

Abstract
Human cytomegalovirus (HCMV) virions are structurally complex, and the mechanisms by which they are assembled are poorly understood. However, several tegument proteins are known to be essential for proper particle assembly and maturation. Despite intense investigation, the function of many tegument proteins remains unknown. The HCMV UL94 gene is conserved among all herpesviruses and encodes a virion protein of unknown function. We demonstrate here that UL94 is a tegument protein that is expressed with true-late kinetics and localizes to the viral assembly complex during infection. To elucidate the function of UL94, we constructed a UL94-null mutant, designated UL94stop. This mutant is completely defective for replication, demonstrating that UL94 is essential. Phenotypic analysis of the UL94stop mutant shows that in the absence of UL94, viral gene expression and genome synthesis occur at wild-type levels. However, analysis of the localization of viral proteins to the cytoplasmic assembly complex shows that the essential tegument protein UL99 (pp28) exhibits aberrant localization in cells infected with the UL94stop mutant. Finally, we show that there is a complete block in secondary envelopment in the absence of UL94. Taken together, our data suggest that UL94 functions late in infection to direct UL99 to the assembly complex, thereby facilitating secondary envelopment of virions.
INTRODUCTION
Human cytomegalovirus (HCMV) is a ubiquitous member of the betaherpesvirus family. HCMV infection is predominantly asymptomatic in healthy individuals but can cause severe disease in individuals with compromised immune function. Moreover, HCMV poses a significant threat to neonates and is the leading infectious cause of birth defects in the United States (17).
HCMV is the largest of the human herpesviruses, with a >230-kbp DNA genome that has been estimated to encode more than 200 open reading frames (ORFs) (15, 16). The HCMV particle is composed of a DNA-containing nucleocapsid, a surrounding layer of virally encoded proteins referred to as the tegument, and a host-derived envelope containing virally encoded glycoproteins. Assembly of these structurally complex particles is poorly understood. This is especially true of the cytoplasmic phase of virion assembly, which includes the majority of tegument acquisition as well as final envelopment.
Many HCMV tegument and glycoproteins localize late in infection to a unique juxtanuclear structure that is referred to as the assembly complex (21). The assembly complex is formed through a dramatic relocalization of various components of the cellular secretory apparatus and is thought to be the site of final virion assembly and envelopment (4, 5, 21). While the formation of the assembly complex is a well-documented phenomenon, the subsequent events that result in the formation of mature virus particles remain elusive. Abundant viral structural proteins that are known to accumulate at the assembly complex include the tegument proteins UL32 (pp150) and UL99 (pp28), as well as the glycoproteins gB, gH, and gM:gN (9, 21, 22, 25). Phenotypic analysis of viral mutants lacking UL32 or UL99 demonstrate that the assembly complex forms normally in the absence of these proteins but that final particle maturation does not occur, indicating that these tegument proteins play essential roles in virion assembly (1, 26).
Although the mechanisms of tegumentation and viral assembly are not well understood, these processes are thought to be mediated at least in part by protein-protein interactions. Our lab and others have reported an interaction between the tegument proteins UL94 and UL99 (7, 11, 18, 30). UL99 is essential for the acquisition of the viral envelope in the cytoplasm. However, the function of UL94 is unknown (23, 26). UL94 is a core herpesvirus gene that is conserved among all members of the herpesvirus family. UL94 was previously shown to be expressed with true-late kinetics and to partition exclusively to the nuclear fraction of infected cells, suggesting a potential role in the regulation of viral or cellular gene expression (33).
We sought to further characterize UL94 and investigate its function during HCMV infection. We confirm that UL94 is expressed with true-late kinetics. However, we found that UL94 localizes almost exclusively to a juxtanuclear structure during infection that is consistent with the assembly complex. We also constructed a UL94-null mutant, designated UL94stop. The UL94stop mutant is completely defective for growth, demonstrating that UL94 is essential for HCMV replication. The UL94 mutant virus shows no defect in viral gene expression or genome synthesis, suggesting that UL94 functions during the late phase of infection, subsequent to DNA replication. Analysis of the subcellular localization of viral proteins to the assembly complex late in infection shows that while several structural proteins localize normally, UL99 displays aberrant localization in the absence of UL94. Finally, we show that there is an accumulation of nonenveloped capsids in the cytoplasm of cells infected with the UL94stop mutant. These data suggest that UL94 functions at least in part to direct the proper localization of UL99 to the assembly complex, a step that is necessary for secondary envelopment of virions.
MATERIALS AND METHODS
Cell culture and virus infections.
Human foreskin fibroblast (HFF) cells were cultured in Dulbecco modified Eagle medium supplemented with 10% (vol/vol) fetal calf serum (HyClone), 100 U of penicillin/ml, and 100 μg of streptomycin/ml in an atmosphere of 5% CO2 at 37°C. All infections were carried out with HCMV strain ADCREGFP derived from AD169 (2). For infections, HFF cells were infected for 2 h at 37°C. After a 2-h incubation period, the infection inoculum was removed and replaced with fresh complete medium.
Western blotting.
Total protein was harvested from HFF cells by trypsinization, centrifugation, and lysis in radioimmunoprecipitation assay lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 0.4 mM EDTA, 1% Ipegal, 0.1% sodium dodecyl sulfate [SDS], 0.5% deoxycholate) containing protease inhibitor cocktail (Roche). Protein concentration was determined by the method of Bradford. Lysates were boiled in 2× SDS sample buffer (62.5 mM Tris-HCl [pH 6.8], 2.5% SDS, 20% glycerol, 1% β-mercaptoethanol). Proteins were separated by SDS-PAGE on 10% polyacrylamide gels and transferred to nitrocellulose membranes (Whatman Optitran). Membranes were blocked in 5% nonfat dry milk and probed with primary and secondary antibodies. Immunoreactive proteins were detected by the ECL chemiluminescence system (Thermo).
Antibodies.
The following antibodies used for Western blotting were obtained from commercial sources: mouse α-HA (16B12; Covance), mouse α-UL94 (6J8; Santa Cruz Biotechnology), rabbit α-dsRed (632496; Clontech), mouse α-UL99 (1207; Rumbaugh-Goodwin Institute), mouse α-UL44 (1202S; Rumbaugh-Goodwin Institute), mouse αIE1/2 (1203; Rumbaugh-Goodwin Institute), mouse α-gB (1201; Rumbaugh-Goodwin Institute), mouse α-EEA1 (14; BD Transduction Labs), and mouse α-tubulin (TU-02; Santa Cruz Biotechnology). Rabbit α-UL86 was kindly provided by W. Gibson (Johns Hopkins) and has been previously described (8). Mouse α-UL32 (36-14) was kindly provided by W. Britt (University of Alabama at Birmingham) and has been previously described (21). The following horseradish peroxidase-conjugated secondary antibodies were used for chemiluminescent detection: goat α-mouse IgG (Molecular Probes), goat α-mouse IgM (Chemicon), and goat α-rabbit IgG (Zymax). Alexa 546-conjugated Fab2 goat α-mouse (Molecular Probes) was used for immunofluorescence analysis.
Trypsin digestion of purified virions.
HFF cells were infected at a multiplicity of 0.1 PFU/cell and supernatant was collected 14 days postinfection when 100% cytopathic effect (CPE) was observed. Cellular debris was removed by centrifugation at 1,500 rpm for 5 min. Supernatant was further cleared by centrifugation at 20,000 rpm for 1 h on a 20% sucrose cushion. Supernatant was decanted, and pelleted virus was resuspended in phosphate-buffered saline (PBS). Equal aliquots of purified virus were subjected to treatment with 1% Triton X-100, 1 μg of trypsin, or both at 37°C for 1 h in digest buffer (50 mM Tris [pH 7.5], 100 mM NaCl). Trypsin inhibitor was added to stop the reactions, and an equal volume of 2× SDS sample buffer was added prior to Western blot analysis.
Recombinant virus generation.
pADCREGFP-UL94stop and pADCREGFP-UL94repair bacterial artificial chromosomes (BACs) were generated using a two-step linear recombination protocol in SW105 Escherichia coli as previously described (19, 32). pADCREGFP-UL94stop was generated via a single nucleotide substitution to introduce a premature stop codon after the seventh codon in the UL94 ORF. The first step in generating pADCREGFP-UL94stop was recombination between the HCMV wild-type pADCREGFP BAC (2) and a linear PCR fragment containing a GalK marker cassette flanked by 50-bp UL94 homology arms. The GalK linear fragment was obtained by PCR using the pGalK plasmid as a template (32) and the oligonucleotide primers UL94stopGalK5′ (5′-TTGCCGTCTCTTCGCGCGTCACTCTTCATGGCTTGGCGCAGCGGGCTTTGCCTGTTGACAATTAATCATCGGCA-3′) and UL94stopGalK3′ (5′-AGCTTCCACATGCATTCCTCTTGCAAGAACTGCTTCAAAGTTCTGGAATCTCAGCACTGTCCTGCTCCTT-3′). The first recombination step resulted in insertion of the GalK cassette after nucleotide 136375 of the AD169 genome (RefSeq X17403.1) downstream of the UL94 start codon to generate pADCREGFPGalKUL94. Recombinants containing the GalK cassette were selected on M63 minimal medium with galactose, leucine, biotin, and chloramphenicol, followed by selection on MacConkey agar plates with galactose and chloramphenicol. BACs were further screened by restriction enzyme analysis. A second recombination step to replace the GalK cassette was carried out using double-stranded oligonucleotides containing a C-to-A mutation at nucleotide 136376 of the genome (RefSeq X17403.1). The oligonucleotides UL94stop sense (5′-GCGGCTTTGCCGTCTCTTCGCGCGTCACTCTTCATGGCTTGGCGCAGCGGGCTTTGAGAGACCGATTCCAGAACTTTGAAGCAGTTCTTGCAAGAGGAATGCATGTGGAAGCTGGTCGG-3′) and UL94stopAS (5′-CCGACAGCTTCCACATGCATTCCTCTTGCAAGAACTGCTTCAAAGTTCTGGAATCGGTCTCTCAAAGCCCGCTGCGCCAAGCCATGAAGAGTGACGCGCGAAGAGACGGCAAAGCCGC-3′) were annealed, purified, and transformed into competent SW105 E. coli cells containing pADCREGFPGalKUL94. Recombinants were selected on 2-deoxy-galactose minimal agar plates to counterselect for GalK. The resulting recombinant BACs were screened by restriction enzyme digestion and PCR. Insertion of the premature stop codon was verified by DNA sequencing.
pADCREGFP-UL94repair was generated as described above using pADCREGFP-UL94stop as the parental BAC. The GalK cassette was amplified with the oligonucleotide primers UL94repGalK5′ (5′-TTGCCGTCTCTTCGCGCGTCACTCTTCATGGCTTGGCGCAGCGGGCTTTGCCTGTTGACAATTAATCATCGGCA-3′) and UL94repGalK3′ (5′-AGCTTCCACATGCATTCCTCTTGCAAGAACTGCTTCAAAGTTCTGGAATCTCAGCACTGTCCTGCTCCTT-3′). Insertion of the GalK cassette by recombination with pADCREGFP-UL94stop resulted in generation of pADCREGFP-UL94repairGalK BAC. The GalK cassette was subsequently removed by recombination of pADCREGFPUL94repairGalK with double-stranded oligonucleotides UL94repair sense (5′-GCGGCTTTGCCGTCTCTTCGCGCGTCACTCTTCATGGCTTGGCGCAGCGGGCTTTGCGAGACCGATTCCAGAACTTTGAAGCAGTTCTTGCAAGAGGAATGCATGTGGAAGCTGGTC-3′) and UL94repair AS (5′-GACCAGCTTCCACATGCATTCCTCTTGCAAGAACTGCTTCAAAGTTCTGGAATCGGTCTCGCAAAGCCCGCTGCGCCAAGCCATGAAGAGTGACGCGCGAAGAGACGGCAAAGCCGC-3′). The resulting recombinant BACs were screened by restriction enzyme digest and PCR. Insertion of the wild-type UL94 sequence was verified by DNA sequencing. Recombinant viruses were generated by transfecting ∼1 μg of BAC DNA and 5 μg of pCGN-pp71 expression plasmid into 5 × 106 HFF cells via electroporation (950 μF, 260 V). Cells were plated, and infectious virus was harvested when 100% CPE was observed. Infectious titers for all viruses were determined by plaque assay on HFF or UL94 complementing HFF cells.
Complementation of UL94stop mutant.
The UL94stop mutant virus was propagated in cells transduced with LEX lentivirus (Open Biosystems) expressing HAUL94. HAUL94 was cloned into the BamHI and XhoI sites of the LentiORF pLEX-MCS vector. The resulting HAUL94 pLEX plasmid was transfected into 293T cells with packaging mix using the Profection mammalian transfection system (Promega). Lentivirus was harvested at 48 and 72 h posttransfection, passed through 0.45-μm-pore-size filters, and used directly or stored at −80°C until transduction. To generate complementing cells, HFFs were transduced with HAUL94-LEX in the presence of 8 μg of Polybrene/ml.
Analysis of viral DNA replication.
A total of 105 HFF cells were plated per well in 12-well culture dishes. Cells were infected with ADCREGFPUL94HA (UL94HA virus) or ADCREGFPUL94stop (UL94stop virus) at a multiplicity of 0.01 PFU/cell. Cells were harvested and lysed in Southern blot lysis buffer (0.5% SDS, 25 mM EDTA, 100 mM NaCl, 10 mM Tris [pH 8], 100 μg of proteinase K/ml) and spiked with 100 ng of pGalK plasmid (NCI). Lysates were incubated at 37°C for 2 h, followed by sequential phenol and chloroform-isoamyl alcohol extractions. DNA was precipitated with 100% ethanol and washed with 70% ethanol. Pellets were rehydrated in water, and 100 ng was used for detection of viral genomes by real-time PCR. Real-time PCR was performed with QuantiFast SYBR green PCR mix (Qiagen) on the iQ5 real-time PCR detection system (Bio-Rad). Viral genomes were detected by amplification of the UL44 ORF with the primers 5′-CAGCTGCACGTTGATACGCATGTT-3′ and 5′-TCCACCGGCCATCAAGTTTATCCT-3′. GalK plasmid was detected using the primers 5′-TCTCATGCCTCTATGCGCGATGAT-3′ and 5′-CACCTTTGTCGCCAATCACAGCTT-3′. Viral DNA present in each sample was calculated by comparison to a standard curve prepared from a viral DNA sample of a known concentration. The data were normalized to the levels of GalK plasmid in each sample to account for variation in DNA yield during purification.
Immunofluorescence analysis.
HFF cells on glass coverslips were washed in PBS and then fixed in 4% paraformaldehyde in PBS at room temperature for 20 min. Fixed cells were washed and permeabilized in PBS-T (PBS, 0.05% Tween 20, 0.1% Triton X-100). Cells were blocked with PBS-T plus 0.5% bovine serum albumin plus 1% goat serum for 30 min, followed by sequential incubation with primary and secondary antibodies in a humidified chamber at 37°C. The cells were incubated with Hoechst stain to visualize nuclei and mounted in 90% glycerol. Fluorescence was visualized on a Zeiss Axiovert 40 CFL microscope. Images were taken using a Jenoptik ProgRes C10 Plus camera equipped with ProgRes CapturePro v.2.8.0 software.
TEM analysis.
A total of 105 HFF cells were infected at a multiplicity of 0.05 PFU/cell and processed for transmission electron microscopy (TEM) 120 h postinfection. Briefly, the cells were trypsinized, pelleted, and washed in PBS. Pellets were rinsed in 0.1 M cacodylate buffer and fixed in 2.5% glutaraldehyde for 45 min at room temperature. Cells were washed and postfixed in 1% osmium tetroxide for 30 min at room temperature. Samples were dehydrated with increasing concentrations of ethanol from 30 to 100% and then infiltrated with increasing concentrations of Poly/Bed 812 resin. Samples were embedded by curing at 40° for 24 h, followed by 60°C for 48 h prior to sectioning for TEM. Embedded samples were cut into 65-nm-thick sections and stained with uranyl acetate and lead citrate. Samples were imaged using a JEOL 1200-EXII transmission electron microscope, and images were acquired using Olympus iTEM imaging platform software.
RESULTS
Characterization of epitope-tagged UL94 protein in infected cells.
Several antibodies raised against UL94 are commercially available. All of these antibodies were raised against a peptide antigen corresponding to amino acids 26 to 40 of UL94. We tested three different UL94 antibody clones and found that none of them specifically recognized UL94 (Fig. 1C and data not shown). Therefore, to characterize the expression and localization of UL94 during infection, we generated recombinant viruses that express UL94 with a C-terminal hemagglutinin (HA) tag, designated UL94HA virus (18), or UL94 fused to the fluorescent mCherry protein, designated UL94mCherry virus. To verify UL94 expression from these recombinant viruses, HFF cells were infected with wild type, UL94HA, or UL94mCherry virus. Cell lysates were harvested at 72 h postinfection and assayed for UL94 expression by Western blotting with an antibody directed against HA, dsRed (mCherry), or UL94 (Santa Cruz Biotechnology, clone 6J8). We were able to detect UL94 with either the HA or dsRed antibody, and these proteins migrated at the expected molecular masses of 39 and 66 kDa, respectively (Fig. 1A and B). In contrast, when an identical blot was incubated with an antibody raised against a peptide antigen corresponding to amino acids 26 to 40 of UL94, we were only able to detect an ∼72-kDa band (Fig. 1C). Because we do not observe the expected size shift corresponding to the fusion of the HA tag or mCherry to the C terminus of UL94, it is likely that the UL94 6J8 antibody is recognizing an infected cell protein other than UL94. Two other commercial antibodies raised against an identical peptide were also tested, and neither antibody specifically detected UL94 (data not shown). The UL94HA virus replicates to wild-type levels (see Fig. 5A). Therefore, we used the UL94HA virus as the wild-type virus in all experiments designed to characterize UL94 during infection.
Fig 1.

Detection of UL94 fusion proteins in infected cell lysates. HFF cells were mock infected or infected at a multiplicity of 3 PFU/cell with wild-type, UL94HA, or UL94mCherry virus. Total protein was harvested at 72 h postinfection, and lysates were analyzed for UL94 expression. Western blots were probed with α-HA antibody (A) α-dsRed antibody, which also recognizes mCherry (B) or α-UL94 antibody clone 6J8 (C).
Fig 5.

Characterization of UL94stop phenotype. (A) The UL94stop virus is completely defective for replication. HFF cells were infected with wild-type virus (●), UL94HA virus (○), UL94 stop virus (▼), or UL94 repair virus (▽) at a multiplicity of 0.01 PFU/cell. Total virus (cells and supernatant) was harvested at the indicated days postinfection. Harvested virus titers were determined by plaque assay on HAUL94 complementing cells. (B) Expression of viral proteins over a time course of infection. HFF cells were infected with UL94HA virus or UL94stop virus at a multiplicity of 0.01 PFU/cell, and total protein was harvested at the indicated times postinfection. Viral proteins were analyzed by Western blotting with the indicated antibodies. (C) Quantitative PCR analysis of viral genome replication over time. HFF cells were infected with UL94HA virus (○) or UL94stop virus (●) at a multiplicity of 0.01 PFU/cell, and total DNA was harvested at the indicated times postinfection. Accumulation of viral DNA was analyzed by quantitative PCR. The results were normalized for both input DNA and variations in DNA recovery introduced during purification. Picograms of viral DNA present in each sample were calculated from a standard curve prepared from purified viral DNA of a known concentration.
UL94 is expressed with true-late kinetics and displays differential localization in transfected and infected cells.
UL94 was previously reported to be expressed with true-late kinetics based on Northern blot analysis of UL94 transcript (33). We sought to confirm these results by analyzing the expression of UL94 protein. Therefore, we performed Western blot analysis of infected-cell lysates over a time course of infection. HFF cells were infected with UL94HA virus at a multiplicity of 3 PFU/cell, and total protein was harvested every 24 h for 5 days. Infected cells were also incubated in the presence of phosphonoacetic acid (PAA) to inhibit viral DNA synthesis. Proteins were separated by SDS-PAGE and subjected to Western blot analysis for expression of representative immediate-early (IE1/2), early (UL44), and late (UL99) genes, as well as UL94 (HA). UL94 was first detected at 48 h postinfection, and its expression continued to increase over the next 48 h (Fig. 2A). In addition, the expression of UL94 was completely inhibited by the addition of PAA to the growth medium (Fig. 2A, lane 5). These data confirm that UL94 is a true-late gene and demonstrate that the addition of the HA tag to the C terminus of UL94 does not affect its expected expression kinetics.
Fig 2.
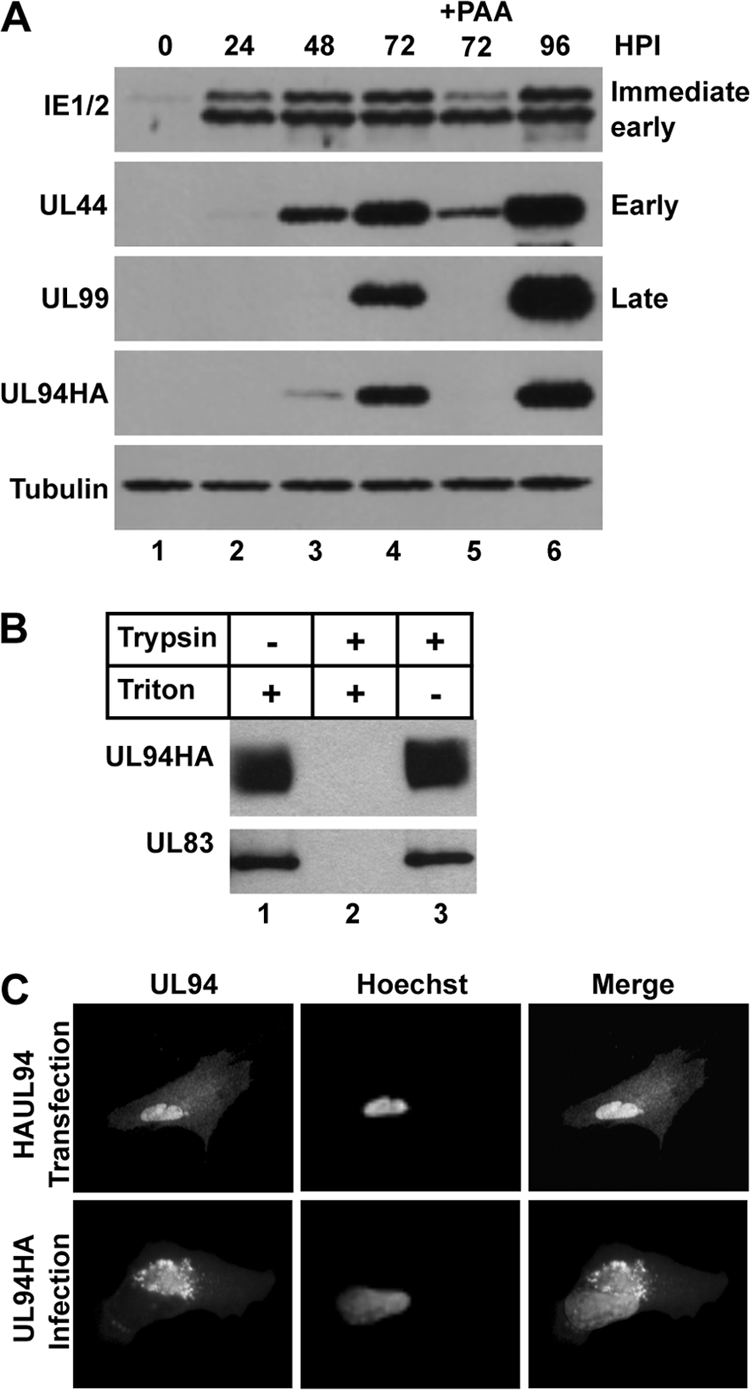
Characterization of UL94 protein. (A) UL94 is expressed with true-late kinetics. HFF cells were infected with UL94HA virus at a multiplicity of 3 PFU/cell in the presence or absence of PAA, and total protein was harvested at the indicated times postinfection. Cell lysates were analyzed by Western blotting with the indicated antibodies. (B) UL94 is incorporated into the virion tegument. Equal amounts of purified virions were subjected to treatment with trypsin and/or Triton X-100 as indicated. Samples were analyzed for the presence of viral proteins by Western blotting. (C) UL94 exhibits a different pattern of localization in transfected and infected cells. HFF cells were transfected with HAUL94-pcDNA3.1 expression plasmid (top row) or infected at a multiplicity of 3 PFU/cell with UL94HA virus (bottom row). UL94 protein was visualized by immunofluorescence staining with anti-HA antibody at 48 h posttransfection or at 96 h postinfection.
UL94 is thought to be virion-associated based on proteomic analysis of HCMV particles. To establish whether UL94 is a tegument protein, HFF cells were infected with UL94HA virus and supernatant was collected when 100% CPE was observed. Virus particles were purified, incubated with trypsin and/or Triton X-100, and assayed by Western blotting for the presence of UL94. As shown in Fig. 2B, UL94 remains associated with the particle after treatment with Triton X-100 (lane 1) or when particles are exposed to trypsin alone (lane 3). However, when particles are treated with both detergent and protease, UL94 is readily degraded (lane 2). The same results were observed when particles were assayed for the abundant tegument protein UL83 (pp65). Therefore, we conclude that UL94 is located within the tegument layer of the virion and that the addition of the HA-tag to the C terminus of UL94 does not prevent its packaging into HCMV virions.
Previous reports analyzing the localization of UL94 in infected cells described conflicting results regarding whether UL94 localizes to the cytoplasm or nucleus of infected cells (11, 33). Therefore, we sought to determine the subcellular localization of UL94 in both transfected and HCMV-infected cells. HFF cells were transfected with a plasmid expressing HA-tagged UL94, and immunofluorescence analysis with an α-HA antibody was performed at 48 h posttransfection. In the absence of other viral proteins, UL94 exhibits a diffuse pattern of localization that can be seen in both the nucleus and the cytoplasm of transfected cells (Fig. 2C, top row). To determine the localization of UL94 during infection, HFF cells were infected with UL94HA virus at a multiplicity of 3 PFU/cell, and immunofluorescence analysis was performed at 96 h postinfection. In contrast to the pattern of localization observed in transfected cells, UL94 is detected predominantly in a juxtanuclear structure consistent with the viral assembly complex during infection (Fig. 2C, bottom row). An identical pattern of localization was observed with the UL94mCherry fusion protein during infection (data not shown). These results suggest that UL94 localizes to the assembly complex late in infection and that this localization requires other infected-cell proteins.
UL94 is essential for HCMV replication.
To investigate the function of UL94 during infection, we generated a UL94-null mutant which we have designated UL94stop. This mutant was generated through a single point mutation that introduces a premature stop codon after the seventh codon of the UL94 ORF. Figure 3A shows the region of the viral genome surrounding the UL94 ORF and the recombination strategy used to generate the UL94stop mutant. The UL93 ORF overlaps the 5′ end of UL94 by 142 nucleotides. Due to this overlap, we generated the UL94stop mutant with a single C-to-A point mutation that introduces a premature stop codon in UL94 without altering the amino acid sequence of UL93. Figure 3B shows verification of the desired recombination events by restriction digest analysis. The shifts in restriction fragment sizes based on the proper recombination events are indicated with arrows. Insertion of the GalK cassette results in a 1,333-bp increase in the size of the EcoRI fragment containing the UL94 ORF. The GalK cassette was then removed by recombination with double-stranded oligonucleotides containing the desired mutation. The restriction pattern of the resulting UL94stop BAC is identical to that of the parental ADCREGFP BAC. The entire UL94 ORF in the UL94stop BAC was sequenced to confirm the presence of the C-to-A point mutation and to verify that no other mutations were incorporated into the UL94 gene. UL94stop BAC DNA was cotransfected into HFF cells with a pp71 expression plasmid to determine whether UL94 is essential for viral replication. The spread of infection was then monitored by GFP expression from the ADCREGFP viral genome (2). Figure 3C shows GFP expression in cell cultures 21 days posttransfection of either wild-type or UL94stop BAC DNA. Cells transfected with the wild-type BAC display significant CPE and >95% of the cells are GFP positive by 21 days posttransfection due to the production of infectious virus. In contrast, we are only able to detect single GFP-positive cells in cultures transfected with the UL94stop BAC. These individual GFP-positive cells represent those that were initially transfected with BAC DNA; however, there is no subsequent spread of infection, indicating that no infectious virus is produced from the UL94stop BAC. This result suggests that UL94 is essential for HCMV replication.
Fig 3.

Generation of UL94stop mutant BAC. (A) Schematic diagram of genomic region containing UL94 and the strategy used to generate the UL94stop BAC containing a premature stop codon from wild-type ADCREGFP. (B) BAC DNA was isolated and digested with EcoRI. Digested DNA fragments were separated by agarose gel electrophoresis. Arrows indicate predicted fragments generated as a result of the desired recombination events. (C) Wild-type or UL94stop BAC DNA was transfected into HFF cells. Spread of infection was visualized 21 days postinfection by GFP expression from the viral genome.
Complementation of the UL94stop mutant.
Because UL94 is likely essential for viral replication, we developed a complementation system that would allow us to propagate the UL94stop mutant. To do this, we used a lentiviral system that allows for the transduction of quiescent HFF cells. UL94 complementing HFF cells were generated by transduction with lentivirus expressing HA-tagged UL94. Expression of UL94 from transduced HFF cells was verified by Western blot analysis with an α-HA antibody and compared to the expression of UL94 from cells infected with the UL94HA virus. The level of UL94 expressed from the UL94 complementing HFF cells was significantly lower than the level of UL94 present after infection with UL94HA virus (Fig. 4A, compare lanes 2 and 3). However, upon infection of UL94 complementing HFF cells with wild-type HCMV, we observe an induction in HAUL94 expression (Fig. 4A, compare lanes 3 and 4). This effect is likely due to the action of viral proteins on the CMV immediate-early promoter that is used to drive expression of UL94 from the lentivirus. However, even after induction, the levels of UL94 observed in the complementing cells are still less than the levels of UL94 observed during infection (compare lanes 2 and 4).
Fig 4.
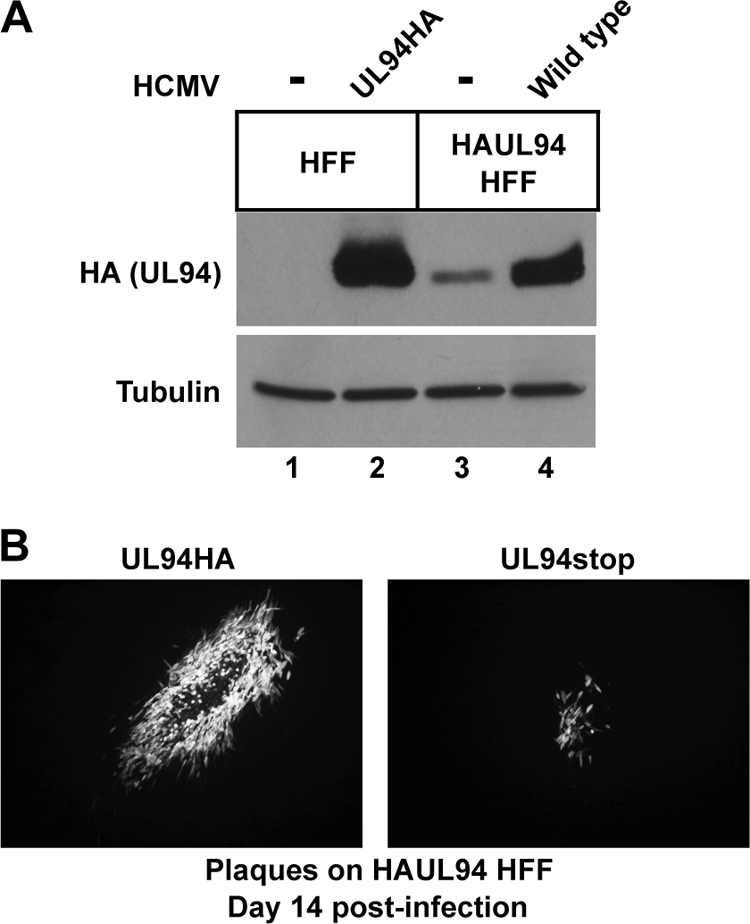
Complementation of UL94stop mutant. (A) Western blot analysis of HAUL94 expression in UL94 complementing cells. Normal HFF or complementing HAUL94 HFF cells were either mock infected (lanes 1 and 3) or infected with UL94HA virus (lane 2) or wild-type virus (lane 4). Total protein was harvested 72 h postinfection and analyzed for expression of HA-tagged UL94. (B) Plaques formed 14 days postinfection by UL94HA virus (left) or UL94stop virus (right) on complementing HAUL94 HFF.
To generate a stock of the UL94stop virus, HFF cells were first transfected with the UL94stop BAC. Cells were then transduced with lentivirus expressing HA-tagged UL94 the day after BAC transfection. Virus produced after BAC transfection was harvested when cells exhibited advanced CPE and then used to infect a fresh culture of UL94 complementing HFF cells. The resulting UL94stop virus titer was determined by plaque assay on UL94 complementing HFF cells. Although we were able to generate a stock of the UL94stop virus, the complementation was inefficient. The UL94stop virus spread very slowly compared to wild-type virus and only produced titers of ∼5 × 103 PFU/ml, ∼3 logs lower than the titers routinely obtained with wild-type virus. In addition, the UL94stop mutant formed plaques that were significantly smaller in size than those produced by UL94HA virus by 14 days after infection (Fig. 4B).
Once a stock of the UL94stop virus was obtained, we performed growth curve analysis (Fig. 5). HFF cells were infected with wild type, UL94HA, UL94stop, or UL94repair viruses at a multiplicity of 0.01 PFU/cell. Total virus was harvested at the indicated times postinfection, and virus titers were determined by plaque assay on UL94 complementing HFF cells. We were unable to detect any infectious virus from cells infected with the UL94stop mutant at any time postinfection (Fig. 5, filled triangles), confirming that UL94 is essential for replication. However, when the UL94stop mutation is repaired, the resulting UL94repair virus replicates to levels similar to those for the wild type (Fig. 5, open triangles). This result demonstrates that the growth defect of the UL94stop mutant can be attributed specifically to the mutation in UL94 and is not due to a secondary mutation elsewhere in the viral genome. The UL94HA virus also replicates to wild-type levels, demonstrating that the addition of the HA tag to the C terminus of UL94 does not interfere with the essential function of UL94 (Fig. 5, open circles).
UL94 is not required for viral gene expression or genome replication.
We next sought to determine where the block in viral replication occurs following infection with the UL94stop virus. We first examined the kinetics of viral gene expression in cells infected with the UL94stop mutant. HFF cells were infected with UL94HA or UL94stop virus at a multiplicity of 0.01 PFU/cell, and total protein was harvested at the indicated times following infection. Cell lysates were analyzed for expression of representative viral immediate-early (IE1/2), early (UL44), and late (UL86) proteins by Western blotting. We observed no defect in the kinetics of immediate-early, early, or late gene expression in cells infected with the UL94stop mutant compared to the UL94HA virus (Fig. 5B). We did observe a modest increase in viral protein levels at late times in cells infected with the UL94HA virus compared to the UL94stop mutant. However, this is likely due to viral spread in cells infected with the UL94HA virus, whereas the UL94stop mutant is unable to produce infectious virus. These results suggest that the absolute growth defect of the UL94stop mutant is not due to a global defect in viral gene expression.
We also analyzed the kinetics and levels of viral genome replication in both UL94HA and UL94stop virus-infected cells. HFF cells were infected at a multiplicity of 0.01 PFU/cell, and total DNA was harvested at the indicated times postinfection. Purified DNA was subjected to real-time PCR analysis for levels of viral DNA. The results were normalized for both input DNA and variations in DNA recovery during purification. Picograms of viral DNA present in each sample were calculated from a standard curve prepared from purified viral DNA of a known concentration. The kinetics and levels of viral genome synthesis are nearly identical for both UL94HA and UL94stop virus infections (Fig. 5C). This result suggests that UL94 does not play an essential role in viral DNA replication.
UL94 directs the proper localization of UL99 to the assembly complex.
Many viral tegument proteins function in assembly by directing proper tegumentation, capsid trafficking, and envelope acquisition at the assembly complex in the cytoplasm (1, 20, 23, 24, 26, 28, 29). We therefore sought to characterize the phenotype of the assembly complex with respect to the localization of viral proteins in cells infected with the UL94stop mutant. HFF cells were seeded on glass coverslips and infected at a multiplicity of 0.01 PFU/cell with either UL94HA or UL94stop virus. Cells were fixed and stained for immunofluorescence analysis of viral proteins 120 h postinfection (Fig. 6A). In cells infected with UL94HA virus we observed the expected localization pattern of UL94, UL32, gB, and UL99 to the juxtanuclear viral assembly complex (Fig. 6A). We saw a similar localization of UL32 and gB to the assembly complex in cells infected with the UL94stop virus. Interestingly, we observed a dramatic difference in the localization of UL99 following infection with the UL94stop mutant. In contrast to the juxtanuclear localization observed in cells infected with the UL94HA virus, UL99 remained predominantly diffuse throughout the cytoplasm in cells infected with the UL94stop mutant.
Fig 6.
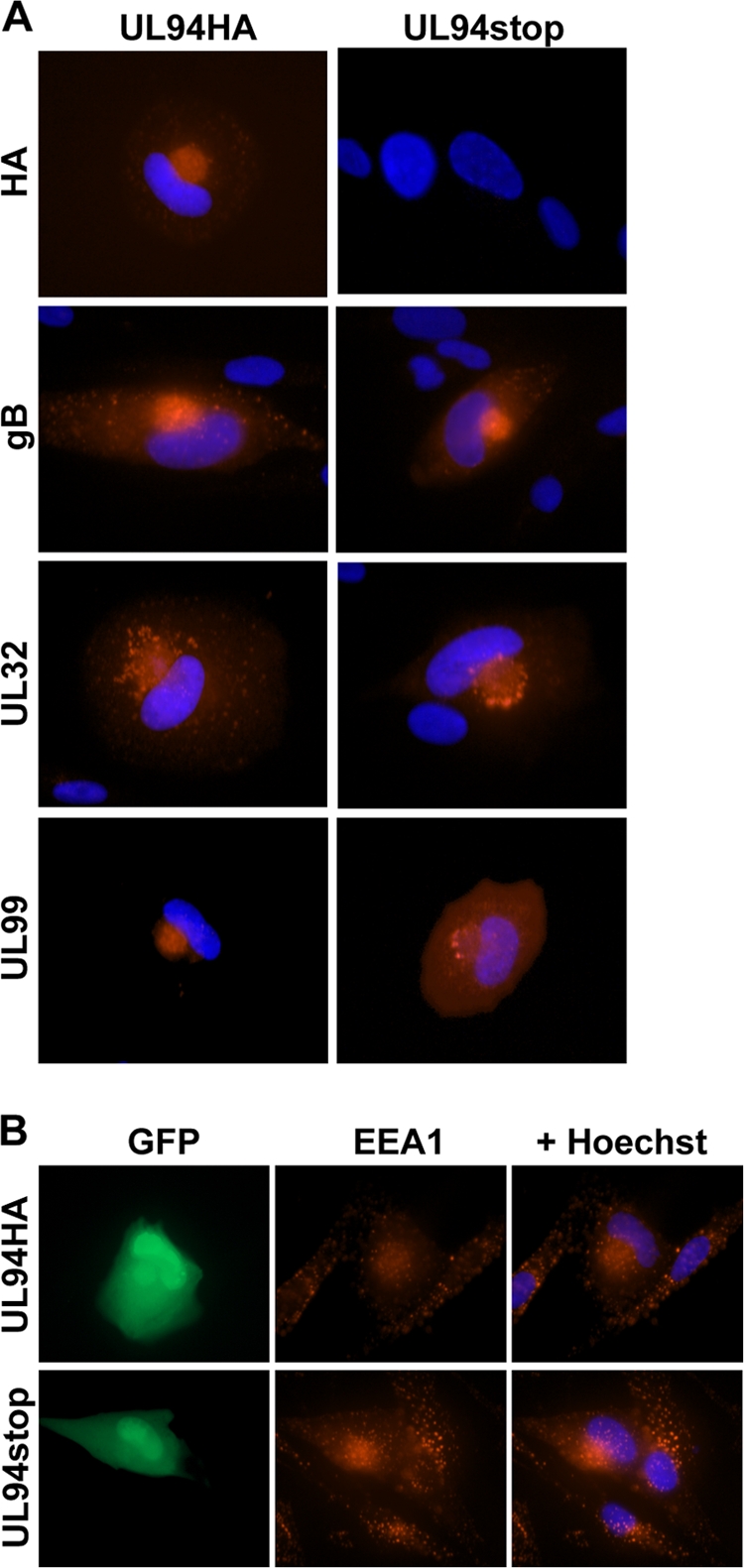
Immunofluorescence analysis of the viral assembly complex. HFF cells were infected with UL94HA or UL94stop virus at a multiplicity of 0.01 PFU/cell. Cells were fixed 120 h postinfection, and immunofluorescence analysis was performed with the indicated antibodies to visualize viral proteins (A) or cellular EEA1 (B). Proteins were visualized using an Alexa 546-conjugated secondary antibody (red). Nuclei are stained with Hoechst (blue). Infected cells are indicated by GFP expression from viral genome.
The assembly complex is composed of many components of the cellular secretory apparatus including early endosomes. To determine whether cellular components of the assembly complex localize properly in the absence of UL94, we determined the localization of the cellular early endosomal marker EEA1 in cells infected with the UL94stop mutant (Fig. 6B). Infected cells are indicated by GFP expression from the viral genome. In cells infected with either UL94HA virus (top row) or UL94stop virus (bottom row), early endosomes become reoriented and accumulate at the juxtanuclear assembly complex as previously described (3–5). In contrast, early endosomes in uninfected cells are observed throughout the cytoplasm. These results show that the reorientation of early endosomes to the assembly complex does not require UL94. Taken together, these data suggest that the absence of UL94 does not result in a global defect in the formation of the assembly complex, but rather that UL94 is required specifically for the proper localization of UL99.
UL94 is required for virion envelopment in the cytoplasm.
To determine the ultrastructural morphology of virus particles in the absence of UL94, we performed TEM analysis. HFF cells were infected with UL94HA or UL94stop virus at a multiplicity of 0.05 PFU/cell, and the cells were collected for TEM analysis at 120 h postinfection. Images of thin sections were collected at a final magnification of ×20,000 (Fig. 7). We observed similar numbers of empty and DNA-containing capsids in the nuclei of both UL94HA and UL94stop virus-infected cells, suggesting that there is no defect in capsid formation or DNA packaging in the absence of UL94 (Fig. 7A). The cytoplasm of cells infected with both UL94HA and UL94stop virus also contained both empty (Fig. 7B, black arrowheads) and DNA-containing capsids (Fig. 7B, white arrows). The majority of cytoplasmic particles in both UL94HA and UL94stop virus-infected cells appeared to be surrounded by a layer of tegument proteins based on their thickness compared to nuclear capsids, suggesting that the initial steps involved in tegument acquisition do not require UL94. We also observed a >3-fold increase in the total number of capsids in the cytoplasm of UL94stop-infected cells compared to cells infected with UL94HA virus. Importantly, we were able to readily detect fully enveloped virions in cells infected with the UL94HA virus (Fig. 7B, black arrows). Approximately 10% of all cytoplasmic particles in UL94HA virus-infected cells were surrounded by an envelope. However, we were unable to detect any enveloped virions in cells infected with the UL94stop mutant. Numerous infected cell sections were examined and after counting more than 600 cytoplasmic capsids in cells infected with the UL94stop virus, we were unable to detect a single enveloped virion. We also observed large spherical electron-dense structures in the cytoplasm of cells infected with the UL94stop mutant that were enlarged compared to those found in the cytoplasm of cells infected with the UL94HA virus. Immature particles formed by the UL94stop mutant were often found on the periphery of these structures but did not acquire an envelope. These results suggest that there is a complete block in secondary envelopment in the absence of UL94.
Fig 7.

TEM analysis of cells infected with UL94HA or UL94stop virus. HFF cells were infected with UL94HA or UL94stop virus at a multiplicity of 0.05 PFU/cell. Cells were fixed and processed for TEM 120 h postinfection as described in Materials and Methods. Representative infected cell nuclear (A) or cytoplasmic (B) regions are shown. Black arrows indicate enveloped virions. White arrows indicate DNA-containing capsids. Black arrowheads indicate empty capsids. Images were taken at a final magnification of ×20,000. Insets show magnified regions containing enveloped, empty, and DNA-containing capsids.
DISCUSSION
The mechanisms that facilitate the assembly of mature HCMV particles in the cytoplasm of infected cells remain poorly understood. Although the events that result in virion maturation remain largely undefined, many reports have demonstrated a role for tegument proteins in virion assembly. Our lab and others have reported an interaction between the tegument proteins UL99 (pp28) and UL94 (7, 11, 18, 30). We have previously demonstrated that this interaction occurs at late times in infection, when infectious progeny are being produced (18). UL99 localizes to the viral assembly complex and is essential for secondary envelopment of virions in the cytoplasm (22, 23, 26). In the absence of UL99, partially tegumented but nonenveloped capsids accumulate in the cytoplasm, and no extracellular infectious virus is produced. Localization of UL99 to the cytoplasmic assembly complex is essential for virus replication and requires the expression of other late viral proteins (23, 25). In contrast, despite being a conserved herpesvirus core gene, little is known about UL94, and its function during infection has not been investigated. Mutagenesis of the HCMV genome has suggested that UL94 is either essential or augmenting depending on the mutagenesis strategy used (6, 35). Further, overlap of ORFs and the presence of coterminal transcripts in the region of the viral genome in which UL94 is located complicate the interpretation of results obtained with these mutants. The role of UL94 for HCMV replication has never been tested by the generation of a specific UL94-null mutant. Therefore, we sought to characterize UL94 and determine whether it is required for HCMV replication.
To circumvent the lack of a specific UL94 antibody, we generated a recombinant virus that expresses UL94 with a C-terminal HA tag (18). This virus expresses a protein of the predicted molecular weight and replicates to wild-type levels (Fig. 1A and 5A). Using the UL94HA virus, we demonstrate that UL94 is a tegument protein that is expressed with true-late kinetics (Fig. 2A and B). We also used this virus to determine the subcellular localization of UL94 in infected cells. Interestingly, we observed a dramatically different pattern of UL94 localization in transfected cells compared to infected cells (Fig. 2C). Whereas UL94 was found throughout both the nucleus and the cytoplasm of transfected cells, we found that UL94 localizes almost exclusively to the viral assembly complex during infection. This demonstrates that the proper localization of UL94 in infected cells requires other infected-cell proteins.
To elucidate the function of UL94 during infection, we generated a UL94-null virus designated UL94stop. The UL94stop mutant is completely defective for replication, which necessitated the development of a complementation system for propagating the mutant virus. We utilized a UL94 lentivirus complementation system to generate HFF cells that stably express HA-tagged UL94. UL94stop virus grown on complementing cells only replicated to titers of ∼5 × 103 PFU/ml. The reason for this poor complementation is unknown. Analysis of the localization of UL94 and UL99 in UL94 complementing cells infected with the UL94stop mutant indicates that both proteins localize properly to the assembly complex in these cells, suggesting that the low level of complementation achieved is not due to improper localization of UL94 or UL99 (data not shown). The most likely explanation is that poor complementation is due to the decreased level of UL94 protein expressed in the complementing cells compared to the levels of UL94 achieved during infection (Fig. 4A). Another possibility is that poor complementation results from constitutive expression of UL94 in the complementing cells. Viral gene expression is precisely controlled, and many structural proteins are expressed with true-late kinetics, meaning that their expression is tightly suppressed until after the onset of viral DNA synthesis. Late viral proteins expressed constitutively in the host cell are therefore temporally dysregulated and may not complement properly or perhaps may even interfere with normal viral replication. Other groups have also reported difficulty in complementing HCMV mutants of essential viral structural proteins (1, 26). Interestingly, the levels of complementation for the UL94stop mutant were similar to those achieved for a UL99 deletion mutant (26).
Phenotypic analysis of the UL94stop mutant suggests that UL94 functions late in infection, subsequent to viral DNA replication. The early phases of viral replication such as immediate-early and early viral gene expression, as well as the synthesis of viral DNA occur at wild-type levels in the absence of UL94 (Fig. 5B and C). We also observe proper localization of the abundant viral proteins UL32 (tegument) and gB (envelope) to the assembly complex following infection with the UL94stop virus (Fig. 6A). Proper formation of the assembly complex was further confirmed by demonstrating reorientation of the early endosomal marker EEA1 to the juxtanuclear site of virion assembly (Fig. 6B). Interestingly, we observe a dramatic defect in the localization of UL99 following infection with the UL94stop mutant. In the absence of UL94, UL99 fails to accumulate at the assembly complex and remains largely diffuse throughout the cytoplasm. These results suggest that UL94 functions at least in part to ensure the proper localization of UL99 to the assembly complex where final virion envelopment is thought to occur. These data are further supported by our ultrastructural analysis, which shows that in the absence of UL94 there is a block in secondary virion envelopment, as indicated by the complete absence of enveloped virions in the cytoplasm of cells infected with the UL94stop virus (Fig. 7B). A very similar phenotype was previously reported for cells infected with a UL99 deletion mutant (26). Taken together, our results suggest that UL94 is required for the proper localization of UL99 to the assembly complex, a step that is necessary for secondary envelopment of virions.
Although we cannot rule out the possibility that UL94 directly influences virion envelopment, we favor a model in which the accumulation of unenveloped particles is secondary to the absence of sufficient amounts of UL99 at the assembly complex. It has been shown that the localization of UL99 to the assembly complex is essential for HCMV replication and that a mutant form of UL99 that traffics to but is not retained in the assembly complex shows a defect in envelopment and delayed growth kinetics (23, 25). Further, UL99 exhibits differential localization in transfected and infected cells, associating with different cellular membranes in the presence of other viral proteins (22, 25). These observations indicate that other viral factors are required for the proper localization of UL99 during infection. Based on the data presented here, we propose that UL94 is the late viral protein that is essential for trafficking of UL99 to the assembly complex.
UL94 and UL99 are both conserved across all members of the herpesvirus family. Although their interaction is also conserved (7, 10, 11, 13, 27, 31), its functional significance remains unclear. In the case of herpes simplex virus type 1 (HSV-1), the interaction between the UL94 homolog (UL16) and the UL99 homolog (UL11) has been proposed to function in virion morphogenesis in the cytoplasm (12, 14, 34). However, neither UL16 nor UL11 is essential for HSV-1 replication, whereas HCMV lacking UL94 or UL99 are completely defective for growth. Thus, it is likely that the mechanisms of cytoplasmic virion assembly are different for betaherpesviruses and alphaherpesviruses.
The data shown here represent the first report of a specific function of the essential UL94 tegument protein during HCMV infection. Our data suggest that UL94 is required for the proper localization of UL99 to the cytoplasmic assembly complex, an event ultimately necessary for final envelopment. Interestingly, the phenotype of the UL94-null virus characterized in the present study is virtually identical to that of a previously characterized UL99 deletion mutant (26). Mutants lacking either UL94 or UL99 exhibit wild-type replication until the phase of cytoplasmic assembly, during which unenveloped particles accumulate in the cytoplasm without subsequent envelope acquisition. Based on the fact that UL94 and UL99 physically interact during infection, it is reasonable to hypothesize that their interaction is required for the proper localization of UL99 and thus for virus replication. However, we cannot rule out alternate mechanisms involving other viral or cellular proteins in this process. For example, it is possible that while the proper localization of UL99 requires UL94, the localization of UL94 may also be influenced by UL99. In addition, the localization of each protein to the assembly complex may also require one or more cellular proteins. We are currently investigating the functional significance of the interaction between UL94 and UL99 during HCMV replication.
ACKNOWLEDGMENTS
We are grateful to W. Gibson (Johns Hopkins) for the rabbit polyclonal antibody recognizing UL86 and to W. Britt (University of Alabama at Birmingham) for the mouse monoclonal antibody recognizing UL32. We are thank Fang Zhou at the University of Minnesota Characterization Facility for assistance with the TEM studies.
This study was supported in part by NIH grant AI059340 (to W.A.B.) and by grant number T32DE007288 from the National Institute of Dental and Craniofacial Research. Parts of this work were carried out at the Characterization Facility, University of Minnesota, which receives partial support from NSF through the MRSEC program.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Dental and Craniofacial Research or the National Institutes of Health.
Footnotes
Published ahead of print 14 December 2011
REFERENCES
- 1. AuCoin DP, Smith GB, Meiering CD, Mocarski ES. 2006. Betaherpesvirus-conserved cytomegalovirus tegument protein ppUL32 (pp150) controls cytoplasmic events during virion maturation. J. Virol. 80:8199–8210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cantrell SR, Bresnahan WA. 2005. Interaction between the human cytomegalovirus UL82 gene product (pp71) and hDaxx regulates immediate-early gene expression and viral replication. J. Virol. 79:7792–7802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cepeda V, Esteban M, Fraile-Ramos A. 2010. Human cytomegalovirus final envelopment on membranes containing both trans-Golgi network and endosomal markers. Cell. Microbiol. 12:386–404 [DOI] [PubMed] [Google Scholar]
- 4. Das S, Pellett PE. 2011. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J. Virol. 85:5864–5879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Das S, Vasanji A, Pellettl PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J. Virol. 81:11861–11869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dunn W, et al. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223–14228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fossum E, et al. 2009. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog. 5:e1000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gibson W, Baxter MK, Clopper KS. 1996. Cytomegalovirus “missing” capsid protein identified as heat-aggregable product of human cytomegalovirus UL46. J. Virol. 70:7454–7461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krzyzaniak MA, Mach M, Britt WJ. 2009. HCMV-encoded glycoprotein M (UL100) interacts with Rab11 effector protein FIP4. Traffic 10:1439–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347–354 [DOI] [PubMed] [Google Scholar]
- 11. Liu YL, et al. 2009. The tegument protein UL94 of human cytomegalovirus as a binding partner for tegument protein pp28 identified by intracellular imaging. Virology 388:68–77 [DOI] [PubMed] [Google Scholar]
- 12. Loomis JS, Courtney RJ, Wills JW. 2003. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 77:11417–11424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maninger S, et al. 2011. M94 is essential for the secondary envelopment of murine cytomegalovirus. J. Virol. 85:9254–9267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meckes DG, Wills JW. 2007. Dynamic interactions of the UL16 tegument protein with the capsid of herpes simplex virus. J. Virol. 81:13028–13036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murphy E, Rigoutsos I, Shibuya T, Shenk TE. 2003. Reevaluation of human cytomegalovirus coding potential. Proc. Natl. Acad. Sci. U. S. A. 100:13585–13590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Murphy E, et al. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. U. S. A. 100:14976–14981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pass RF. 2001. Cytomegalovirus, p 2675–2706 In Knight DM, Howley PM. (ed), Fields virology, 4th ed Lippincott-Raven Publishers, Philadelphia, PA [Google Scholar]
- 18. Phillips SL, Bresnahan WA. 2011. Identification of binary interactions between human cytomegalovirus virion proteins. J. Virol. 85:440–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qian ZK, Xuan BQ, Hong TT, Yu D. 2008. The full-length protein encoded by human cytomegalovirus gene UL117 is required for the proper maturation of viral replication compartments. J. Virol. 82:3452–3465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sampaio KL, Cavignac Y, Stierhof YD, Sinzger C. 2005. Human cytomegalovirus labeled with green fluorescent protein for live analysis of intracellular particle movements. J. Virol. 79:2754–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanchez V, Greis KD, Sztul E, Britt WJ. 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J. Virol. 74:975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sanchez V, Sztul E, Britt WJ. 2000. Human cytomegalovirus pp28 (UL99) localizes to a cytoplasmic compartment which overlaps the endoplasmic reticulum-Golgi-intermediate compartment. J. Virol. 74:3842–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seo JY, Britt WJ. 2007. Cytoplasmic envelopment of human cytomegalovirus requires the postlocalization function of tegument protein pp28 within the assembly compartment. J. Virol. 81:6536–6547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seo JY, Britt WJ. 2008. Multimerization of tegument protein pp28 within the assembly compartment is required for cytoplasmic envelopment of human cytomegalovirus. J. Virol. 82:6272–6287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Seo JY, Britt WJ. 2006. Sequence requirements for localization of human cytomegalovirus tegument protein pp28 to the virus assembly compartment and for assembly of infectious virus. J. Virol. 80:5611–5626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Silva MC, Yu QC, Enquist L, Shenk T. 2003. Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J. Virol. 77:10594–10605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stellberger T, et al. 2010. Improving the yeast two-hybrid system with permutated fusion proteins: the varicella-zoster virus interactome. Proteome Sci. 8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tandon R, Mocarski ES. 2008. Control of cytoplasmic maturation events by cytomegalovirus tegument protein pp150. J. Virol. 82:9433–9444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tandon R, Mocarski ES. 2011. Cytomegalovirus pUL96 is critical for the stability of pp150-associated nucleocapsids. J. Virol. 85:7129–7141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. To A, et al. 2011. Yeast two hybrid analyses reveal novel binary interactions between human cytomegalovirus-encoded virion proteins. PLoS One 6:e17796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vittone V, et al. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566–9571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using gaIK selection. Nucleic Acids Res. 33:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wing BA, Lee GCY, Huang ES. 1996. The human cytomegalovirus UL94 open reading frame encodes a conserved herpesvirus capsid/tegument-associated virion protein that is expressed with true late kinetics. J. Virol. 70:3339–3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yeh PC, Meckes DG, Wills JW. 2008. Analysis of the interaction between the UL11 and UL16 tegument proteins of herpes simplex virus. J. Virol. 82:10693–10700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396–12401 [DOI] [PMC free article] [PubMed] [Google Scholar]