

Abstract
Herpesviruses are thought to be highly genetically stable, and their use as vaccine vectors has been proposed. However, studies of the human gammaherpesvirus, Epstein-Barr virus, have found viral isolates containing mutations in HLA class I-restricted epitopes. Using murine gammaherpesvirus 68 expressing ovalbumin (OVA), we examined the stability of a gammaherpesvirus antigenic locus under strong CD8 T cell selection in vivo. OVA-specific CD8 T cells selected viral isolates containing mutations in the OVA locus but minimal alterations in other genomic regions. Thus, a CD8 T cell response to a gammaherpesvirus-expressed antigen that is not essential for replication or pathogenesis can result in selective mutation of that antigen in vivo. This finding may have relevance for the use of herpesvirus vectors for chronic antigen expression in vivo.
TEXT
Viruses utilize diverse mechanisms for escaping the host immune response. Many RNA viruses can rapidly alter immunogenic viral antigens, whereas large double-stranded DNA (dsDNA) viruses, such as herpesviruses, are thought to rely on subversion and evasion instead. Factors that contribute to this difference include the longer replication cycle for herpesviruses, greater fidelity of DNA-dependent DNA polymerases, and the ability of herpesviruses to encode immunomodulatory molecules in their large genomes. Herpesviruses can also establish latent infections during which virus-infected cells are less effectively recognized by the host immune response. These strategies facilitate establishment of the lifelong persistence in hosts that is a characteristic of herpesvirus infection. Thus, herpesviruses are believed to remain genetically stable in spite of a strong host adaptive immune response. Because the use of herpesviruses in vaccine protocols as a means of expressing heterologous antigens from persistent viral vectors has been previously proposed (11, 12), the stability of herpesvirus genomes under immune pressure is an important consideration.
There is evidence for mutation of immunodominant herpesvirus epitopes in response to immune pressure. A bone marrow transplant patient with Epstein-Barr virus (EBV)-associated lymphoproliferative disease who was treated with adoptively transferred EBV-specific cytotoxic T lymphocytes (CTLs) developed progressive disease and died. Tumor cells from this patient were found to be resistant to cytolysis by the EBV-specific CTLs, and sequence analysis revealed a mutation that deleted the two immunodominant epitopes recognized by the CTLs (10). Studies have also examined EBV sequence variations in human populations with differing prevalences of specific human leukocyte antigen (HLA) alleles. EBV isolates from populations with a high frequency of the HLA A11 allele were found to contain mutations in an immunodominant A11-restricted viral epitope (7, 8, 18). These mutations abrogate both peptide binding to the A11 molecule and CTL recognition of lymphoblastoid cell lines (LCL) carrying these EBV mutants. Although these data are consistent with the hypothesis that gammaherpesviruses can evolve in response to host CTL selection, this has been challenged by other studies that found no correlation between mutations in viral epitopes and the prevalence within a population of the HLA alleles to which those epitopes are restricted (5, 14, 15).
A limitation of studying human gammaherpesviruses is that they are strictly species specific and that, consequently, examination of their evolution in response to host immune pressure is restricted to correlative studies in humans. We and others have therefore studied murine gammaherpesvirus 68 (γHV68 or MHV68) as a model for gammaherpesvirus in vivo infection. Previous studies of T cell recognition of γHV68 infection in vivo have utilized a model system involving a recombinant virus expressing ovalbumin (OVA; γHV68.OVA) and transgenic mice expressing OVA-specific T cell receptors (TCR) on CD8 and CD4 T cells (OTI and OTII mice, respectively) (4, 22). Breeding of the relevant TCR-transgenic mouse onto the RAG-deficient (RAG) background generates an artificially optimized model system in which the effect of preexisting T cells with a single specificity on a defined virus-expressed antigen can be examined.
Using this system, we show here that the presence of OVA-specific OTI CD8 T cells resulted in the in vivo selection of viral escape mutants that were no longer controlled by OTI T cells due to either loss of OVA expression or expression of a mutated OVA protein. The mutations in these viruses ranged from large deletions in the OVA locus to a single nucleotide mutation that changed the SIINFEKL epitope recognized by OTI T cells to NIINFEKL, which was no longer recognized. Moreover, we show using full-genome sequencing of multiple viruses that the mutations in viruses selected in vivo are confined to the OVA locus, indicating that they likely resulted from selection exerted by the OTI T cells rather than from random mutation. These data provide direct evidence for in vivo mutation of a gammaherpesvirus in response to CD8 T cell selection.
Selection of gammaherpesvirus escape mutants by CD8 T cells specific to a virus-expressed antigen.
Infection of OTI mice bred to the RAG background (OTI-RAG mice) with γHV68.OVA has shown that OTI T cells are sufficient for controlling γHV68.OVA acute infection and for limiting latent infection (4). Despite this, OTI-RAG mice still exhibit 100% lethality following infection but with delayed kinetics relative to infected RAG mice (4). Whereas all infected RAG mice died by 17 days postinfection (dpi), all but one of the infected OTI-RAG mice survived until 25 dpi and they did not universally succumb to lethality until 68 dpi (Fig. 1A). The lethality in OTI-RAG mice was associated with productive viral replication, as 11 of 16 moribund mice examined between 30 and 68 dpi had detectable viral titers in spleen and/or lung tissues, with eight mice having at least 105 PFU/ml (not shown). Since OTI T cells limit acute replication of γHV68.OVA to below detectable levels at 8 and 16 dpi (4), the presence of infectious virus in these animals at later times postinfection suggested that the observed lethality was due to viral recrudescence.
Fig 1.

Selection of viral mutants with increased virulence in OTI-RAG mice. Briefly, γHV68.OVA was generated by inserting an expression cassette containing OVA fused to the transferrin receptor (OVA-Tfn) and driven by the CMV promoter, followed by an internal ribosome entry site (IRES) and the enhanced green fluorescent protein (EGFP) gene, as previously described (4). This insertion removes most of ORF 72, leaving behind only 281 nucleotides at its 3′ end, but does not alter ORFs upstream of ORF 72. γHV68.OVA replicates with kinetics similar to those of wild-type γHV68 in vitro (4), and disruption of ORF 72 via gene insertion does not impair either viral acute replication or establishment of latent infection in vivo (24). (A) OTI-RAG and RAG mice were infected intraperitoneally (i.p.) with γHV68.OVA at 4 × 104 PFU of virus per mouse and were followed for lethality. At 7 to 8 weeks postinfection, when infected OTI-RAG mice had died or were ill, tissues were harvested from moribund mice and 12 individual viruses were isolated via propagation in NIH 3T12 cells. These new viral isolates were designated γHV68.OVA.1 through γHV68.OVA.12. γHV68.OVA.1 was used to infect additional naïve OTI-RAG mice and was reisolated from a moribund mouse at 15 dpi and plaque purified three times using previously described methods (6). Stocks of these 12 viruses were made, and the titers were determined by a standard plaque assay as previously described (27). (B) OTI-RAG (filled squares) and RAG (filled circles) mice were infected i.p. with 4 × 104 PFU of each of the 12 new viral isolates per mouse and were followed for lethality. Lethality curves for γHV68.OVA in OTI-RAG (open squares) and RAG (open circles) mice (Fig. 1A) are overlaid in gray for comparison. These data represent the results of at least two independent experiments, and the total numbers of mice analyzed are indicated.
Two explanations for loss of OTI T cell control of viral infection are mutation of the virus and loss of OTI T cell function, either from anergy or loss of the T cells. To test whether the virus had mutated to escape OTI T cell control, we extracted 12 individual viral isolates, designated γHV68.OVA.1 through γHV68.OVA.12, from tissues of infected OTI-RAG mice at time points when other infected mice had died or were ill. We infected OTI-RAG and RAG mice with each virus and followed the mice for lethality. All 12 viruses killed RAG mice with the same kinetics as the parental γHV68.OVA (Fig. 1B). But whereas OTI-RAG mice infected with γHV68.OVA exhibited the expected delay in lethality, OTI-RAG mice infected with 11 of the 12 new viral isolates died with kinetics similar to those of infected RAG control mice. The exception was γHV68.OVA.3, whose phenotype was similar to that of parental γHV68.OVA. The fact that these new viral isolates exhibited increased virulence compared to γHV68.OVA when reinoculated into OTI-RAG mice, and that OTI-RAG mice infected with these viruses died with kinetics similar to those of infected RAG mice, demonstrates that replication of these viruses cannot be controlled by OTI T cells.
To investigate the alternative explanation that the observed viral recrudescence was due to OTI T cell loss or anergy, we evaluated the presence and activation status of OTI T cells from infected OTI-RAG mice on various dpi. The number of live splenocytes was determined by counting in the presence of trypan blue, and the splenocytes were examined by flow cytometry for expression of cell surface markers associated with T cell activation. We found no significant changes in the numbers of live splenocytes, and the splenocytes expressed markers consistent with those for antigen-stimulated T cells (not shown). Together, these data indicate that the lethality observed in γHV68.OVA-infected OTI-RAG mice at late times postinfection is due not to the loss of functional OTI T cells but to the selection of viral mutants that have escaped OTI T cell control.
Viral escape mutants have alterations in the genomic locus encoding OVA.
One explanation for the inability of OTI T cells to control replication of the viral mutants is the loss of OVA expression by these viruses. To assess this, we infected 3T12 cells with each virus and performed Western blot analyses on cell lysates to detect OVA expression. Productive infection of the 3T12 cells by each virus was confirmed by Western blot analysis for the γHV68 M3 protein. As expected, γHV68.OVA expressed OVA whereas wild-type γHV68 did not (Fig. 2A). Among the new viral isolates, γHV68.OVA.3 expressed OVA at levels similar to those seen with γHV68.OVA, whereas isolates 2 and 4 to 12 did not express detectable levels of OVA, consistent with their in vivo phenotypes. Surprisingly, γHV68.OVA.1 expressed OVA even though it was not effectively controlled by OTI T cells in vivo. To further investigate this, we sequenced the genomic locus containing the OVA expression cassette in 8 viral isolates, including γHV68.OVA.1 and γHV68.OVA.3. The six viruses that did not express detectable amounts of OVA all had large deletions in the OVA-Tfn gene (Fig. 2B), with some deletions extending into the upstream cytomegalovirus (CMV) promoter and/or the downstream enhanced green fluorescent protein (EGFP) gene and open reading frame (ORF) 72 fragment. In contrast, γHV68.OVA.3, which resembled γHV68.OVA in both OVA expression and in vivo phenotype, had no sequence alterations in the OVA-IFN gene. Interestingly, γHV68.OVA.1, which behaved like an escape mutant in vivo but still expressed OVA in Western blot analysis, had a single nucleotide change in the OVA-Tfn gene. This resulted in mutation of the immunodominant OVA 257-264 peptide recognized by OTI T cells in the context of H2-Kb from SIINFEKL to NIINFEKL (Fig. 2C). The phenotype of γHV68.OVA.1 suggests that mutation of SIINFEKL to NIINFEKL does not alter OVA expression but impairs OTI T cell responses to cells infected with this virus. Thus, a strong CD8 T cell response to a gammaherpesvirus-expressed antigen can select for mutant viruses in which the antigen is mutated.
Fig 2.

Viral mutants no longer express OVA due to mutations in the OVA locus. (A) Western blot analysis of whole-cell lysates from 3T12 cells that were subjected to mock infection or were infected with wild-type γHV68, γHV68.OVA, or one of the 12 new viral isolates. The blots were probed with a polyclonal antibody to OVA (catalog number ab1211; Abcam, Cambridge, MA), an antiserum to the γHV68 M3 protein (23), or a monoclonal antibody to β-actin (Sigma-Aldrich, St. Louis, MO). (B) Schematic showing multiple-sequence alignment of the OVA locus of γHV68.OVA and eight of the new viral isolates. The OVA expression cassette, including the CMV promoter, OVA-Tfn gene, IRES and EGFP genes, and the remaining fragment of ORF 72, is shown above the γHV68.OVA sequence. Deletions relative to the γHV68.OVA sequence are represented by horizontal black lines, and insertions and nucleotide changes are represented by vertical black lines. (C) Sequence alignment of the OVA-Tfn gene from γHV68.OVA and the γHV68.OVA.1 isolate, showing the single nucleotide mutation in γHV68.OVA.1 (represented by a vertical black line) and the resulting amino acid sequence change from SIINFEKL to NIINFEKL. All sequence alignments were generated using Geneious software (Biomatters Ltd., Auckland, New Zealand).
NIINFEKL mutation results in loss of OTI T cell activation and impaired control of viral infection.
The P1 residue of the SIINFEKL peptide is important for its ability to stabilize H2-Kb expression (3, 13), suggesting that the phenotype of γHV68.OVA.1 might be due to low-affinity binding of the NIINFEKL peptide to H2-Kb, resulting in poor presentation to CD8 T cells. To test this, we performed major histocompatibility complex (MHC) class I stabilization assays (20) using the SIINFEKL and NIINFEKL peptides and the previously characterized null peptide, EIINFEKL, which binds to MHC class I but does not activate OVA-specific T cells (13). Surface expression of MHC class I was detected by flow cytometry using the monoclonal antibody 25.D1.16, which recognizes all three peptides in complex with H2-Kb (17, 19). Cells pulsed with each of the three peptides exhibited similar levels of MHC class I cell surface expression (Fig. 3A), and similar results were obtained when MHC class I cell surface expression was detected using a monoclonal antibody to H2-Kb instead (not shown). To test whether the NIINFEKL peptide can activate OTI T cells when presented by H2-Kb, we performed an agonist assay (13) in which OTI T cells were incubated with splenocytes pulsed with SIINFEKL, NIINFEKL, or EIINFEKL. 3H-thymidine incorporation was quantitated as a readout for OTI T cell proliferation. Whereas the SIINFEKL peptide stimulated OTI T cells to proliferate, the NIINFEKL and EIINFEKL peptides did not (Fig. 3B). This is consistent with a previous report that incubation of NIINFEKL peptide with target cells does not result in OVA 257-264-specific CTL lysis of the target cells (3). Thus, the NIINFEKL peptide can bind to H2-Kb with affinity comparable to that of SIINFEKL and EIINFEKL but does not activate OTI T cells.
Fig 3.
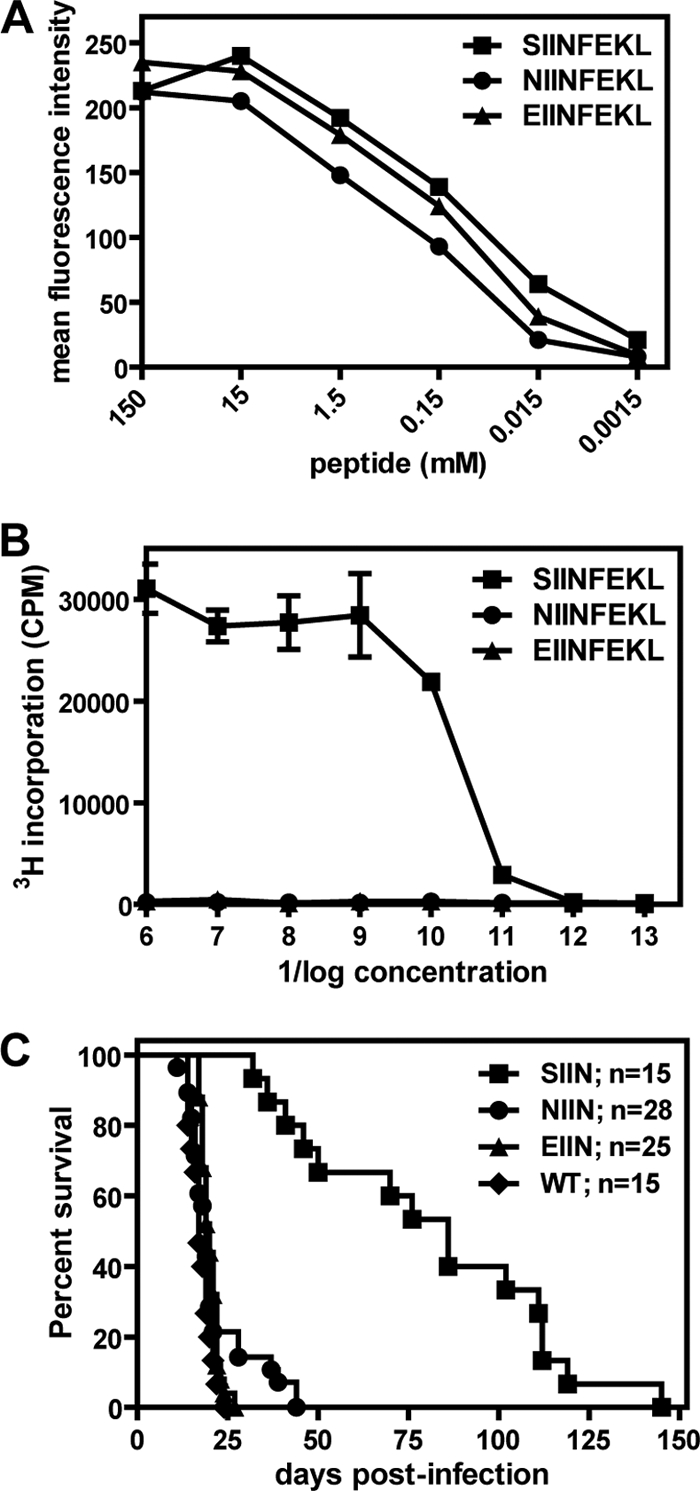
NIINFEKL peptide binds to MHC class I but fails to stimulate OTI T cells. (A) RMA-S cells were pulsed with various concentrations of the SIINFEKL, NIINFEKL, or EIINFEKL peptide and stained with the 25.D1.16 monoclonal antibody. H2-Kb cell surface expression was assessed by flow cytometry. The data are representative of results from two independent experiments. (B) OTI T cells were incubated with γ-irradiated splenocytes that had been pulsed with various concentrations of the SIINFEKL, NIINFEKL, or EIINFEKL peptide. After 24 h of coincubation, 3H-thymidine was added for an additional 12 h of incubation. 3H-thymidine incorporation was quantitated as a measure of OTI T cell proliferation. The samples were analyzed in triplicate, and these data are representative of the results of 2 independent experiments. (C) OTI-Rag mice were infected with BAC-derived wild-type γHV68 (WT) or OVA-expressing viruses containing the SIINFEKL (SIIN), NIINFEKL (NIIN), or EIINFEKL (EIIN) epitope. Insertion of the OVA expression cassette into each BAC recombinant virus, as well as the lack of gross mutations elsewhere in the viral genomes, was confirmed by Southern blot analyses (not shown). The mice were infected i.p. with either 4 × 103 or 5 × 103 PFU/mouse and were followed for lethality. These data represent the results of at least 2 independent experiments, and the total numbers of mice analyzed are indicated.
These data suggest that the increased virulence of the γHV68.OVA.1 mutant virus in OTI-RAG mice compared to that of γHV68.OVA is due to a lack of OTI T cell activation when presented with the mutant NIINFEKL peptide. This increased virulence should therefore be observed only in OTI-RAG mice. To confirm this, we infected RAG mice transgenic for the irrelevant 2C TCR (2C-RAG mice) with γHV68.OVA or γHV68.OVA.1. These mice all died with kinetics similar to those of infected RAG mice, and there were no differences observed between the two groups of infected mice (not shown). We also infected OTII-RAG mice, which have CD4 T cells specific to the OVA 323-339 peptide presented by the MHC class II molecule I-Ab (2), with γHV68.OVA or γHV68.OVA.1. Again, we observed no differences in lethality between OTII-RAG mice infected with γHV68.OVA and those infected with γHV68.OVA.1 (not shown). This confirms that the increased virulence phenotype of γHV68.OVA.1 is specific to the OTI-RAG host and that recognition of the mutated OVA protein is specifically compromised in the CD8 T cell compartment.
To formally demonstrate that the SIINFEKL-to-NIINFEKL mutation is the cause of the increased virulence phenotype of γHV68.OVA.1, we generated recombinant γHV68.OVA viruses expressing the SIINFEKL, NIINFEKL, or EIINFEKL epitope via allelic exchange (21) in the γHV68 bacterial artificial chromosome (BAC) system (1). All four BAC-derived viruses killed RAG mice with the same kinetics as observed for γHV68.OVA and the mutant viral isolates (not shown). For OTI-RAG mice, only those infected with the SIINFEKL-expressing virus exhibited the delay in lethality observed with γHV68.OVA (Fig. 3C). Mice infected with the NIINFEKL-expressing virus died with kinetics similar to those of infected RAG mice, as did mice infected with the EIINFEKL-expressing and wild-type γHV68 BAC-derived viruses. Thus, specific introduction of the NIINFEKL mutation reproduces the increased virulence phenotype of the γHV68.OVA.1 virus in infected OTI-RAG mice, indicating that the SIINFEKL-to-NIINFEKL mutation is sufficient for the in vivo phenotype of the γHV68.OVA.1 virus.
Mutation of a gammaherpesvirus in vivo is restricted to the genomic locus under immune selection.
Given that many of the γHV68.OVA mutant viruses contained large deletions in the OVA locus, it was possible that the development of mutations in these viruses was not specific to the OVA locus under immune selection. We therefore completely sequenced the genomes of seven γHV68.OVA mutant viruses, including γHV68.OVA.1, γHV68.OVA.3, and five other γHV68.OVA mutants that had large deletions in the OVA locus. For three of the viruses (γHV68.OVA.6, γHV68.OVA.7, and γHV68.OVA.9), we were unable to achieve sequence assembly across the 40-bp and 100-bp repeat regions, resulting in assembly of three contigs separated by those two repeat regions for each virus. We therefore compared sequences of the mutant viruses to the γHV68.OVA sequence for the left region of the genome up to the 40-bp repeat (Fig. 4A), the middle region of the genome between the 40-bp and 100-bp repeats (Fig. 4B), and the right region of the genome starting after the 100-bp repeat (Fig. 4C). We found that none of the viruses had large deletions or insertions outside the OVA locus. For the three mutant viruses for which we were unable to obtain full-length assemblies, we generated additional assemblies using the γHV68.OVA theoretical sequence as the reference genome and showed that none of the sequence reads mapped to the regions of these genomes in which large deletions were found (not shown). This confirms that the OVA locus deletions observed in γHV68.OVA.6, γHV68.OVA.7, and γHV68.OVA.9 (Fig. 2B) were not due to assembly errors or low coverage.
Fig 4.

Mutations in the new viral isolates are confined to the OVA locus. The schematic shows multiple sequence alignments of γHV68.OVA (black bar) and the seven viral isolates that were fully sequenced (light gray bars). The figure shows (A) the left end of the genomes up to the 40-bp repeat region, (B) the middle of the genomes between the 40-bp and 100-bp repeat regions, and (C) the right end of the genomes from the 100-bp repeat region onward. γHV68 ORFs are represented by gray arrows; ORFs and other features of the OVA expression cassette are represented by black arrows. ORF labels that did not fit within their respective arrows are shown outside the arrows. Deletions relative to the γHV68.OVA sequence are represented by horizontal black lines, while insertions and nucleotide changes relative to the γHV68.OVA sequence are represented by vertical black lines. Due to the compression of the genome sequences, mutations that are close together may appear as single horizontal or vertical black lines in this schematic. Briefly, genomic DNA for each virus was prepared as previously described (25) and sequenced on the 454 GS-FLX platform (454 Life Sciences) according to the manufacturer's instructions. For each virus, randomly selected sets containing different numbers of sequence reads were assembled using Newbler as previously described (16) and the largest contig generated is reported here. The estimated fold coverage for each assembled genome was as follows: for γHV68.OVA.1, 42-fold; for γHV68.OVA.2, 53-fold; for γHV68.OVA.3, 52-fold; for γHV68.OVA.5, 41-fold; for γHV68.OVA.6, 12-fold; for γHV68.OVA.7, 11-fold; and for γHV68.OVA.9, 10-fold. All sequence alignments were generated using Geneious software (Biomatters Ltd., Auckland, New Zealand).
Moreover, there were very few nucleotide differences between the mutant viruses and γHV68.OVA outside the OVA locus. Across the seven mutant viruses sequenced, we identified a total of 26 nucleotide changes (excluding the nucleotide change in γHV68.OVA.1 that resulted in the SIINFEKL-to-NIINFEKL mutation) that were not within the large deletions observed. Of these, eight were still within the OVA expression cassette. Among the 19 that were not within the OVA expression cassette, only six were within ORFs. One of these, located in γHV68.OVA.3, was in the fragment of ORF 72 that remained after the insertion of the OVA expression cassette into the ORF 72 locus of γHV68 to generate γHV68.OVA. The remaining five were all in γHV68.OVA.9, which had a large number of nucleotide changes downstream of the OVA locus deletion and extending into M11 and ORF 73 (Fig. 4C). Three of these five nucleotide changes were in M11 (amino acid changes of the initiating M to I, K to N, and D to H), one was in ORF 73 (amino acid change of P to T), and one was in the region of overlap between the rightward M11 and the leftward ORF 73 (amino acid changes of stop codon to L for M11 and Q to K for ORF 73). As these nucleotide changes in γHV68.OVA.9 were within 1.5 kb of its OVA locus deletion, it is possible that these changes were related to an as-yet-undefined molecular process that resulted in the mutation of OVA and loss of responsiveness to OTI T cell control.
Together, these data demonstrate that mutation of these viruses, particularly the large deletions, was specific to the OVA locus. This observation is consistent with the hypothesis that alterations in the OVA locus were due to immune selection by the OTI T cells.
Discussion.
We show here that infection of OTI-RAG mice containing OVA-specific CD8 T cells with an OVA-expressing γHV68 virus resulted in the selection of mutant viruses with sequence alterations specific to the OVA locus, leading to loss of OVA expression and resistance to control by OTI T cells. These results are consistent with previous studies of EBV suggesting that gammaherpesviruses can mutate immunodominant viral epitopes in response to host immune pressure. In vivo mutation of a betaherpesvirus in response to immune selection has previously been shown for murine cytomegalovirus (MCMV) recognized by Ly49H-expressing natural killer (NK) cells (9, 26). The data presented here show that a similar phenomenon can occur for a gammaherpesvirus under CD8 T cell-mediated immune pressure.
The fact that one of the viral escape mutants, γHV68.OVA.1, had a single nucleotide change resulting in mutation of the SIINFEKL peptide to the null NIINFEKL peptide suggests that mutation of gammaherpesviruses in response to immune selection can be extremely specific. Again, this is consistent with previous reports of mutations in EBV isolates that abrogate CTL recognition of immunodominant viral epitopes via alteration of only one or two amino acids within the epitopes (7, 8, 18). In theory, specific mutations in regions of OVA that are important for the efficient processing and/or presentation of the SIINFEKL peptide could also lead to viral escape from OTI T cells, although we did not observe such mutations in our study. Given that the majority of the mutant viruses we analyzed had large deletions in the OVA locus, it is possible that subtle mutations such as these occur at a lower frequency and that analysis of a larger number of mutant viruses would have been necessary to identify mutant viruses with such mutations.
In contrast to the previous studies performed with EBV, the majority of the viral escape mutants we analyzed had large deletions in the OVA locus. This difference is likely due in part to the strength of the immune selection in our model system, in which all the preexisting CD8 T cells were specific to OVA. Consistent with this idea, a 245-bp deletion that eliminated two immunodominant epitopes was observed in the EBV mutant virus from the patient with lymphoproliferative disease who received EBV-specific CTLs as adoptive immunotherapy (10). Additionally, the immune selection in our model system was against an exogenous protein that had no function for the virus and was therefore completely dispensable. It is likely that immune selection against an essential viral protein would result in changes that would be more conservative due to the necessity of preserving the function of the protein. Although there may be selective pressure other than immune selection against the retention of the OVA cassette in the γHV68 genome, we did not detect the development of deletions in the OVA locus of the γHV68.OVA and γHV68.OVA.1 viruses during these studies despite multiple passages in fibroblasts in vitro (not shown). This suggests that, at least for in vitro viral replication, there is no significant selection against retention of the OVA cassette.
These findings may have implications for the use of herpesvirus-derived vectors in vaccines, since the antigens expressed by vaccine vectors are, in general, nonessential for viral replication or latency. However, we note that our findings result from an optimized model system in which a heterologous and nonessential antigen is recognized by preexisting, antigen-specific T cells that are present at an artificially high frequency. While such a model system is useful for studying viral mutagenesis events that may normally occur at low frequencies, it may not be representative of immune selection that occurs during a natural viral infection. In addition, studies similar to those reported here have not been performed for α- or betaherpesviruses under T cell immune pressure. Thus, the degree to which our findings can be generalized to other herpesviruses is uncertain. Despite these caveats, it is clear that, at least for a gammaherpesvirus, significant mutations in the expression of inserted antigens can occur under selective immune pressure in vivo.
The fact that we were able to isolate viruses containing mutations in the OVA locus in multiple independent experiments using different stocks of γHV68.OVA (not shown) strongly suggests that in our studies, the viral mutants arose de novo during in vivo infection. However, we cannot rule out the possibility that the viral mutants represent outgrowth of sequence variants that were part of the original γHV68.OVA virus population. This would not alter our primary finding, since the viruses with altered antigen recognition were selected and grew in vivo in the presence of OTI T cells. Regardless of the mechanism for generation of viral escape mutants, the observation that their genomic sequences outside the OVA locus were virtually identical to that of the parental γHV68.OVA indicates that the mutations in the OVA locus were not the result of random genetic drift. Although these data do not prove that evolution of gammaherpesviruses due to immune pressure occurs in humans, they demonstrate that such evolution could occur and suggest that the γHV68 mouse model could be useful for further studies of gammaherpesvirus evolution in vivo.
ACKNOWLEDGMENTS
H.W.V. was supported by grant R01CA96511. D.L.P. was supported by the Division of Dermatology at Washington University. D.C.B. was supported by a fellowship from the Leukemia and Lymphoma Society and by NIH training grants T32 AI07172-22 and -21.
We thank Darren Kreamalmeyer for expert assistance with the generation and breeding of mouse lines.
Footnotes
Published ahead of print 14 December 2011
REFERENCES
- 1. Adler H, Messerle M, Wagner M, Koszinowski UH. 2000. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J. Virol. 74:6964–6974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barnden MJ, Allison J, Heath WR, Carbone FR. 1998. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 76:34–40 [DOI] [PubMed] [Google Scholar]
- 3. Barnden MJ, Heath WR, Rodda S, Carbone FR. 1994. Peptide antagonists that promote positive selection are inefficient at T cell activation and thymocyte deletion. Eur. J. Immunol. 24:2452–2456 [DOI] [PubMed] [Google Scholar]
- 4. Braaten DC, Sparks-Thissen RL, Kreher S, Speck SH, Virgin HW. 2005. An optimized CD8+ T-cell response controls productive and latent gammaherpesvirus infection. J. Virol. 79:2573–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burrows JM, et al. 1996. Unusually high frequency of Epstein-Barr virus genetic variants in Papua New Guinea that can escape cytotoxic T-cell recognition: implications for virus evolution. J. Virol. 70:2490–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clambey ET, Virgin HW, Speck SH. 2000. Disruption of the murine gammaherpesvirus 68 M1 open reading frame leads to enhanced reactivation from latency. J. Virol. 74:1973–1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Campos-Lima PO, et al. 1993. HLA-A11 epitope loss isolates of Epstein-Barr virus from a highly A11+ population. Science 260:98–100 [DOI] [PubMed] [Google Scholar]
- 8. de Campos-Lima PO, et al. 1994. T cell responses and virus evolution: loss of HLA A11-restricted CTL epitopes in Epstein-Barr virus isolates from highly A11-positive populations by selective mutation of anchor residues. J. Exp. Med. 179:1297–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. French AR, et al. 2004. Escape of mutant double-stranded DNA virus from innate immune control. Immunity 20:747–756 [DOI] [PubMed] [Google Scholar]
- 10. Gottschalk S, et al. 2001. An Epstein-Barr virus deletion mutant associated with fatal lymphoproliferative disease unresponsive to therapy with virus-specific CTLs. Blood 97:835–843 [DOI] [PubMed] [Google Scholar]
- 11. Hansen SG, et al. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hansen SG, et al. 2009. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat. Med. 15:293–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hogquist KA, et al. 1994. T cell receptor antagonist peptides induce positive selection. Cell 76:17–27 [DOI] [PubMed] [Google Scholar]
- 14. Khanna R, et al. 1997. Evolutionary dynamics of genetic variation in Epstein-Barr virus isolates of diverse geographical origins: evidence for immune pressure-independent genetic drift. J. Virol. 71:8340–8346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee SP, et al. 1995. Epstein-Barr virus isolates with the major HLA B35.01-restricted cytotoxic T lymphocyte epitope are prevalent in a highly B35.01-positive African population. Eur. J. Immunol. 25:102–110 [DOI] [PubMed] [Google Scholar]
- 16. Loh J, et al. 2011. Identification and sequencing of a novel rodent gammaherpesvirus that establishes acute and latent infection in laboratory mice. J. Virol. 85:2642–2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Messaoudi I, LeMaoult J, Nikolic-Zugic J. 1999. The mode of ligand recognition by two peptide:MHC class I-specific monoclonal antibodies. J. Immunol. 163:3286–3294 [PubMed] [Google Scholar]
- 18. Midgley RS, et al. 2003. HLA-A11-restricted epitope polymorphism among Epstein-Barr virus strains in the highly HLA-A11-positive Chinese population: incidence and immunogenicity of variant epitope sequences. J. Virol. 77:11507–11516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. 1997. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity 6:715–726 [DOI] [PubMed] [Google Scholar]
- 20. Schumacher TN, et al. 1990. Direct binding of peptide to empty MHC class I molecules on intact cells and in vitro. Cell 62:563–567 [DOI] [PubMed] [Google Scholar]
- 21. Smith GE, Fraser MJ, Summers MD. 1983. Molecular engineering of the Autographa californica nuclear polyhedrosis virus genome: deletion mutations within the polyhedrin gene. J. Virol. 46:584–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sparks-Thissen RL, Braaten DC, Kreher S, Speck SH, Virgin HW. 2004. An optimized CD4 T-cell response can control productive and latent gammaherpesvirus infection. J. Virol. 78:6827–6835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Berkel V, et al. 2000. Identification of a gammaherpesvirus selective chemokine binding protein that inhibits chemokine action. J. Virol. 74:6741–6747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Dyk LF, Virgin HW, Speck SH. 2000. The murine gammaherpesvirus 68 v-cyclin is a critical regulator of reactivation from latency. J. Virol. 74:7451–7461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Virgin HW, et al. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71:5894–5904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Voigt V, et al. 2003. Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. Proc. Natl. Acad. Sci. U. S. A. 100:13483–13488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weck KE, Barkon ML, Yoo LI, Speck SH, Virgin HW. 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 70:6775–6780 [DOI] [PMC free article] [PubMed] [Google Scholar]