effects of gefitinib at early stages but switches autophagy to apoptosis at late stages of the treatment
To investigate the mechanisms underlying the synergistic cytocidal effect between gefitinib and MK-2206, we compared apoptotic and autophagic activity in cells treated with gefitinib alone or with combination of gefitinib and MK-2206. As shown in Fig. 3A, co-treatment with gefitinib and MK-2206 for 60 h increased the percentage of cells with Annexin V staining, as compared with treatment with gefitinib alone, indicating an increase in apoptosis. Augmentation of apoptosis by this combined drug treatment was further evidenced by a down-regulation of the anti-apoptotic protein, survivin, and an up-regulation of the pro-apoptotic protein, Bim (Fig. 3B). Autophagic activity, as determined by Western blot of LC3 II and microscopic observation of GFP-LC3 localization, was also higher in the cells co-treated with gefitinib and MK-2206, as compared to those treated with gefitinib alone (Fig. 3C and D), indicating that MK-2206 also augments the gefitinib-induced autophagy.
Figure 3. Effects of MK-2206 on the gefitinib-induced apoptosis and autophagy.
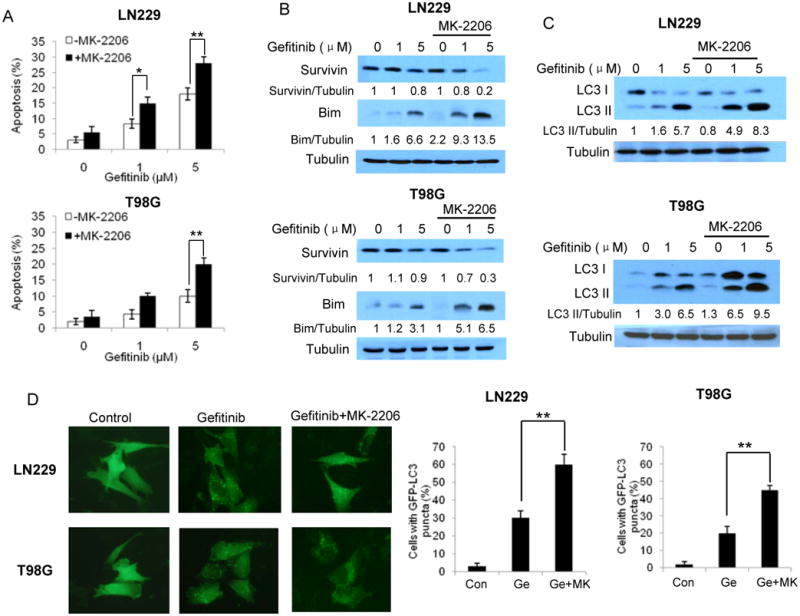
(A) LN229 and T98G cells were treated with various concentrations of gefitinib for 60 h in the absence or presence of 0.5 μM of MK-2206. At the end of treatment, apoptosis was examined by measuring Annexin V staining using flow cytometry. Each bar represents the mean ± SE of three experiments. *p < 0.05, **p < 0.01. (B) LN229 and T98G cells were treated with various concentrations of gefitinib for 24 h in the absence or presence of 0.5 μM of MK-2206. The levels of survivin and Bim were examined by Western blot. Tubulin was used as a loading control. (C) LN229 and T98G cells were treated with gefitinib for 24 h in the absence or presence of 0.5 μM MK-2206, and the levels of LC3 were examined by Western blot. Tubulin was used as a loading control. (D) LN229 and T98G cells were transfected with a GFP-LC3 plasmid, and then treated with gefitinib for 24 h in the absence or presence of 0.5 μM of MK-2206. At the end of treatment, the cells were examined by fluorescence microscopy. Bars are the quantification of the percentage of cells with 10 or more GFP-LC3 puncta. At least 100 cells were scored in each treatment. * *, P < 0.01, t-test.
To delineate the relationship between autophagy and apoptosis activated by gefitinib and MK-2206, we extended our observation of these two cellular processes to 96 h. We found that activation of autophagy by gefitinib, as indicated by the levels of LC3 II, lasted up to 96 h (Fig. 4A); by contrast, in the presence of MK-2206, autophagy levels reached a peak at 48 h, but began to decline thereafter (Fig. 4B). Notably, with the decrease of autophagy (LC3 II) after 48 h, the level of the pro-apoptotic protein Bim was further increased (Fig. 4B). In contrast, no detectable increase in the Bim level was observed in the cells treated with gefitinib alone (Fig. 4A). Elevations of Bim in the tumor cells subjected to combinatory treatment were accompanied by increases in apoptotic cell death (Fig. 4C). The importance of Bim in induction of apoptosis was further demonstrated in the experiments showing that knocked down of Bim by siRNA reduced apoptotic rate in those treated cells (Fig. 4D). These results suggest that excessive autophagy promoted by MK-2206 may trigger a switch from autophagy to apoptotic cell death.
Figure 4. Prolonged co-treatment with gefitinib and MK-2206 promotes the switch from autophagy to apoptosis.

(A) LN229 and T98G cells were treated with 5 μM of gefitinib for different time periods, and the levels of LC3 and Bim were examined by Western blot. Tubulin was used as a loading control. (B) LN229 and T98G cells were treated with 5 μM gefitinib for different time periods in the absence or presence of 0.5 μM of MK-2206, and the levels of LC3 and Bim were examined by Western blot. Tubulin was used as a loading control. (C) LN229 and T98G cells were treated with 5 μM of gefitinib for different periods of time in the absence or presence of 0.5 μM of MK-2206. Apoptosis was examined by measuring Annexin V staining using flow cytometry. (D) LN229 and T98G cells were transfected with a Bim siRNA or a non-targeting siRNA, and then treated with gefitinib for 60h in the absence or presence of 0.5 μM of MK-2206. Apoptosis was examined by measuring Annexin V staining using flow cytometry.
Suppression of autophagy enhances the efficacy of combined treatment with gefitinib and MK-2206
As autophagy can play either a pro-survival or pro-death role under various stressful conditions, we next wanted to understand how activation of autophagy affected viability of glioma cells treated with gefitinib alone or with the combination of gefitinib and MK-2206. In these experiments, we first suppressed autophagy by silencing the expression of Beclin 1, a key autophagy-regulatory gene, and then determined the viability of tumor cells subjected to the treatments. Fig. 5A and Fig. 5B show that inhibition of autophagy via transfection with a Beclin 1-targeted siRNA further decreased viability of the cells treated with the combinatorial treatment, as compared to transfection with a non-targeting RNA. The reduced cellular viability seen in Fig. 5A and Fig. 5B appears to result from apoptosis, as suppression of autophagy by silencing Beclin 1 further increased apoptotic cell death rate of the cells subjected to the combinatorial treatment (Fig. 5C and Fig. 5D). The effect of autophagy on cytocidal effect of the combinatorial treatment was further verified through use of the autophagy inhibitor, 3-MA (1 mM) (Supplementary Fig. 1A and Figure 1B). These results suggest that induction of autophagy serves as a compensatory mechanism in tumor cells in response to the targeted therapies.
Figure 5. Suppression of autophagy by silencing of Beclin 1 expression sensitizes glioma cells to the combinatorial treatment with gefitinib and MK-2206, and the effects of gefitinib on activity of mTOR and S6K in the absence or presence of MK-2206.
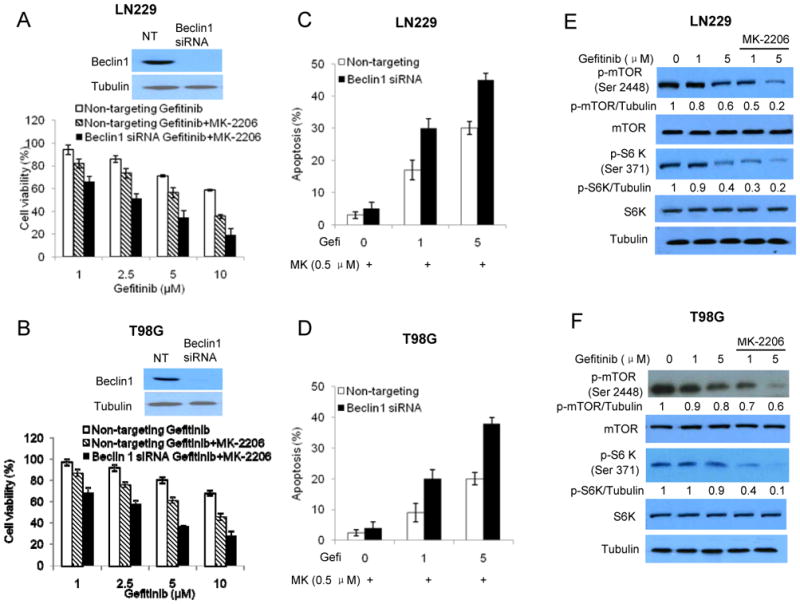
(A, B) LN229 and T98G cells were transfected with a Beclin1 siRNA or a non-targeting siRNA, and then treated with a series of concentrations of gefitinib for 72h in the absence or presence of 0.5 μM of MK-2206. Cell viability was measured by MTT assay. (C, D) LN229 and T98G cells were transfected with a Beclin1 siRNA or a non-targeting siRNA, and then treated with gefitinib (1 and 5 μM) for 60h in the absence or presence of 0.5 μM of MK-2206. Apoptosis was examined by measuring Annexin V staining using flow cytometry. (E, F) LN229 and T98G cells were treated with various concentrations of gefitinib for 24 h in the absence or presence of 0.5 μM of MK-2206. The levels of phospho-mTOR (Ser2448), mTOR, phospho-S6K (Ser371) and S6K were examined by Western blot. Tubulin was used as a loading control.
Regulation of autophagy in glioma cells co-treated with gefitinib and MK-2206 is mediated via the mTOR-S6 kinase pathway
To explore the signaling pathway involved in the modulation of autophagy by combinatorial treatment with gefitinib and MK-2206, we examined the activities of mTOR and S6 kinase, two components downstream of the EGFR-PI3 kinase-Akt cascade with known roles in the regulation of autophagy in response to cellular stress. As shown in Fig. 5E and Fig. 5F, in comparison to treatment with gefitinib alone, the combinatorial treatment with gefitinib and MK-2206 markedly decreased the levels of phospho-mTOR and phospho-S6 kinase, indicating a deactivation of the mTOR-S6 kinase pathway. Since inhibition of mTOR and S6 kinase is known to induce autophagy, the decreases in the activity of this pathway by dual inhibition of EGFR and Akt may account for the activation of autophagy by gefitinib and MK-2206, as shown in Fig. 3C.
Combinatorial treatment with gefitinib and MK-2206 produces a stronger anti-tumor- activity in an intracranial glioma mouse model
To determine the in vivo relevance of the above observations, we tested the therapeutic benefits of the combinatorial treatment with gefitinib and MK-2206 in an orthotopic glioma mouse model in which the LN229 human glioma cells were implanted intracranially. In these experiments, the tumor-bearing mice were either treated with vehicle, gefitinib (80 mg/kg) or MK-2206 (100 mg/kg) alone, or with combination of the two drugs. Fig. 6A shows that as compared to gefitinib treatment alone, co-administration of this EGFR inhibitor with MK-2206 demonstrated a better therapeutic benefit, as indicated by a significant increase in the survival of the tumor - bearing mice (p = 0.0155). The greater anti-tumor effect of this combinatorial treatment than gefitinib alone was also evidenced by histologic examinations of the brain tissues on the day 17 following tumor inoculations. As shown in Fig. 6B, although gefitinib and MK-2206 individually showed inhibitory effects on tumor growth in comparison to vehicle, the brain tissues of the tumor - bearing mice treated with the combination of gefitinib and MK-2206 contained even smaller tumor masses and less glioma cells as compared to those treated with gefitinib or MK-2206 alone, suggesting that the prolonged survival of the mice receiving combination therapy might be attributed to the less brain damage caused by glioma.
Figure 6. Effect of the combinatorial treatment with gefitinib and MK-2206 on tumor growth in an intracranial glioma model.

The human glioma cells LN229 (1 × 105 cells in 15 μl of DMEM medium) were injected into the brains of 6-week-old male BALB/c nude mice at 4 mm depth under anesthesia with chloralic hydras (4%, 2ml/kg, ip). Three days after tumor cell implantation, mice were randomly divided into four groups (15 mice/group). Treatments were begun on day 4, according to the following regimens: gefitinib, 80 mg/kg, p.o., 5 consecutive days; MK-2206, 100 mg/kg, p.o., three times per week for 2 weeks; gefitinib + MK-2206, same as above; control, 10% DMSO in saline, p.o. (A) The kaplan-Meier survival curves, n = 10; gefitinib + MK-2206 vs. gefitinib, p = 0.0155, log-rank statistic analysis; (B) At day 17 after tumor cell implantation, the mice were euthanized, and the brains were fixed in 10% buffered formalin, embedded in paraffin, and then stained with hematoxillin-eosin (H&E). The images shown are the representative of 5 mice from each group.
Discussion
The EGFR-initiated pathway is considered an attractive therapeutic target in malignant glioma due to its frequent dysregulation in this disease. Thus, small-molecule inhibitors of EGFR tyrosine kinase such as gefitinib are being evaluated as anti-glioma therapies (28). Also, it has been noticed that sensitivity of tumor cells to gefitinib is closely correlated to their dependence on Akt activation for survival and proliferation (29), and this suggests that inhibiting Akt may serve as an approach to improving the anti-tumor efficacy of EGFR inhibitors. In the current study, we evaluated whether or not combining the novel allosteric Akt inhibitor, MK-2206, with gefitinib could enhance the anti-glioma activity of this targeted therapy. Our results demonstrated that gefitinib in combination with MK-2206 produced a synergistic effect against glioma cells (Fig. 2) and enhanced the antitumor activity in an orthotopic glioma mouse model (Fig. 6). It was reported that the responsiveness to EGFR inhibitors is associated with the co-expressions of EGFRvIII and functional PTEN in glioma cells (30), and that downstream inhibition of the PI3K pathway can be combined with EGFR inhibitors to promote responsiveness in patients with PTEN-deficient tumors (31). Indeed, the synergistic effect reported here was observed not only in the glioma cells harboring wild-type PTEN (LN229 cells) but also in the tumor cells with PTEN mutant (T98G cells) or PTEN null (U87MG cells). PTEN is a tumor suppressor that acts as a critical negative regulator of PI3K-Akt-mTOR signaling axis. Loss of PTEN is one of the most common genetic lesions, and occurs in 60-80% of malignant gliomas. A previous study suggests that GBM patients who have high levels of EGFR expression and low levels of phosphorylated Akt had better response to EGFR inhibitor erlotinib treatment than those with low levels of EGFR expression and high levels of phosphorylated Akt (32), but these results have not been confirmed in larger studies. Although the synergistic effect reported here may have potential impacts in the treatment of GBM, we are aware that this synergism was observed only in three glioma cell lines and in a mouse glioma model; whether this synergistic action is truly unaffected by PTEN status would need further pre-clinical and clinical investigation. Additionally, successful treatment of malignant glioma may depend on tailoring cocktails of targeted drugs to individual patients. For instance, combinations of temozolomide with inhibitors of mTOR, PI3K, Akt or with reticulum stress inhibitors have been proposed (33).
The synergism between MK-2206 and gefitinib appears to be associated with the modulation of autophagy and apoptosis in the cells subjected to the combinatory treatment of gefitinib and MK-2206 (Fig. 3 and Fig. 4). Apoptosis and autophagy are the two cellular processes known to affect efficacies of numerous cancer therapeutic agents. To explore the mechanism underlying the synergism between gefitinib and MK-2206 in killing tumor cells, we examined the activity of both apoptosis and autophagy in the glioma cells subjected to combinatorial treatment of the two agents. We observed that gefitinib simultaneously induced apoptosis and autophagy (Fig. 1C and Fig. 1D), and that in the presence of MK-2206, both of apoptosis and autophagy were increased in the tumor cells treated with gefitinib for 48 h. As autophagy may contribute to either cell survival or cell death, we then assessed the role of autophagy in determining the efficacy of the treatments. We found that suppression of autophagy significantly enhanced the cytocidal activity of the combinatorial treatments, suggesting that induction of autophagy is a compensatory response to therapeutic stress. Interestingly, 48 h after the co-treatments, autophagic activity began to decrease but apoptosis was further activated (Fig. 4B), and this shift did not occur in the glioma cells treated with gefitinib alone (Fig. 4A). Co-treatment of gefitinib with MK-2206 also caused a reduction of anti-apoptotic protein, survivin, but led to an elevation of pro-apoptotic protein, Bim (Fig. 3B). It has been reported that Bim is required for apoptosis induced by certain targeted therapy in cancer cells with oncogenic EGFR (34), and that the down-regulation of survivin plays a pivotal role in the gefitinib-induced apoptosis (35). Thus, our results are consistent with those reported observations. More importantly, our study suggests that Akt inhibitors such as MK-2206 may play a therapeutically beneficial role in promoting functional switch from autophagy to apoptosis, and this switch may account for the synergistic effect of MK-2206 on cytocidal activity of gefitinib. Evidence of a switch from autophagy to apoptosis was previously reported for temozolomide (36), a chemotherapeutic drug commonly used in treatment of GBM, and targeting autophagy has been proposed as a new approach to treating apoptosis-resistant GBM (37). Additionally, we found that mTOR-S6K pathway, which locates down-stream of Akt and normally plays a role in suppressing autophagy (38), was involved in the induction of autophagy in the cells co-treated with gefitinib and MK-2206, as evidenced by a decrease in the levels of p-mTOR and p-S6K (Fig. 5E and Fig. 5F). These observations were consistent with our previous results showing the involvement of mTOR-S6K pathway in altering autophagic response in tumor cells treated with MK-2206 (39). As mTOR and S6K are down-stream effectors of Akt, and their inhibition might cause de-inhibition of a negative feedback loop, how precisely this pathway affects the targeted therapy might be worth further study.
The need and importance for combining gefitinib with other molecular targeted agents in treating glioblastoma is also underscored by a recent phase II study showing that gefitinib exerts little effects on the activity of the down-stream pathways despite its inhibitory effect on EGFR phosphorylation (10). Therefore, multi-target based new therapeutic strategies may be necessary for better suppression of the EGFR-initiated oncogenic pathways. It has now become increasingly appreciated that manipulating autophagy and apoptosis may achieve better therapeutic outcomes in cancer treatment; hence, the molecular mechanisms and pathways involved in the regulation of the cross-talk between autophagy and apoptosis, and the approaches to exploiting these cellular process for killing cancer cells, are undergoing extensive exploration. The results reported here demonstrate that the novel allosteric Akt inhibitor, MK-2206, can synergize with gefitinib in inhibiting malignant glioma by promoting a switch from autophagy to apoptosis, suggesting that this dual targeted therapy may make these drugs more useful and valuable in the treatment of malignant glioma or other types of cancer. Considering that the majority of human malignant gliomas harbor mutations that result in activation of PI3K/Akt pathway, and that aberrant EGFR signaling is a primary contributor to the pathogenesis of gliomas, our results provide a rationale and basis for further clinical investigation of the combinatorial use of MK-2206 and gefitinib in the treatment of this disease, and thus may have potential impact in cancer therapy.
Supplementary Material
Footnotes
Supported by grants from the US Public Health Service R01CA135038, and from Merck & Co. Inc.
References
- 1.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–25. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 2.Ritter CA, Arteaga CL. The epidermal growth factor receptor-tyrosine kinase: a promising therapeutic target in solid tumors. Semin Oncol. 2003;30:3–11. doi: 10.1053/sonc.2003.50027. [DOI] [PubMed] [Google Scholar]
- 3.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358:1160–74. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 4.Sathornsumetee S, Desjardins A, Vredenburgh JJ, McLendon RE, Marcello J, Herndon JE, et al. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro Oncol. 2010;12:1300–10. doi: 10.1093/neuonc/noq099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thiessen B, Stewart C, Tsao M, Kamel-Reid S, Schaiquevich P, Mason W, et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother Pharmacol. 2010;65:353–61. doi: 10.1007/s00280-009-1041-6. [DOI] [PubMed] [Google Scholar]
- 6.Giannopoulou E, Dimitropoulos K, Argyriou AA, Koutras AK, Dimitrakopoulos F, Kalofonos HP. An in vitro study, evaluating the effect of sunitinib and/or lapatinib on two glioma cell lines. Invest New Drugs. 2010;28:554–60. doi: 10.1007/s10637-009-9290-0. [DOI] [PubMed] [Google Scholar]
- 7.Guillamo JS, de Bouard S, Valable S, Marteau L, Leuraud P, Marie Y, et al. Molecular mechanisms underlying effects of epidermal growth factor receptor inhibition on invasion, proliferation, and angiogenesis in experimental glioma. Clin Cancer Res. 2009;15:3697–704. doi: 10.1158/1078-0432.CCR-08-2042. [DOI] [PubMed] [Google Scholar]
- 8.Griffero F, Daga A, Marubbi D, Capra MC, Melotti A, Pattarozzi A, et al. Different response of human glioma tumor-initiating cells to epidermal growth factor receptor kinase inhibitors. J Biol Chem. 2009;284:7138–48. doi: 10.1074/jbc.M807111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thaker NG, Pollack IF. Molecularly targeted therapies for malignant glioma: rationale for combinatorial strategies. Expert Rev Neurother. 2009;9:1815–36. doi: 10.1586/ern.09.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hegi ME, Diserens AC, Bady P, Kamoshima Y, Kouwenhoven MC, Delorenzi M, et al. Pathway Analysis of Glioblastoma Tissue after Preoperative Treatment with the EGFR Tyrosine Kinase Inhibitor Gefitinib--A Phase II Trial. Mol Cancer Ther. 2011;10:1102–12. doi: 10.1158/1535-7163.MCT-11-0048. [DOI] [PubMed] [Google Scholar]
- 11.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riemenschneider MJ, Betensky RA, Pasedag SM, Louis DN. AKT activation in human glioblastomas enhances proliferation via TSC2 and S6 kinase signaling. Cancer Res. 2006;66:5618–23. doi: 10.1158/0008-5472.CAN-06-0364. [DOI] [PubMed] [Google Scholar]
- 13.Degtyarev M, De Maziere A, Orr C, Lin J, Lee BB, Tien JY, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008;183:101–16. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng Y, Li H, Ren X, Niu T, Hait WN, Yang J. Cytoprotective effect of the elongation factor-2 kinase-mediated autophagy in breast cancer cells subjected to growth factor inhibition. PLoS One. 2010;5:e9715. doi: 10.1371/journal.pone.0009715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han W, Pan H, Chen Y, Sun J, Wang Y, Li J, et al. EGFR Tyrosine Kinase Inhibitors Activate Autophagy as a Cytoprotective Response in Human Lung Cancer Cells. PLoS One. 2011;6:e18691. doi: 10.1371/journal.pone.0018691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eskelinen EL. Doctor Jekyll and Mister Hyde: autophagy can promote both cell survival and cell death. Cell Death Differ. 2005;12:1468–72. doi: 10.1038/sj.cdd.4401721. [DOI] [PubMed] [Google Scholar]
- 17.Balasis ME, Forinash KD, Chen YA, Fulp WJ, Coppola D, Hamilton AD, et al. Combination of Farnesyltransferase and Akt Inhibitors Is Synergistic in Breast Cancer Cells and Causes Significant Breast Tumor Regression in ErbB2 Transgenic Mice. Clin Cancer Res. 2011;17:2852–62. doi: 10.1158/1078-0432.CCR-10-2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meng J, Dai B, Fang B, Bekele BN, Bornmann WG, Sun D, et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PLoS One. 2010;5:e14124. doi: 10.1371/journal.pone.0014124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9:1956–67. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 20.Premkumar DR, Arnold B, Pollack IF. Cooperative inhibitory effect of ZD1839 (Iressa) in combination with 17-AAG on glioma cell growth. Mol Carcinog. 2006;45:288–301. doi: 10.1002/mc.20141. [DOI] [PubMed] [Google Scholar]
- 21.Hofer S, Frei K. Gefitinib concentrations in human glioblastoma tissue. J Neurooncol. 2007;82:175–6. doi: 10.1007/s11060-006-9257-3. [DOI] [PubMed] [Google Scholar]
- 22.Hofer S, Frei K, Rutz HP. Gefitinib accumulation in glioblastoma tissue. Cancer Biol Ther. 2006;5:483–4. doi: 10.4161/cbt.5.5.2653. [DOI] [PubMed] [Google Scholar]
- 23.McKillop D, Partridge EA, Kemp JV, Spence MP, Kendrew J, Barnett S, et al. Tumor penetration of gefitinib (Iressa), an epidermal growth factor receptor tyrosine kinase inhibitor. Mol Cancer Ther. 2005;4:641–9. doi: 10.1158/1535-7163.MCT-04-0329. [DOI] [PubMed] [Google Scholar]
- 24.Fury MG, Solit DB, Su YB, Rosen N, Sirotnak FM, Smith RP, et al. A phase I trial of intermittent high-dose gefitinib and fixed-dose docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2007;59:467–75. doi: 10.1007/s00280-006-0286-6. [DOI] [PubMed] [Google Scholar]
- 25.Wang S, Guo P, Wang X, Zhou Q, Gallo JM. Preclinical pharmacokinetic/pharmacodynamic models of gefitinib and the design of equivalent dosing regimens in EGFR wild-type and mutant tumor models. Mol Cancer Ther. 2008;7:407–17. doi: 10.1158/1535-7163.MCT-07-2070. [DOI] [PubMed] [Google Scholar]
- 26.Wang S, Zhou Q, Gallo JM. Demonstration of the equivalent pharmacokinetic/pharmacodynamic dosing strategy in a multiple-dose study of gefitinib. Mol Cancer Ther. 2009;8:1438–47. doi: 10.1158/1535-7163.MCT-09-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 28.Halatsch ME, Schmidt U, Behnke-Mursch J, Unterberg A, Wirtz CR. Epidermal growth factor receptor inhibition for the treatment of glioblastoma multiforme and other malignant brain tumours. Cancer Treat Rev. 2006;32:74–89. doi: 10.1016/j.ctrv.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Ono M, Hirata A, Kometani T, Miyagawa M, Ueda S, Kinoshita H, et al. Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol Cancer Ther. 2004;3:465–72. [PubMed] [Google Scholar]
- 30.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–24. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez J, de Groot J. Combination therapy for malignant glioma based on PTEN status. Expert Rev Anticancer Ther. 2008;8:1767–79. doi: 10.1586/14737140.8.11.1767. [DOI] [PubMed] [Google Scholar]
- 32.Haas-Kogan DA, Prados MD, Tihan T, Eberhard DA, Jelluma N, Arvold ND, et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005;97:880–7. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- 33.Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol. 2005;23:2411–22. doi: 10.1200/JCO.2005.03.089. [DOI] [PubMed] [Google Scholar]
- 34.Gong Y, Somwar R, Politi K, Balak M, Chmielecki J, Jiang X, et al. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007;4:e294. doi: 10.1371/journal.pmed.0040294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okamoto K, Okamoto I, Okamoto W, Tanaka K, Takezawa K, Kuwata K, et al. Role of survivin in EGFR inhibitor-induced apoptosis in non-small cell lung cancers positive for EGFR mutations. Cancer Res. 2010;70:10402–10. doi: 10.1158/0008-5472.CAN-10-2438. [DOI] [PubMed] [Google Scholar]
- 36.Roos WP, Batista LF, Naumann SC, Wick W, Weller M, Menck CF, et al. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene. 2007;26:186–97. doi: 10.1038/sj.onc.1209785. [DOI] [PubMed] [Google Scholar]
- 37.Lefranc F, Facchini V, Kiss R. Proautophagic drugs: a novel means to combat apoptosis-resistant cancers, with a special emphasis on glioblastomas. Oncologist. 2007;12:1395–403. doi: 10.1634/theoncologist.12-12-1395. [DOI] [PubMed] [Google Scholar]
- 38.Pan T, Rawal P, Wu Y, Xie W, Jankovic J, Le W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience. 2009;164:541–51. doi: 10.1016/j.neuroscience.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 39.Cheng Y, Ren X, Zhang Y, Patel R, Sharma A, Wu H, et al. eEF-2 kinase dictates cross-talk between autophagy and apoptosis induced by Akt Inhibition, thereby modulating cytotoxicity of novel Akt inhibitor MK-2206. Cancer Res. 2011;71:2654–63. doi: 10.1158/0008-5472.CAN-10-2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.