

Abstract
Understanding the complex mechanisms by which infectious agents can disrupt behavior represents a major challenge. The Borna disease virus (BDV), a potential human pathogen, provides a unique model to study such mechanisms. Because BDV induces neurodegeneration in brain areas that are still undergoing maturation at the time of infection, we tested the hypothesis that BDV interferes with neurogenesis. We showed that human neural stem/progenitor cells are highly permissive to BDV, although infection does not alter their survival or undifferentiated phenotype. In contrast, upon the induction of differentiation, BDV is capable of severely impairing neurogenesis by interfering with the survival of newly generated neurons. Such impairment was specific to neurogenesis, since astrogliogenesis was unaltered. In conclusion, we demonstrate a new mechanism by which BDV might impair neural function and brain plasticity in infected individuals. These results may contribute to a better understanding of behavioral disorders associated with BDV infection.
INTRODUCTION
Epidemiological analyses of human neuropsychiatric illness, as well as studies conducted in animal models, have suggested that infection underlies a wide range of neuropsychiatric disorders. It has been hypothesized that persistent viral infection plays a role in human mental disorders of unclear etiology ((5, 24, 39). However, establishing a causal relationship between infection and a behavioral disturbance can be difficult. In these chronic disorders, Koch's postulate (i.e., proof of a causative relationship by isolation, propagation outside the original host, and reintroduction into a new host resulting in disease) may never be demonstrated. Nevertheless, it is of great interest to investigate the complex mechanisms by which infectious agents can disrupt behavior. The observation that about 0.5 to 1% of the worldwide population is affected by a mental illness, such as schizophrenia, underscores the importance of this research.
Borna disease virus (BDV) is a highly neurotropic virus which persists in the central nervous system (CNS) of infected individuals for their entire life span. It is a nonsegmented, negative-sense, single-stranded RNA virus belonging to the Bornaviridae family within the order Mononegavirales (3, 10). BDV was originally described as an agent of nonpurulent encephalomyelitis in horses in Germany (36) but later was identified in a wide range of vertebrates, including sheep, cattle, dogs, cats, shrews, ostriches, and nonhuman primates (2, 15, 17, 22, 26). Infected hosts develop a wide spectrum of neurological disorders, ranging from immune-mediated disease to behavioral alteration without inflammation, including deficits in learning and social behavior, which are reminiscent of symptoms observed in human psychiatric diseases such as schizophrenia, mood disorders, and autism (18, 32). Epidemiologic studies have further suggested that BDV infection can occur in humans and that it is related to certain psychiatric diseases (6, 25). In support of this hypothesis, BDV infection was demonstrated in the brain of a schizophrenic patient (28). However, the role of BDV infection in human pathology still is under debate (23). Nevertheless, owing to its implication in neurobehavioral disorders in animals and its suspected role in mental diseases in humans, BDV is of great interest for investigating the mechanisms by which viral infection alters behavior.
BDV primarily infects neurons of the limbic system, notably the cortex and the hippocampus (14). Other cellular types, however, such as astrocytes (4) and neural progenitor cells (37, 38), have been shown to be infected and might be involved in BDV-induced neuropathogenesis. Indeed, astrocyte dysfunction can play a key role in the pathogenesis of CNS disorders (9), and striking neurobehavioral abnormalities have been reported in mice expressing BDV phosphoprotein (BDV-P) selectively in glial cells (20). The alteration of progenitor cells and neurogenesis also would critically affect brain function. In humans, it has been hypothesized that the impairment of adult neurogenesis plays a role in the etiopathogenesis of neuropsychiatric disorders (13, 21). The demonstration of a significant reduction in the proliferation of neural stem cells (NSC) found in schizophrenic patients has provided support for this new theory (35). In newborn rats, BDV infection is responsible for intensive neurodegeneration that is restricted to areas of the rat brain that are still maturing at the time of infection (1, 5, 33). This suggested that the function of immature neural cells was impaired by BDV. The development of neural stem/progenitor cell (NSPC) cultures of human origin has offered the possibility of tackling this question. Such cultures allow the investigation of whether BDV, like some other neurotropic viruses, including DNA viruses like cytomegalovirus (29) and retroviruses like HIV (27), can infect and damage human NSPCs (HNPCs).
To gain an understanding of the involvement of neural stem/progenitors in BDV-induced neuropathogenesis, we used a cell culture system consisting of primary human brain-derived neural stem/progenitor cells, which can differentiate into neurons and astrocytes. We report that BDV persistently infects these cells without altering their survival or undifferentiated phenotype. In contrast, upon the induction of differentiation, BDV is capable of severely impairing neurogenesis by interfering with the survival of newly formed neurons. Thus, our study highlights a new mechanism by which BDV can impair neural function and brain plasticity in infected individuals. These results may help us understand the behavioral disorders associated with BDV infection.
MATERIALS AND METHODS
HNPC cultures.
HNPCs used in this study were prepared from the CNS of first-trimester human fetuses. Procedures for the procurement and use of this human fetal CNS tissue were approved and monitored by the Comité Consultatif de Protection des Personnes dans la Recherche Biomedicale of Henri Mondor Hospital, France. Briefly, the cortex was dissected and cut into 1-mm3 tissue pieces. After mechanical dissociation, single-cell suspensions were cultured in Dulbecco's modified Eagle medium-F12 (DMEM-F12; 1/1; Invitrogen Life Technologies) supplemented with B27 (Invitrogen Life Technologies) and containing epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) at 20 ng/ml (R&D Systems), heparin (5 μg/ml, Sigma), 100 U penicillin, and 1,000 U streptomycin (Invitrogen Life Technologies). This cell suspension generated proliferating clones containing HNPCs in floating spheres (termed neurospheres). Cells were further expanded and maintained in suspension as neurospheres in uncoated tissue culture dishes in advanced Dulbecco's modified Eagle medium-F12 (DMEM-F12 Adv.; Invitrogen Life Technologies) supplemented with l-glutamine (2 mM; Gibco), apotransferrin (0.1 mg/ml; Sigma), insulin (25 μg/ml; Sigma), and progesterone (6.3 ng/ml; Sigma). Medium, referred to as N2A medium, was changed 3 times a week, and the growth factors EGF and bFGF (both at 20 ng/ml; Abcys) were added to maintain undifferentiated cells. For infection, cells were cultured as monolayers by seeding them in matrigel-coated dishes (1/1,000; BD Biosciences) in N2A medium. They were subcultured using TrypLE (Invitrogen Life Technologies) when 80% confluence was reached. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Virus infection.
Cell-free BDV (strain He80) was prepared by the osmotic shock treatment of persistently infected Vero cells and titrated (in focus-forming units [FFU]/ml) on Vero cells using an immunofocus assay as described in Danner et al. (7). For both kinetic studies and the establishment of a persistently infected HNPC culture, adherent cells were infected at a multiplicity of infection (MOI) of 0.05 FFU/cell. Cells were incubated with the virus for 1 h at 37°C to allow adsorption. The inoculum then was removed and, after one washing step, replaced by fresh N2A medium supplemented with growth factors. Persistently infected cells were maintained with one or two subcultures per week, and BDV infection then was verified after 2 to 3 weeks of culture for each experiment by immunofluorescence using an anti-BDV nucleoprotein serum (BDV-p40; 1/800; a generous gift from D. Gonzalez-Dunia). By 3 weeks postinfection, more than 95% of HNPCs were infected, and differentiation was initiated as described below. The infection of primary cultures of human astrocytes was performed according to a similar protocol.
Neuronal and glial differentiation.
To obtain mixed cultures of human neurons and astrocytes, BDV-infected and matching noninfected HNPCs were seeded in 24-well and 6-cm plates at a density of 1.5 × 105 cells/well or 1.4 × 106 cells/plate, respectively. Differentiation was initiated 1 or 2 days after plating by replacing N2A medium with N2ANBC (50% N2A plus 50% Neurobasal supplemented with l-glutamine and B27 without vitamin A; Invitrogen Life Technologies) and withdrawing growth factors. Differentiation conditions were maintained for up to 21 days, during which time medium was changed 3 times a week. Twenty-four-well plates were used for immunocytofluorescence, and 6-cm plates were used to prepare lysates for the analysis of protein and RNA. To obtain primary cultures of human astrocytes, HNPCs were induced to differentiate in DMEM-F12 medium supplemented with 2 mM l-glutamine and 10% fetal bovine serum for 3 weeks. Medium was changed every 2 to 3 days. Their status as glial fibrillary acidic protein (GFAP)-positive cells was verified before infection using an antibody directed against GFAP.
Immunocytofluorescence analysis.
Standard immunocytofluorescence was performed. Undifferentiated and differentiated HNPCs grown in 24-well plates were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) in phosphate-buffered saline (PBS) for 20 min at room temperature (RT), rinsed 3 times with PBS, and blocked with 0.1% PBS–Triton X-100 plus 3% bovine serum albumin (BSA; Sigma) for 45 min at RT. Primary antibodies were incubated in 0.1% PBS–Triton X-100 plus 0.3% BSA for 1.5 h at RT, washed 3 times with PBS, and incubated for 1 h at RT with secondary antibodies in 0.1% PBS–Triton X-100 plus 0.3% BSA. After rinsing with PBS, nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; 0.1 μg/ml; Sigma) in PBS for 10 min at RT. Primary antibodies used were directed against nestin (a neural stem cell marker; 1/1,000; AbD Serotec, France), β-tubulin isotype III (a neuronal precursor marker; 1/1,000; Sigma-Aldrich, France), microtubule-associated protein 2 (MAP2; a dendritic marker of postmitotic neurons; 1/1,500; Sigma, France), GFAP (1/1,000; Dako, France), cleaved caspase 3 (an apoptotic marker; 1/200; Cell Signaling Technology), and BDV nucleoprotein (BDV-p40; as described above). Secondary antibodies were anti-rabbit Alexa Fluor 488 and anti-mouse Alexa Fluor 546 (1/1,000; Molecular Probes-Invitrogen Life Technologies, France). For the assessment of cell death, terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining was performed according to the manufacturer's instructions (Promega, France). Images were acquired using an ApoTome microscope (Zeiss) equipped with a 20× objective and AxioVision software. For each experiment, images were acquired from 3 to 4 different wells for each condition and 4 to 9 fields per well. Neurons, astrocytes, total cells, and apoptotic cells were counted in whole fields based on β-tubulin III, GFAP, DAPI, and TUNEL staining. For TUNEL staining, the percentage of apoptotic events referred to the number of apoptotic events versus the number of nonapoptotic cells per field. On average, 600 to 800 cells were counted per well.
Proliferation test.
Proliferation was quantified using the Wst1 kit (Roche) according to the manufacturer's instructions. Briefly, HNPCs were infected as described previously. BDV-infected cells at the 3rd (PI3) and 10th (PI10) passage following infection and their matching noninfected cells were plated in 6 wells of a 48-well plate precoated with matrigel. Wst1 reagent was added to noninfected and BDV-infected cultures at days 0, 2, 4, and 7 after plating. After 1 h of incubation, 100 μl of supernatant was transferred to a 96-well plate to allow the measurement of absorbance using an enzyme-linked immunosorbent assay (ELISA) reader.
Western blot analysis.
Differentiated and nondifferentiated HNPCs were rapidly lysed in protein lysis buffer containing 20 mM Tris-HCl, pH 8, 137 mM NaCl, 10% glycerol, 2 mM EDTA, 1% NP-40, and a protease inhibitor cocktail (Sigma). Insoluble material was removed by centrifugation (14,000 rpm for 15 min at 4°C), and the protein concentration was determined by using the MicroBCAssay protein quantification kit (Uptima-Interchim) in accordance with the manufacturer's instructions. Twenty micrograms of protein per lane was separated by SDS-polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane (ECL Amersham Hybond membrane; GE Healthcare). Blots were blocked in Tris-buffered saline containing 0.1% Tween 20 (TBST) supplemented with 3% BSA for 1 h at RT, followed by incubation with the primary antibody overnight at 4°C. After washing with TBST, the blots were incubated for 1 h with either horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibody (1/25,000; GE Healthcare). After 3 washes with TBST, peroxidase activity was revealed by incubation with Pierce Super Signal West Pico substrate (Thermoscientific). All blots were analyzed using a fusion fx5 molecular imager (Vilber Lourmat, France) and fusion-capt software. The quantification of protein was carried out by measuring the density of the bands and normalizing for actin expression. For reprobing, blots were stripped for 90 min at RT in 0.1 M acetic acid and 0.15 M NaCl. Primary antibodies used were anti-β tubulin isotype III (1/1,000; Sigma), anti-GFAP (1/2,000; Dako), anti-β-actin (1/500; Sigma), and rabbit polyclonal antibodies raised against the BDV nucleoprotein (BDV-p40) and the BDV phosphoprotein (BDV-p24) (1/20,000 and 1/10,000, respectively; generous gifts from D. Gonzalez-Dunia).
Reverse transcription and quantitative real-time PCR.
Total RNA was extracted from differentiated and nondifferentiated HNPCs by using the RNeasy kit for RNA purification (Qiagen) according to the manufacturer's instructions. Isolated RNA was quantified with a NanoDrop spectrophotometer, and 500 ng of RNA was reverse transcribed with SuperScript II reverse transcriptase (Invitrogen Life Technologies), using either random hexamers or a primer specific for viral genomic RNA (5′-TGTTGCGCTAACAACAAACCAATCAC-3′). Two μl of cDNA was used to perform real-time PCR with a LightCycler 1.5 (Roche Applied Science) using QuantiTect SYBR green PCR master mix (Qiagen) in a 20-μl reaction mixture. For PCR, samples were held for 15 min at 94°C and then subjected to 45 amplification cycles consisting of incubation at 94°C for 15 s, 60°C for 15 s, and 72°C for 10 s, with a final extension step at 72°C for 10 min. Except for those for BDV-p40, primers have been described previously by Hong et al. (17a) and were the following: F-nestin, 5′-CAGCGTTGGAACAGAGGTTGG-3′; R-nestin 5′-TGGCACAGGTGTCTCAAGGGTAC-3′; F-cd133, 5′-CAAAAGGAGTCGGAAACTGG-3′; R-cd133, 5′-GATCTGTGAACGCCTTGTCC-3′; F-β tubulin III, 5′-CAACAGCACGGCCATCCAGG-3′; R-β tubulin III, 5′-CTTGGGGCCCTGGGCCTCCGA-3′; F-GFAP, 5′-GGCACGTGCGGGAGGCGGCC-3′; R-GFAP, 5′-TCTCATCACATCCTTGTGC-3′; F-MBP, 5′-AAGGACTCACACCACCCGGC-3′; and R-MBP, 5′-TTTCAGCGTCTAGCCATGGG-3′. For BDV-p40, primers were the following: F-BDVp40, 5′-AGGAACGCAGTGGCATTGT-3′; and R-BDVp40, 5′-GCAGCGTGCAGTCCTGGGATTA-3′.
Statistical analysis.
Data are presented as means ± standard errors of the means (SEM). Statistical analyses were performed by employing the Student's t test. P > 0.1 was considered not significant.
RESULTS
HNPCs are multipotent cells capable of differentiating into mixed cultures of neurons and astrocytes.
We first characterized the HNPCs utilized in this study. Neurospheres in suspension were grown for 15 passages and were further subcultured up to 10 additional passages as adherent cells on matrigel-coated dishes. Cells maintained an undifferentiated or early differentiated state. Immunofluorescence demonstrated that more than 95% of cells expressed the neuroepithelial stem cell marker nestin (Fig. 1A, left). Upon growth factor withdrawal, cells grown in matrigel-coated plates differentiated into neurons and astrocytes. The two phenotypes were distinguished on the basis of morphology and cell marker expression (Fig. 1A, right, and B). In our cultures, very few cells had the typical morphology of oligodendrocytes, and the myelin basic protein (mbp) gene was not detectable by quantitative real-time PCR (data not shown).
Fig 1.

HNPCs differentiate into neurons and astrocytes (A and B) and are fully permissive to BDV infection (C, D, and E). (A) Immunofluorescence labeling of human neural progenitor cells (HNPCs) and their derivatives. On the left is a photomicrograph of the undifferentiated cells immunostained with nestin antibody (orange). On the right is a photomicrograph of cells differentiated into neurons (N) and astrocytes (As) immunostained with antibodies against β-tubulin III (β-tub III; orange) and GFAP (green). Nuclei were stained with DAPI (blue). (B) Western blot showing an increase in neuronal and astroglial markers upon differentiation. N + As, mixed cultures of neurons and astrocytes. (C) The susceptibility of HNPCs to BDV infection was analyzed with an antibody directed toward the viral nucleoprotein p40 at day 2, 4, 8, and 12 postinfection. Nuclei were stained with DAPI. (D) Based on BDV immunostaining, the percentage of infected HNPCs was determined from day 2 to day 12 following infection. Data represent means ± SEM from one experiment performed in triplicate. Similar results were obtained in two independent experiments. NI, noninfected. (E) The expression of the gene coding for viral nucleoprotein p40 was analyzed by quantitative real-time reverse transcription-PCR on 500 ng of total RNA. Data are representative of two independent experiments performed in duplicate. Statistical analysis was performed by employing the Student's t test. ***, P < 0.001.
HNPCs are fully permissive to BDV infection.
To evaluate the susceptibility of HNPCs to BDV infection, we examined whether cells were capable of supporting the full viral cycle from entry into the cells to replication and propagation. Cells were plated on matrigel-coated 96-well plates and infected at a low MOI of 0.05. The capacity of the virus to infect, to replicate, and to disseminate in HNPCs then was evaluated by immunofluorescence on days 2, 4, 8, and 12 following infection using an antibody directed toward the viral nucleoprotein p40 (Fig. 1C). On day 2, a number of cells, albeit a small number, stained positive for BDV-p40, revealing their permissivity to BDV infection. The observation of cultures from days 2 to 12 showed that while only 2.6% ± 2.85% of the cells were infected by day 2, a large proportion of cells, 75.5% ± 11.7%, was infected by day 12 (Fig. 1D), thus demonstrating that the virus replicates in HNPCs and disseminates in a very efficient manner. The relative content of BDV-p40 RNA (genomic and transcripts) was examined next (Fig. 1E). Viral RNA increased dramatically from day 2 (1 arbitrary unit) until day 12 (396 ± 108 arbitrary units), at which time the virus had disseminated throughout most of the culture.
BDV infection persists in HNPCs without altering their undifferentiated phenotype or capacity for survival.
HNPCs were infected at low MOI (0.05) as described above and further subcultured, with an average of one passage a week. After infection, cells were analyzed at each passage for survival, at passages 2, 3, 4, 6, and 10 for infection, protein level, and morphology, and at passages 3 and 8 for proliferation. After 2 to 3 subcultures, which corresponded to approximately 12 days of infection, more than 96% of the cells were infected as determined by the detection of the viral nucleoprotein p40 by immunofluorescence (Fig. 2A, BDV-PI2). The percentage of infected cells then remained stable during the duration of the experiment up to 10 passages (Fig. 2A, BDV-PI10), which corresponded to about 70 days of infection. The content of viral proteins, as determined by Western blotting using antibodies directed against phosphoprotein p24 and nucleoprotein p40, was stable in fully infected cultures from passages 2 to 10 (Fig. 2B). These results demonstrated that BDV establishes a persistent infection in HNPCs. At every passage at which cells were examined using phase-contrast microscopy, BDV infection had no cytopathic effect. Also, BDV infection did not alter the morphology of HNPCs. Early after infection, at passage 2 (PI2), both BDV-infected and noninfected cells stained with an anti-nestin antibody presented the typical morphology of neural progenitor cells (Fig. 2C). At later passages, it was not unusual to observe cells with a more differentiated morphology. However, this was observed in both infected and noninfected cultures and therefore was not due to BDV infection but rather to natural differentiation occurring in HNPCs grown on matrigel-coated plates. The level of expression of nestin, as determined by Western blotting, did not differ in BDV-infected and noninfected cells. This was observed from passage 3 following infection (Fig. 2D) to passage 10 (not shown). However, a general decrease in nestin expression was observed in later passages (not shown), confirming the morphological observation. Thus, the immature state of HNPCs was affected by serial passaging on matrigel-coated plates, but no alteration of the undifferentiated phenotype, based on morphology or expression of nestin level, was observed in relation to BDV infection. Moreover, mitochondrial dehydrogenase activity was similar in noninfected and BDV-infected cultures (Fig. 2E), showing that BDV infection did not alter the capacity of HNPCs to proliferate. Therefore, although BDV establishes a full viral cycle in HNPCs, it does not alter its undifferentiated phenotype.
Fig 2.

BDV infection persists in HNPCs without altering their undifferentiated phenotype, survival, and proliferation. HNPCs were infected and analyzed at different passages postinfection (PI2, PI3, and PI10) for (A) permissivity to infection, by immunofluorescence using an anti-p40 antibody; (B) viral protein level, by Western blotting; (C) morphology, by immunofluorescence using an anti-nestin antibody; (D) nestin level, by Western blotting; and (E) proliferation. Data represent the means ± SEM from 3 independent experiments performed in quintuplicate. Statistical analysis was performed by employing the Student's t test. ns, not significant.
Initial steps of neural differentiation are not altered by BDV infection.
We then sought to determine whether BDV could impair the process of neural differentiation by examining the initial steps when, upon growth factor withdrawal, proliferation is arrested and differentiation is initiated. At this time, nestin and cd133, two neural stem cells markers, are downregulated. Fully infected HNPCs (after passage 2 postinfection) and matched noninfected controls were seeded at the same density on matrigel-coated 6-cm dishes and induced to differentiate for 4 days by growth factor withdrawal. Nestin and cd133 mRNA were quantified by quantitative real-time PCR. As expected, the levels of nestin and cd133 mRNA decreased in our noninfected cultures (Fig. 3A and B). A similar decrease was observed in BDV-infected cultures (Fig. 3A and B), revealing that BDV infection does not alter HNPCs in their capacity to lose their immature stage and commence a differentiation program.
Fig 3.
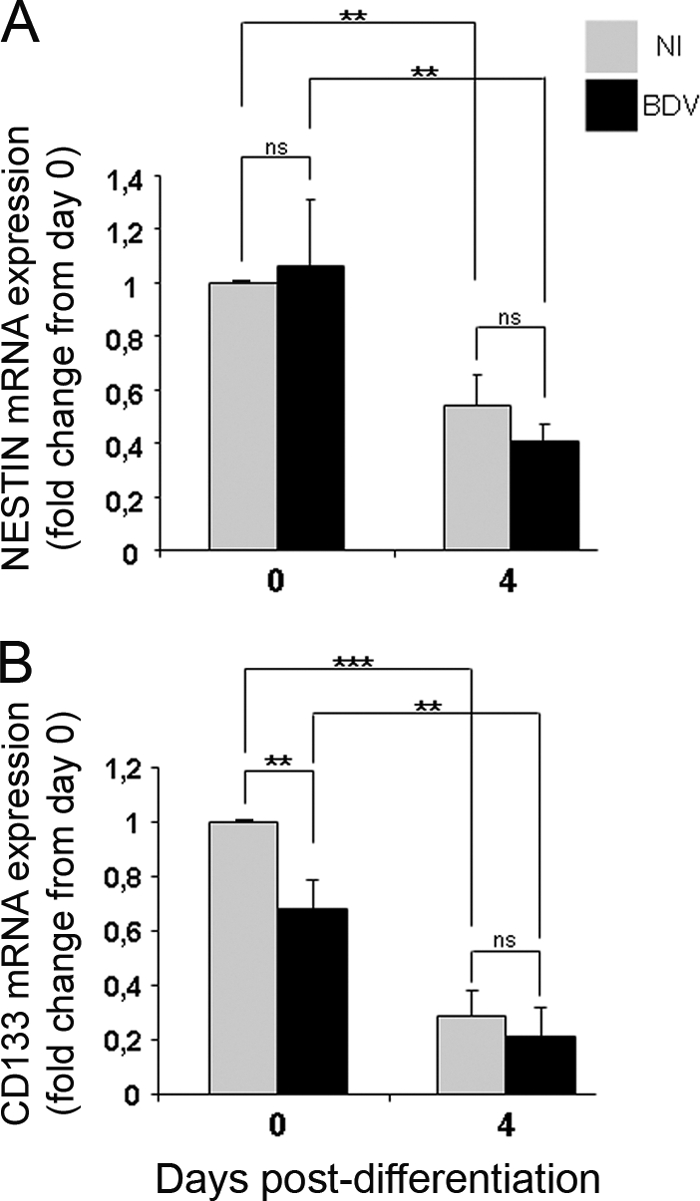
BDV-infected HNPCs lose their immature phenotype upon the initiation of differentiation. Fully infected HNPCs and their matched noninfected controls were induced to differentiate for 4 days by growth factor withdrawal. The expression of nestin (A) and CD133 (B) mRNA was analyzed by quantitative real-time PCR at day 0 and day 4 of differentiation. Data represent the means ± SEM from three independent experiments performed in duplicate. Statistical analysis was performed by employing the Student's t test. ns, not significant. **, P < 0.01; ***, P < 0.001.
BDV impairs neural differentiation by inducing cell death.
We then wondered whether BDV could impair differentiation at later steps. Fully infected HNPCs (after PI2) and their matched noninfected controls were induced to differentiate for 14 days by growth factor withdrawal (Fig. 4A). In noninfected cultures, differentiated cells exhibited a typical pattern of mixed cultures of astrocytes, visible as gray and protoplasmic cells, and neurons, visible as small-sized refringent cells, as observed by phase-contrast microscopy. The two cell types were uniformly distributed in culture dishes. In contrast, in infected cultures an atypical pattern was observed, with numerous dying cells being present in groups and being surrounded by healthy cells of both neuronal and astrocytic morphologies. Cell death was further observed by TUNEL and DAPI stainings. DAPI staining revealed nuclei of two sizes in noninfected cultures, corresponding to astrocytes (14.80 ± 1.27 μm) and neurons (8.25 ± 0.98 μm). In BDV-infected cultures, atypical nuclei of a smaller size (3.28 ± 0.43 μm) were numerous and colocalized with dying cells observed with phase-contrast light and TUNEL staining. The occurrence of cell death in infected cultures was confirmed by the determination of the total number of cells based on DAPI staining (Fig. 4C), as well as by the determination of the percentage of apoptotic events based on TUNEL staining (Fig. 4D). At 14 days of differentiation, a 26.7% decrease in total cell number was observed in BDV-infected cultures (165 ± 7.8 cells per field in noninfected cultures and 121 ± 7.1 in BDV-infected cultures). We verified that a similar number of cells had been plated in noninfected and BDV-infected cells by counting them at day 0 (not shown). Therefore, the decrease in cell number could not be attributed to an experimental artifact but rather demonstrates that BDV diminishes cell survival during the process of neural differentiation. This was confirmed by the dramatic increase in the percentage of apoptotic events in BDV-infected cells (89.4% ± 20%) compared to that of the noninfected one (5.1% ± 0.9%). By examining cells at an earlier time point, the kinetics of cell death could be established. At day 4 of differentiation (Fig. 4B, C, and D), the number of cells in noninfected and BDV-infected cultures was similar (Fig. 4C). At this time point, a fraction of the cell population was apoptotic (Fig. 4B and D). This was independent of BDV infection and was due to natural death occurring in the initial step of neural differentiation; this led to a general decrease in cell population by day 14. The percentage of apoptotic events was higher in BDV-infected cultures, showing that BDV infection contributed to cell death at this early time point. Thus, while apoptosis was transitory in uninfected cells, apoptotic events in BDV-infected cultures markedly increased between days 4 and 14, indicating that BDV-induced death occurred mainly after the initial steps of neural differentiation.
Fig 4.

BDV interferes with cell survival during neural differentiation. Fully infected HNPCs and their matched noninfected controls were induced to differentiate for 14 (A) or 4 (B) days by growth factor withdrawal. Differentiated cells were observed by phase-contrast microscopy, TUNEL, and DAPI staining. Similar observations (bright-field/DAPI) were made in 7 independent experiments at 14 days of differentiation and 3 independent experiments at 4 days of differentiation. Note that TUNEL staining matches with fragmented nuclei stained with DAPI. (C) Total number of cells based on DAPI staining. Four to 9 fields and a minimum of 700 cells per well were counted. Data represent mean values ± SEM from one experiment performed in triplicate. Similar results were obtained from 3 independent experiments. Statistical analysis was performed by employing the Student's t test. **, P < 0.01; ns, not significant. d4, day 4; d14, day 14. (D) Percentage of apoptotic events based on TUNEL staining. Three to 4 fields and a minimum of 400 cells per well were counted. Data represent mean values ± SEM from one experiment performed in triplicate. Statistical analysis was performed by employing the Student's t test. *, P < 0.1; **, P < 0.05.
BDV infection strongly impairs neuronal differentiation.
We next examined whether BDV impaired neurogenesis or gliogenesis. To evaluate the effect of BDV on neurogenesis, HNPCs that had undergone differentiation for 4 and 14 days were fixed and immunostained with an antibody directed against the neuronal marker β-tubulin III. Four days following the onset of differentiation, the examination of noninfected and BDV-infected cultures revealed that numerous β-tubulin III-positive cells had been generated at this early time point (Fig. 5A, a and c). Gross examination did not reveal major morphological differences between the two cultures. β-Tubulin III-positive cells were heterogeneous, presenting either no neurites, short neurites, or more elongated ones, showing that cells were at different states of neuronal differentiation. Alteration in neurite length or branching was not observed in infected cultures, indicating that BDV does not interfere with neuritogenesis. The enumeration of β-tubulin III-positive cells showed an average of 84 ± 2.0 neurons per field in noninfected cultures (Fig. 5B), whereas 66 ± 7.3 neurons were present in BDV-infected cultures (Fig. 5B). Thus, although numerous neurons had been generated in both noninfected and BDV-infected cultures, a 22% loss occurred in infected cultures. At that time, no difference was observed in the level of either β-tubulin III mRNA or protein in the two cultures (Fig. 5C and D). Fourteen days after the induction of differentiation, the number of β-tubulin III-positive cells had increased by 20% in noninfected cultures (106 ± 7.4 neurons per field) (Fig. 5B), showing that neurons had continued to be generated. In the meantime, extensive neurite outgrowth had occurred, as shown by β-tubulin III immunostaining (Fig. 5A, b), indicating that neurons had acquired a more mature morphology. At that time, a lower density of β-tubulin III-positive cells was clearly observed in BDV-infected cultures (Fig. 5A, d). Quantification confirmed that a dramatic decrease in the number of neurons had occurred (Fig. 5B). The average per field was 54 ± 5.4, showing that 50% of the neurons had been lost compared to levels in noninfected cultures. The decrease in the quantity of mRNA encoding β-tubulin III (Fig. 5C) and of the β-tubulin III protein (Fig. 5D), as shown by quantitative real-time PCR and Western blotting, respectively, confirmed the loss of β-tubulin III-positive cells in infected cultures 21 days after the onset of differentiation. The careful examination of double DAPI and β-tubulin III staining in BDV-infected cultures revealed apoptotic nuclei surrounded by β-tubulin III staining (Fig. 6A), demonstrating that BDV-induced neuronal loss was attributable to neuronal apoptosis. To increase precision concerning the stage of neuronal differentiation at which apoptosis was initiated, we performed immunostaining for MAP2, a dendritic marker of postmitotic neurons (Fig. 6B). Four days following the induction of differentiation, the overall MAP2 staining was weak in both noninfected and BDV-infected cultures, except for a few cells in which neuritic extensions were highly stained (Fig. 6B, a and c). This recapitulated the observation made with β-tubulin III staining and further confirmed that, at that time, the majority of neuronal cells were poorly differentiated. At 14 days of differentiation, all neuronal cells had acquired neuritic extensions which formed a network immunostained with MAP2. Neuronal cells had matured and acquired a postmitotic phenotype. However, almost all of them exhibited a uni- or bipolar morphology (Fig. 6B, b, arrowheads, and C, top), showing that they had not reached a fully matured stage, which is characterized by a complex multipolar morphology. At that time, double MAP2- and TUNEL-positive staining was observed in BDV-infected cultures but not in the noninfected one, demonstrating that dying cells were postmitotic neurons. Our results, therefore, showed that BDV-induced death observed at 4 days of differentiation (Fig. 4B and D) occurred when neurons were poorly differentiated. As neuronal death became massive (Fig. 4A and D), by day 14 of differentiation more matured postmitotic neurons were affected. To further elucidate the mechanism of neuronal death, noninfected and BDV-infected cells differentiated for 14 days were analyzed for cleaved caspase 3, another marker of apoptosis. In infected cultures, all small-sized atypical nuclei, as revealed by DAPI, expressed cleaved caspase 3 (Fig. 6C), demonstrating that caspase 3 activation was substantially higher in infected than in noninfected cultures, in which only a few cells were positive for cleaved caspase 3. This confirmed that cells were dying by an apoptotic process that was caspase dependent. Furthermore, cleaved caspase 3 was also observed along degenerating neurites (Fig. 6C), which provided additional evidence that, at 14 days of differentiation, dying neurons had grown elongated neurites.
Fig 5.

BDV impairs neurogenesis. Fully infected HNPCs and their matched noninfected controls were induced to differentiate for 4 and 14 days by growth factor withdrawal. (A) Neuronal cells immunostained for β-tubulin III (orange) and DAPI (blue; nuclear staining). (B) Total number of β-tubulin III-positive cells in noninfected and BDV-infected cultures. Data represent mean values ± SEM from one experiment performed in triplicate. Similar results were obtained from 3 independent experiments. β-Tubulin III mRNA and protein were analyzed by quantitative real-time PCR (C) and Western blotting (D). Data represent mean values ± SEM from three independent experiments performed in duplicate (C) or two independent experiments performed in duplicate (D). Statistical analyses were performed by employing the Student's t test. ns, not significant; *, P < 0.1; ***, P < 0.005.
Fig 6.

Neurons die by a caspase-3 dependent pathway. Fully infected HNPCs and their matched noninfected controls were induced to differentiate for 4 (B) and 14 (A, B, and C) days by growth factor withdrawal. (A) Cells were immunostained for β-tubulin III (orange) and DAPI (blue; nuclear staining). Arrows show small-sized atypical nuclei surrounded by β-tubulin III immunostaining, indicating that dying cells have a neuronal phenotype. (B) Cells were immunostained for MAP2 (red), TUNEL (green), and DAPI (blue). Arrowheads show neurons with typical uni- or bipolar morphology. (b) Arrows show double MAP2/TUNEL-positive staining, indicating that dying cells were postmitotic neurons. (C) Cells were immunostained for β-tubulin III (orange), cleaved caspase 3 (green), and DAPI (blue). Note that cleaved caspase 3 staining colocalizes with small-sized atypical nuclei (arrowhead) and that damaged neurites are positive for cleaved caspase 3 (arrows).
BDV infection does not alter glial differentiation.
To examine whether BDV could also influence glial differentiation, we analyzed HNPCs induced to differentiate using an antibody directed against GFAP, an astroglial marker. Immunostaining revealed that numerous astrocytes were formed 14 days after the onset of differentiation in both noninfected and BDV-infected cultures. No morphological differences were observed in the two cultures (Fig. 7A). This suggested that astroglial differentiation occurred normally in infected cultures. This was confirmed by the enumeration of cells, which showed that a similar number of astrocytes was generated in infected (58.8 ± 9.82 per field) and noninfected cultures (53.9 ± 5.61 per field) (Fig. 7B). In addition, the quantification of GFAP at the mRNA level by quantitative real-time PCR (Fig. 7C) or at the protein level by Western blotting (Fig. 7D) showed a strong upregulation of transcripts and proteins by 21 days of differentiation but no statistically significant difference between infected and noninfected cells. Clearly, BDV infection did not alter astroglial differentiation. Whether this could be explained by a lack of permissivity to the BDV infection of cells differentiated into astrocytes was evaluated. The evidence that viral replication, as assessed by the quantification of viral genomic RNA, is slightly increased during the differentiation process (Fig. 7F) did not support this hypothesis. More importantly, we found that all progenies of infected HNPCs, whether they were committed to glial or neuronal lineages, were themselves highly infected, as shown by BDV-p40 immunostaining 14 days after the induction of differentiation (Fig. 7E). Similar observations were made at 4 and 21 days of differentiation (not shown). Finally, the full permissivity of astrocytes to BDV infection was confirmed in primary cultures of human astrocytes. Cells were infected at an MOI of 0.05, and the capacity of the virus to infect, replicate, and disseminate was evaluated by immunofluorescence on days 4 and 9 following infection using an antibody directed against the viral nucleoprotein p40 (Fig. 7G). On day 4, few cells were immunostained for BDV-p40 (Fig. 7G, a), revealing that the virus could enter the cells. Their numbers had considerably increased by day 9 (Fig. 7G, b), demonstrating that the virus could replicate and disseminate in human astrocytes.
Fig 7.

Astroglial differentiation is unaltered by BDV infection. Differentiation of fully infected HNPCs was induced by growth factor withdrawal for 14 to 21 days (A to F). (A) Cells were fixed after 14 days of differentiation and immunostained for GFAP (green). (B) Determination of astrocyte number in noninfected and BDV-infected cultures at day 14 of differentiation. Data represent mean values ± SEM from one experiment performed in triplicate. Similar results were obtained from 3 independent experiments. (C and D) The expression level of GFAP mRNA (C) and proteins (D) was analyzed by quantitative real-time PCR and Western blotting. (E) Cells were fixed after 14 days of differentiation and immunostained with an antibody directed against the viral nucleoprotein p40 (green) and β-tubulin III (orange). Nuclei were stained with DAPI (blue). Arrows indicate infected astrocytes, and arrowheads show infected neurons. Note that all cells are highly infected. (F) The expression level of the viral genomic RNA was analyzed by quantitative real-time PCR. Statistical analyses were performed by employing the Student's t test. ns, not significant. Data represent mean values ± SEM from three independent experiments performed in duplicate (C and F) or two independent experiments performed in duplicate (D). (G) Human astrocytes were infected for 4 and 9 days before fixation and immunostained for BDV-p40 (green). Nuclei were stained with DAPI (blue).
DISCUSSION
The mechanisms by which BDV alters the behavior of infected individuals are poorly understood. It has been proposed that BDV interferes with neuronal communication through the alteration of synaptic plasticity (34, 40, 41), but it is highly probable that behavior impairment also results from BDV-induced neuronal loss. Such a loss has been well documented in one experimental model, infected newborn rats (19), but the underlying mechanisms are unclear, especially because BDV is a noncytolytic virus. Since neuronal loss is restricted to brain areas that are still undergoing maturation at the time of infection, we suspected that BDV was capable of interfering with neurogenesis. In this study, we took advantage of the availability of human brain-derived progenitor cells to investigate whether BDV can impair neurogenesis.
In infected newborn rats, BDV-induced cell death occurs at a time when neurons continue to be generated from precursor cells. This observation led to the hypothesis that immature cells are highly vulnerable to BDV infection (32, 42). Consistently with this hypothesis, progenitor cells had been shown to be infected in different brain areas (37). In the present study, using HNPCs, we investigated whether neural progenitors were indeed particularly vulnerable to BDV infection. We showed that HNPCs are highly permissive to BDV. The virus enters the cells, replicates, and propagates very efficiently. The rapidity of propagation is comparable to that observed in primary cultures of neurons from newborn rats (16) and is much greater than propagation in Vero cells (our unpublished observation), which are commonly used for virus titration or purification. However, although HNPCs are highly permissive to the virus, they did not appear to be particularly vulnerable. As has been observed in other cell types, including primary cultures of neurons and astrocytes from rats (16, 44) and other cell lines, BDV infection proceeds without an overt pathogenic effect in HNPCs, impairing neither their survival nor their undifferentiated phenotype. Therefore, our results argue against a specific vulnerability of immature neural cells to BDV infection.
Although BDV-infected HNPCs were not damaged at an immature stage, a substantial fraction of cells died in infected cultures upon the induction of differentiation by growth factor withdrawal. BDV infection may have inhibited the differentiation of neural progenitors, inducing an instability which culminated in cell death, as has been observed for another neurotropic virus, human cytomegalovirus, which blocks neuronal differentiation and induces apoptosis in human neural precursor cells (29). Our results, however, demonstrate that BDV infection did not block the differentiation of HNPCs, as a similar decrease in the expression of the neural stem cell markers cd133 and nestin was observed in noninfected and BDV-infected cultures. The absence of alteration in astroglial differentiation confirmed that progenitors were not blocked in an undifferentiated stage and further indicated that only cells engaged along the neuronal pathway were affected by the presence of the virus. Thus, neuronal cells were generated and died sometime after their birth. BDV-induced death was first observed as early as 4 days after the induction of differentiation. At that time, a large fraction of neurons had been generated but they were poorly differentiated, as shown by the paucity of neuritic extension immunostained with β-tubulin III and MAP2 antibodies. Neuronal death then was dramatically increased in the next days, as neurons matured and acquired a postmitotic phenotype, as shown by extensive neurite outgrowth and MAP2 immunostaining in neuritic extensions. These data argue that BDV-induced neuronal death occurs in a progressive manner beginning at an early time of neuronal differentiation and being highly intensified through neuronal maturation. In our experimental settings, neurons do not reach a fully mature stage, as shown by morphological immaturity. Human neurons, indeed, require longer culture times to spontaneously acquire such a phenotype. Thus, BDV-induced death in our cultures proceeds during neuronal maturation, before the acquisition of a fully differentiated stage. Our results led us to conclude that the presence of the virus during neuronal differentiation induces neuronal death by interfering with signaling pathways which are important for the proper maturation of neurons. Importantly, the effect of BDV on neurons is restricted to developmental stages, since it has been repeatedly shown in previous works that fully differentiated neurons in cultures were not affected by BDV infection (16, 31). The BDV impairment of neurogenesis was already suggested in a study by Friedl et al. (12), who used an elegant ex vivo model of organotypic hippocampal slice cultures from newborn rat pups. In that study, infection was performed early, before neuronal maturation. The authors found that BDV-induced neuronal death was an event that started late in neuronal maturation, after axons and synapses were formed. Taking these results together with ours, we demonstrate that the BDV impairment of neurogenesis is a robust mechanism in neurons of both rat and human origins. The differences in the timing at which neuronal apoptosis is initiated might be due either to species or environmental particularities. It is possible that, in a complex environment such as the one provided by organotypic slices, survival signals compete with apoptotic signals, allowing infected differentiating neurons to survive longer than in our in vitro system. In our cultures, the exact mechanisms involved in the BDV impairment of neurogenesis remain to be elucidated. Further studies will be needed to determine whether neuronal apoptosis results from an altered neuron-glial cross-talk or whether an intrinsic program of neuronal differentiation is impaired. As both neuronal and glial cells derived from infected HNPCs are themselves highly infected, one or the other mechanism might occur.
The selective loss of neurons in experimentally infected newborn rats has been puzzling, since BDV has been repeatedly shown to be noncytolytic in differentiated neurons in cultures (16, 31). Our findings support the hypothesis that BDV-induced neurodegeneration is due to BDV interference with neurogenesis. In support of this theory, similar caspase 3-dependent apoptotic mechanisms have been demonstrated in vitro and in vivo (43). We cannot, however, exclude that other mechanisms also contribute to BDV-induced neuropathogenesis in vivo. The observation of activated microglia in areas of intense neurodegeneration (42), preceding neuronal apoptosis (31), suggested that microgliosis could trigger the demise of infected neurons. Although this hypothesis was not supported by an in vitro study in which the BDV infection of a coculture of differentiated neurons, astrocytes, and microglia led to microglial activation without neurotoxicity (30), it is plausible that microglia exacerbates dysfunction initiated by direct virus infection in the in vivo setting. Further studies will be needed to determine the precise mechanisms involved in vivo. Nevertheless, HNPCs in culture offer a unique in vitro model to unravel at least some of the mechanisms leading to BDV-induced neurodegeneration.
Several neurotropic viruses are capable of modulating the fate of NPCs both in vivo and in vitro (for a review, see reference 8). Damage might occur either through common pathways, as was suggested by De Miranda et al. (11), or through specific interactions, such as the inhibition of proliferation, the inhibition of neuronal or glial differentiation, progenitor death or persistence, or, as for BDV, the impairment of the survival of newly formed neurons. As there is increasing evidence that the alteration of neurogenesis contributes to behavioral disorders (13, 21), we speculate that a basis of BDV-induced neurobehavioral disorder is the impairment of neurogenesis.
ACKNOWLEDGMENTS
This work was supported financially by the French National Institute for Agricultural Research (INRA). D.B. and V.S. were supported by grants from the French Embassy in Croatia.
We are most grateful to D. Gonzalez-Dunia for his generous gift of antiviral phosphoprotein and nucleoprotein antibodies and his critically reading the manuscript.
Footnotes
Published ahead of print 21 December 2011
REFERENCES
- 1. Bautista JR, Rubin SA, Moran TH, Schwartz GJ, Carbone KM. 1995. Developmental injury to the cerebellum following perinatal Borna disease virus infection. Brain Res. Dev. Brain Res. 90:45–53 [DOI] [PubMed] [Google Scholar]
- 2. Bode L, Durrwald R, Ludwig H. 1994. Borna virus infections in cattle associated with fatal neurological disease. Vet. Rec. 135:283–284 [DOI] [PubMed] [Google Scholar]
- 3. Briese T, et al. 1994. Genomic organization of Borna disease virus. Proc. Natl. Acad. Sci. U. S. A. 91:4362–4366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carbone KM, Moench TR, Lipkin WI. 1991. Borna disease virus replicates in astrocytes, Schwann cells and ependymal cells in persistently infected rats: location of viral genomic and messenger RNAs by in situ hybridization. J. Neuropathol. Exp. Neurol. 50:205–214 [DOI] [PubMed] [Google Scholar]
- 5. Carbone KM, Park SW, Rubin SA, Waltrip RW, Vogelsang GB. 1991. Borna disease: association with a maturation defect in the cellular immune response. J. Virol. 65:6154–6164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chalmers RM, Thomas DR, Salmon RL. 2005. Borna disease virus and the evidence for human pathogenicity: a systematic review. QJM 98:255–274 [DOI] [PubMed] [Google Scholar]
- 7. Danner K, Heubeck D, Mayr A. 1978. In vitro studies on Borna virus. I. The use of cell cultures for the demonstration, titration and production of Borna virus. Arch. Virol. 57:63–75 [DOI] [PubMed] [Google Scholar]
- 8. Das S, Basu A. 2011. Viral infection and neural stem/progenitor cell's fate: implications in brain development and neurological disorders. Neurochem. Int. 59:357–366 [DOI] [PubMed] [Google Scholar]
- 9. De Keyser J, Mostert JP, Koch MW. 2008. Dysfunctional astrocytes as key players in the pathogenesis of central nervous system disorders. J. Neurol. Sci. 267:3–16 [DOI] [PubMed] [Google Scholar]
- 10. de la Torre JC. 1994. Molecular biology of Borna disease virus: prototype of a new group of animal viruses. J. Virol. 68:7669–7675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De Miranda J, et al. 2010. Induction of Toll-like receptor 3-mediated immunity during gestation inhibits cortical neurogenesis and causes behavioral disturbances. mBio 1:e00176–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Friedl G, et al. 2004. Borna disease virus multiplication in mouse organotypic slice cultures is site-specifically inhibited by gamma interferon but not by interleukin-12. J. Virol. 78:1212–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fuchs E. 2007. Neurogenesis in the adult brain: is there an association with mental disorders? Eur. Arch. Psychiatry Clin. Neurosci. 257:247–249 [DOI] [PubMed] [Google Scholar]
- 14. Gonzalez-Dunia D, Sauder C, de la Torre JC. 1997. Borna disease virus and the brain. Brain Res. Bull. 44:647–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hagiwara K, et al. 2008. Borna disease virus RNA detected in Japanese macaques (Macaca fuscata). Primates 49:57–64 [DOI] [PubMed] [Google Scholar]
- 16. Hans A, et al. 2004. Persistent, noncytolytic infection of neurons by Borna disease virus interferes with ERK 1/2 signaling and abrogates BDNF-induced synaptogenesis. FASEB J. 18:863–865 [DOI] [PubMed] [Google Scholar]
- 17. Hilbe M, et al. 2006. Shrews as reservoir hosts of borna disease virus. Emerg. Infect. Dis. 12:675–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17a. Hong S, Kang UJ, Isacson O, Kim KS. 2008. Neural precursors derived from human embryonic stem cells maintain long-term proliferation without losing the potential to differentiate into all three natural lineages, including dopaminergic neurons. J. Neurochem. 104:316–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hornig M, Solbrig M, Horscroft N, Weissenbock H, Lipkin WI. 2001. Borna disease virus infection of adult and neonatal rats: models for neuropsychiatric disease. Curr. Top. Microbiol. Immunol. 253:157–177 [DOI] [PubMed] [Google Scholar]
- 19. Hornig M, Weissenbock H, Horscroft N, Lipkin WI. 1999. An infection-based model of neurodevelopmental damage. Proc. Natl. Acad. Sci. U. S. A. 96:12102–12107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kamitani W, et al. 2003. Glial expression of Borna disease virus phosphoprotein induces behavioral and neurological abnormalities in transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 100:8969–8974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kempermann G, Krebs J, Fabel K. 2008. The contribution of failing adult hippocampal neurogenesis to psychiatric disorders. Curr. Opin. Psychiatry 21:290–295 [DOI] [PubMed] [Google Scholar]
- 22. Kinnunen PM, et al. 2007. Serological evidence for Borna disease virus infection in humans, wild rodents and other vertebrates in Finland. J. Clin. Virol. 38:64–69 [DOI] [PubMed] [Google Scholar]
- 23. Lieb K, Staeheli P. 2001. Borna disease virus–does it infect humans and cause psychiatric disorders? J. Clin. Virol. 21:119–127 [DOI] [PubMed] [Google Scholar]
- 24. Lipkin WI, Hornig M. 2004. Psychotropic viruses. Curr. Opin. Microbiol. 7:420–425 [DOI] [PubMed] [Google Scholar]
- 25. Lipkin WI, Schneemann A, Solbrig MV. 1995. Borna disease virus: implications for human neuropsychiatric illness. Trends Microbiol. 3:64–69 [DOI] [PubMed] [Google Scholar]
- 26. Lundgren AL, et al. 1995. Staggering disease in cats: isolation and characterization of the feline Borna disease virus. J. Gen. Virol. 76:2215–2222 [DOI] [PubMed] [Google Scholar]
- 27. McCarthy M, Vidaurre I, Geffin R. 2006. Maturing neurons are selectively sensitive to human immunodeficiency virus type 1 exposure in differentiating human neuroepithelial progenitor cell cultures. J. Neurovirol. 12:333–348 [DOI] [PubMed] [Google Scholar]
- 28. Nakamura Y, et al. 2000. Isolation of Borna disease virus from human brain tissue. J. Virol. 74:4601–4611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Odeberg J, et al. 2006. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J. Virol. 80:8929–8939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ovanesov MV, Moldovan K, Smith K, Vogel MW, Pletnikov MV. 2008. Persistent Borna disease virus (BDV) infection activates microglia prior to a detectable loss of granule cells in the hippocampus. J. Neuroinflammation 5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ovanesov MV, et al. 2006. Activation of microglia by Borna disease virus infection: in vitro study. J. Virol. 80:12141–12148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pletnikov MV, Moran TH, Carbone KM. 2002. Borna disease virus infection of the neonatal rat: developmental brain injury model of autism spectrum disorders. Front. Biosci. 7:d593–d607 [DOI] [PubMed] [Google Scholar]
- 33. Pletnikov MV, Rubin SA, Moran TH, Carbone KM. 2003. Exploring the cerebellum with a new tool: neonatal Borna disease virus (BDV) infection of the rat's brain. Cerebellum 2:62–70 [DOI] [PubMed] [Google Scholar]
- 34. Prat CM, et al. 2009. Mutation of the protein kinase C site in borna disease virus phosphoprotein abrogates viral interference with neuronal signaling and restores normal synaptic activity. PLoS Pathog. 5:e1000425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reif A, Schmitt A, Fritzen S, Lesch KP. 2007. Neurogenesis and schizophrenia: dividing neurons in a divided mind? Eur. Arch. Psychiatry Clin. Neurosci. 257:290–299 [DOI] [PubMed] [Google Scholar]
- 36. Rott R, Becht H. 1995. Natural and experimental Borna disease in animals. Curr. Top. Microbiol. Immunol. 190:17–30 [DOI] [PubMed] [Google Scholar]
- 37. Solbrig MV, Adrian R, Baratta J, Lauterborn JC, Koob GF. 2006. Kappa opioid control of seizures produced by a virus in an animal model. Brain 129:642–654 [DOI] [PubMed] [Google Scholar]
- 38. Solbrig MV, Hermanowicz N. 2008. Cannabinoid rescue of striatal progenitor cells in chronic Borna disease viral encephalitis in rats. J. Neurovirol. 14:252–260 [DOI] [PubMed] [Google Scholar]
- 39. van den Pol AN. 2009. Viral infection leading to brain dysfunction: more prevalent than appreciated? Neuron 64:17–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Volmer R, Monnet C, Gonzalez-Dunia D. 2006. Borna disease virus blocks potentiation of presynaptic activity through inhibition of protein kinase C signaling. PLoS Pathog. 2:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Volmer R, Prat CM, Le Masson G, Garenne A, Gonzalez-Dunia D. 2007. Borna disease virus infection impairs synaptic plasticity. J. Virol. 81:8833–8837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weissenbock H, Hornig M, Hickey WF, Lipkin WI. 2000. Microglial activation and neuronal apoptosis in Bornavirus infected neonatal Lewis rats. Brain Pathol. 10:260–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Williams BL, Hornig M, Yaddanapudi K, Lipkin WI. 2008. Hippocampal poly(ADP-Ribose) polymerase 1 and caspase 3 activation in neonatal bornavirus infection. J. Virol. 82:1748–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yamashita M, et al. 2005. Persistent Borna disease virus infection confers instability of HSP70 mRNA in glial cells during heat stress. J. Virol. 79:2033–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]