

Abstract
Chronic hepatitis C virus (HCV) infection can persist even in the presence of a broadly neutralizing antibody response. Various mechanisms that underpin viral persistence have been proposed, and one of the most recently proposed mechanisms is the presence of interfering antibodies that negate neutralizing responses. Specifically, it has been proposed that antibodies targeting broadly neutralizing epitopes located within a region of E2 encompassing residues 412 to 423 can be inhibited by nonneutralizing antibodies binding to a less conserved region encompassing residues 434 to 446. To investigate this phenomenon, we characterized the neutralizing and inhibitory effects of human-derived affinity-purified immunoglobulin fractions and murine monoclonal antibodies and show that antibodies to both regions neutralize HCV pseudoparticle (HCVpp) and cell culture-infectious virus (HCVcc) infection albeit with different breadths and potencies. Epitope mapping revealed the presence of overlapping but distinct epitopes in both regions, which may explain the observed differences in neutralizing phenotypes. Crucially, we failed to demonstrate any inhibition between these two groups of antibodies, suggesting that interference by nonneutralizing antibodies, at least for the region encompassing residues 434 to 446, does not provide a mechanism for HCV persistence in chronically infected individuals.
INTRODUCTION
Hepatitis C virus (HCV) has infected approximately 180 million people worldwide (2). Following infection, most people fail to clear the virus, and a chronic infection, often with serious sequelae, ensues (1, 38). HCV-related end-stage liver disease is the leading indication for liver transplantation, and reinfection of the grafted liver occurs rapidly (32). A systematic review of the research literature recently suggested that there is little, if any, benefit gained by the treatment of liver transplant recipients with standard antiviral regimens (24), and possible adverse effects associated with newly emerging direct-acting antivirals may limit their usefulness in this clinical setting. Antibodies are usually well tolerated, and the successful administration of anti-hepatitis B virus immunoglobulin (Ig) (HBIG) (50, 59) sets an important precedent for HCV. The administration of HCV-neutralizing antibodies during the anhepatic phase and following transplantation could likewise prevent the reinfection of the grafted liver; the reduced incidence of HCV in individuals receiving HBIG containing anti-HCV antibodies (20) supports this notion. However, to date, the therapeutic administration of serum immunoglobulin or monoclonal antibodies targeting HCV has been disappointing (10, 51), indicating that further studies of the polyclonal response are needed, if we are to harness the opportunity that antibody therapy offers.
There is also an urgent need for the development of safe and effective HCV vaccines to prevent infection. Significant progress has been made toward T-cell-based vaccines (22), but these vaccines will not be sufficient to elicit sterilizing immunity. Consequently, the development of an antibody-targeted vaccine is still a priority. Protective vaccines will have to overcome significant viral antigenic diversity. HCV can be classified into seven genetically distinct genotypes and can be further subdivided into at least 70 subtypes, which differ by approximately 30% and 15% at the nucleotide level (29, 53). Within an infected individual, the virus exists as a quasispecies composed of genetically related yet distinct variants, and this variability allows the virus to escape host immunity (52). The envelope glycoproteins E1 and E2 are the natural targets of the neutralizing antibody response and are two of the most variable HCV proteins (8). E1 and E2 are N-linked glycosylated trans-membrane proteins with an N-terminal ectodomain and a C-terminal hydrophobic membrane anchor (reviewed in reference 25) that mediate interactions with a range of cell surface molecules that result in entry via endocytosis. HCV entry requires interactions with a number of cell receptors, which include CD81, scavenger receptor class B type I (SR-BI), and members of the claudin and occludin tight-junction family of proteins (3, 5, 6, 16, 48a, 64, 67).
The development of a potent early neutralizing antibody response is associated with a spontaneous resolution of acute infection (12, 48). Antibodies that target both restricted and broadly neutralizing epitopes have been described. In general, antibodies with a restricted range target the first hypervariable region of E2, and quasispecies evolution leads to rapid escape (68). A number of more broadly neutralizing antibodies that target linear and conformational epitopes overlapping discontinuous regions of E2 involved in CD81 binding have been described (28, 31, 34, 44, 47, 56). One of these regions, encompassing amino acids (aa) 412 to 423 (in reference to the sequence of the H77c molecular clone), is targeted by the broadly neutralizing monoclonal antibodies (MAbs) AP33 and 3/11 (56). Antibodies that target this region are rare in natural infection, suggesting that this region is poorly immunogenic (55).
A number of mechanisms have been proposed to explain how HCV can persist in the presence of neutralizing antibodies. These proposed mechanisms include genetic escape (61), the occlusion of neutralizing epitopes through glycan shielding (18, 26) and lipid associations (23, 54), infection enhancement via serum components such as high-density lipoprotein (HDL) and apolipoproteins (13, 14, 60), and cell-to-cell transmission (7, 63). An additional mechanism was recently proposed by Zhang and colleagues, who reported that a linear region of E2 encompassing amino acids 434 to 446 (the so-called epitope II) elicits nonneutralizing antibodies in humans and chimpanzees that can inhibit the neutralizing activity of antibodies targeting aa 412 to 423 (65, 66). However, previous reports suggested that murine monoclonal antibodies, for example, MAbs 2/69a, 7/16b, and 11/20c, whose epitopes overlap the region encompassing aa 434 to 446, neutralize autologous H77c pseudoparticle (HCVpp) infection and CD81 binding (9, 27). If both findings are true, then this suggests that antibodies that target the region encompassing aa 434 to 446 differ in phenotype according to their epitope specificity or that antibodies that target this region exhibit a dual phenotype.
To better understand the interplay between these groups of antibodies, we isolated human Ig fractions by peptide immunoaffinity techniques and characterized the resulting antibodies together with defined murine MAbs. The neutralizing phenotype, degree of cross-competition, and epitope specificity were assessed. Contrary to previous reports, we failed to observe inhibition or competition between antibodies specific for these two groups. Our data show that human antibodies that target the region encompassing aa 434 to 446 neutralize HCVpp and HCVcc infection. Importantly, these antibodies augment neutralization mediated by antibodies that target the region encompassing aa 412 to 423.
MATERIALS AND METHODS
Peptides and MAbs.
Biotinylated peptides were synthesized by using Synphase PA lanterns (Mimotopes, Melbourne, Australia), using a C-terminal biocytin residue separated from the epitope sequence by a Gly-Ser-Gly spacer sequence. Peptide purity was assessed by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) spectrometry. Peptides were initially dissolved in dimethyl sulfoxide (DMSO) and then dissolved in phosphate-buffered saline (PBS) to a final concentration of 1 mg/ml in PBS. The peptides corresponded to amino acids 412 to 423 (QLINTNGSWHIN [peptide I]) and amino acids 434 to 446 (NTGWLAGLFYQHK [peptide II]) of the HCV H77c polyprotein and a control peptide corresponded to a seroreactive region of the rabies virus glycoprotein (VNLHDFRSDEIE) (11). Where appropriate, anti-E2 MAbs 2/69a (21) and AP33 (43) were used as control antibodies.
Patient samples.
Patient samples were obtained with ethics committee approval from the Trent HCV Cohort Study. Sera from 557 HCV antibody-positive (third-generation enzyme-linked immunosorbent assay [ELISA]; Ortho Diagnostics) individuals chronically infected with diverse HCV genotypes were inactivated by heating to 56°C for 30 min before use.
Peptide capture immunoassay.
Neutravidin (Pierce) was coated onto Maxisorp plates (Nunc) at 5 μg ml−1 in 50 mM carbonate-bicarbonate buffer (pH 9.8; Sigma). Unbound Neutravidin was removed by washing with PBS containing 0.05% Tween 20 (PBS-T), and nonspecific binding sites were blocked by using PBS-T containing 3% bovine serum albumin (PBS-TB). Peptide solutions (1 μg ml−1 in PBS-T) were incubated at room temperature for 1 h. Unbound peptide was removed, and human sera, diluted 1/50 in PBS-T, were bound to the capture peptide for 2 h at room temperature. Unbound sera were removed with three washes, and bound immunoglobulin was detected with an alkaline phosphatase-conjugated anti-human IgG antibody (Sigma) and diluted 1/2,000 in a Tris-buffered saline–0.05% Tween 20 solution (TBS-T), followed by p-nitrophenol phosphate (pNPP) substrate (Sigma). The absorbance at 405 nm was determined by using a BMG Labtech Optima plate reader. The negative cutoff was calculated as twice the mean of three HCV antibody-negative human serum control wells. Assays using purified immunoglobulin were performed essentially as described above, with defined concentrations of Ig.
Peptide-based immunoaffinity purification.
Streptavidin-coated M280 Dynabeads (Invitrogen) were coated with biotin-labeled peptides, representing the amino acid sequences of strain H77c encompassing amino acids 412 to 423 or 434 to 446, according to the manufacturer's instructions. Beads were incubated with 400 μl of patient sera for 4 h at room temperature, washed six times, and incubated with antibody elution buffer (Thermo) for 10 min. Following the removal of the beads, antibody-containing solution was neutralized with 1 M Tris (pH 8.5). As a negative control, serum was incubated with streptavidin-coated Dynabeads in the absence of peptide. Antibody-containing solutions were aliquoted and stored at −20°C for further assays.
Epitope mapping by enrichment of random peptide phage display libraries.
Purified Ig fractions were used to selectively enrich a 12-mer random peptide phage display library (NEB), as described previously (56). Following three rounds of enrichment, individual phage clones were isolated, and the peptide insert was determined by using a −96 sequencing primer (NEB). Sequencing products were resolved by using an ABI 3100 capillary sequencer. The deduced peptide insert amino acid sequences and the sequence corresponding to amino acids 412 to 423 or 434 to 446 of the H77c strain were aligned by using ClustalX (58). Enzyme immunoassays (EIAs) were also performed to determine the reactivities of the Ig fractions and control MAbs to the enriched phage clones. Approximately 1 × 1011 phage particles were added to microtiter plate wells coated with 5 μg ml−1 of MAb or purified Ig, and bound phages were detected by an anti-fd antibody (Sigma), followed by an alkaline phosphatase-conjugated anti-mouse antibody (Sigma) and Sigmafast pNPP substrate. Absorbance values were determined at 405 nm.
HCVpp and HCVcc neutralization assays.
Huh-7 human hepatoma cells (42) and HEK293T human embryonic kidney cells (ATCC CRL-1573) were propagated as described previously (9). cDNA sequences encoding full-length E1E2 were previously cloned into the pcDNA3.1 V5his D-TOPO expression vector (Invitrogen) (33). HCVpp were produced essentially as previously reported (5). Pseudoparticles generated in the absence of the E1E2 plasmid were used as a negative control. For neutralization assays, sucrose cushion-purified HCVpp were mixed with dilutions of purified Ig or MAb, incubated for 1 h at 37°C, and then added to Huh-7 cells plated into a 96-well Optilux plate (BD Biosciences) containing 100 μl medium, and the plates were incubated for 4 h before an additional 100 μl of medium was added. Cultures were incubated at 37°C for 3 days in the presence of 5% CO2. Following the removal of medium, cells infected with HCVpp were lysed with 20 μl of cell lysis buffer (Promega), and 50 μl of luciferase substrate (Promega) was added. Luminescence was measured by using a BMG Labtech Optima plate reader.
Huh7.5 cells were grown in Dulbecco's modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% fetal calf serum and 0.1 mM nonessential amino acids. Plasmids containing JFH-1, H77c/JFH-1, and JFH-1GND genome cDNAs were used to produce HCVcc as described previously (30, 35, 62). Cell supernatants were harvested and filtered through a 0.45-μm nitrocellulose membrane. Following staining for the presence of NS5A, infectious-virus titers were determined according to the method of Reed and Muench (49). For neutralization assays, 100 focus-forming units of virus was incubated with serial dilutions of antibodies and added to Huh7.5 cells following 1 h of incubation, and infection was revealed as described above. Inhibition was determined by comparison with an uninhibited control. In all experiments, a negative control was prepared by using the GND replication-defective HCVcc mutant.
PCR amplification of E1E2 coding sequences from patient sera.
E1E2 sequences were amplified following RNA isolation from infected sera, as previously described (30). Briefly, RNA was extracted by using a Viral RNA kit (Qiagen), and cDNA was generated by using Thermoscript reverse transcriptase (Invitrogen). E1E2 sequences were amplified by using LongAmp Taq (NEB) and sequenced with an internal sequencing primer using BigDye sequencing chemistry (Applied Biosystems).
Statistical analysis.
Differences between means were assessed by using t tests and one-way analysis of variance (ANOVA) with Bonferroni posttest correction, as appropriate. All analyses were performed by using Graphpad Prism, version 5, software.
RESULTS
Individuals with antibodies that target the region of E2 encompassing aa 412 to 423 frequently harbor antibodies that recognize the region of E2 encompassing aa 434 to 446.
Extending our previously reported analyses (55), a total of 557 distinct sera obtained from individuals infected with diverse genotypes of HCV were screened for the presence of antibodies directed to a peptide representing amino acids 412 to 423 of strain H77c (peptide I). Seventeen (2.3%) sera possessed peptide I-reactive antibodies. To investigate the presence of antibodies directed to the peptide representing amino acid residues 434 to 446 (peptide II), peptide binding assays were performed with those sera that were reactive with peptide I. Twelve of the 17 sera (70%) also reacted with peptide II, suggesting that these two epitopes are often coimmunogenic despite being recognized by the sera of only a small subset of patients. We then analyzed a subset of the peptide I-negative sera for binding to peptide II, which showed that the prevalence of reactivity to peptide II was 14.6% in this population. Although relatively low, the prevalence of antibodies to the region encompassing aa 434 to 446 was much higher than the prevalence of antibodies to the region encompassing aa 412 to 423. Despite the fact that the target peptides were based upon a genotype 1a strain (H77c), peptide-reactive antibodies were detected in sera from individuals infected with a range of genotypes. This suggested some degree of epitope conservation despite the observed sequence heterogeneity observed across aa 434 to 446 (Fig. 1). Based upon the availability of sufficient amounts of stored serum, independent sera from patients 1A76 (genotype 1a infection) and 3A64 (genotype 3a infection) were selected to further characterize the neutralizing potency and specificity of the antibody response to amino acid regions encompassing residues 412 to 423 and 434 to 446.
Fig 1.

Amino acid variability in the region encompassing aa 409 to 450 in circulating HCV isolates. An amino acid alignment of the region of E2 derived from diverse genotypes of HCV encompassing residues 409 to 450 was performed by using ClustalX. Peptide sequences used in this study are highlighted by underlined sequences in strain H77c.
To identify an appropriate high-titer serum sample for subsequent studies, and to define the longitudinal peptide I and peptide II binding profiles, sequential sera were tested for their abilities to bind to immobilized peptide I and peptide II (Fig. 2). While some differences in the relative binding were observed for each serum and peptide combination, the antibody response to both peptides in sera from both patients was maintained over a period of several years. The earliest available serum sample from patient 3A64 possessed high levels of peptide II-reactive antibodies but much lower levels of antibody reactivity to peptide I, while the other sera from the same patient had comparable reactivities to both peptides. One sample from each patient that exhibited detectable reactivity to both peptides was chosen for further study.
Fig 2.

The serum antibody response to peptide I and peptide II is maintained over time. Sequential serum samples obtained from patients 1A76 and 3A64 were tested for reactivity to immobilized peptide I (corresponding to the region of E2 encompassing aa 412 to 423) and peptide II (E2 region encompassing aa 434 to 446) in an enzyme immunoassay. (A) 1A76-Ig reactivity to peptide I (■) or peptide II (□). (B) 3A64-Ig reactivity to peptide I (●) or peptide II (○). Arrows indicate the samples used as a source of immunoglobulin fractions for subsequent analyses. OD405, optical density at 405 nm.
Affinity-enriched antibodies directed to peptide I and peptide II display specific binding to their respective peptides.
Peptides corresponding to each E2 region were immobilized onto streptavidin-coated magnetic beads and used to affinity purify antibodies from sera from patients 1A76 and 3A64. This approach recovered between 10 μg and 40 μg of antibody per milliliter of serum. The resulting antibody fractions, as well as MAb AP33 and MAb 2/69a, demonstrated a level of dose-dependent binding to the target peptide that was at least 4-fold higher than that of binding to the reciprocal peptide (Fig. 3).
Fig 3.
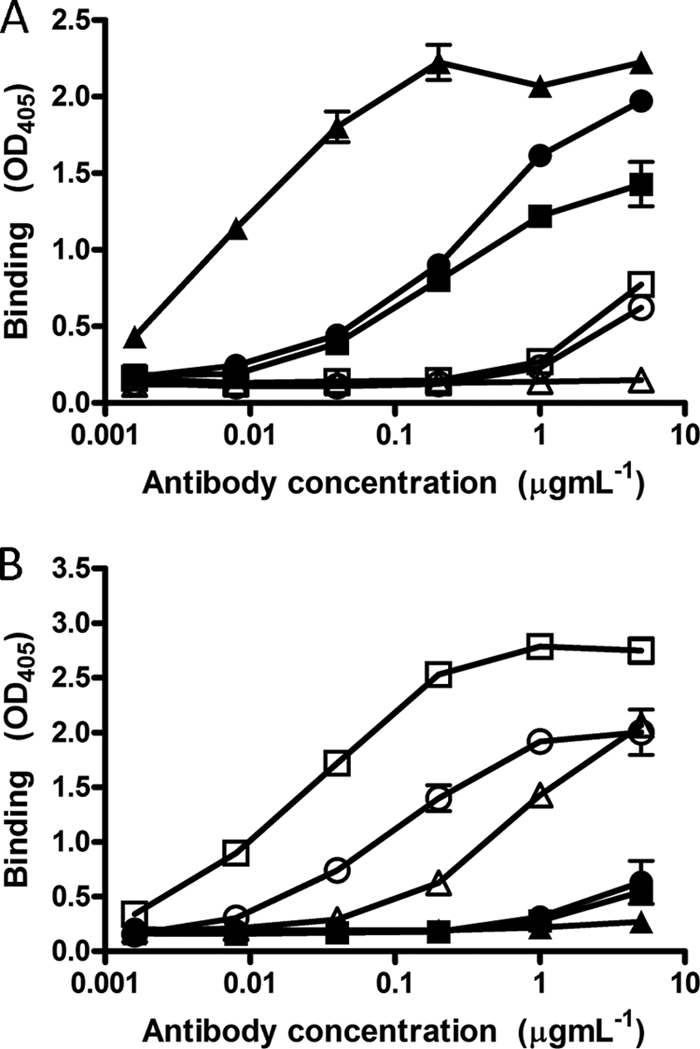
Affinity-purified immunoglobulins demonstrate dose-dependent and specific binding to their selecting peptides. Immunoglobulin fractions were obtained from sera from patients 1A76 and 3A64 by affinity purification using peptide I (peptide I-Ig) and peptide II (peptide II-Ig), which correspond to E2 regions encompassing aa 412 to 423 and aa 434 to 446, respectively. Affinity-purified immunoglobulin and monoclonal antibodies AP33 and 2/69a were tested by an immunoassay for reactivity to immobilized peptide I (A) or peptide II (B). ■, 1A76 peptide I-Ig; ●, 3A64 peptide I-Ig; □, 1A76 peptide II-Ig; ○, 3A64 peptide II-Ig; ▲, MAb AP33; △, MAb 2/69a.
Peptide I-Ig and peptide II-Ig do not cross-compete for E2 binding.
Having purified peptide I- and peptide II-specific immunoglobulin fractions (here termed peptide I-Ig and peptide II-Ig), the accessibilities of their epitopes on H77c E1E2 were assessed (Fig. 4). MAb AP33 and MAb 2/69a recognized Galanthus nivalis agglutinin (GNA) captured E1E2, although the latter MAb bound with a lower relative affinity. Similarly, affinity-purified Ig fractions also bound H77c E1E2 but with differing affinities. Peptide I-Ig derived from sera of patient 3A64 (3A64 peptide I-Ig) showed greater binding to E1E2 than did peptide II-Ig. However, the opposite was true for the Ig derived from sera of patient 1A76 (Fig. 4A). To assess whether the antibodies that target the region encompassing aa 434 to 446 are capable of interfering with the neutralizing potential of antibodies that target E2 residues 412 to 426 (65, 66), we first performed competition assays, where the binding of peptide I-Ig and peptide II-Ig to E2 was detected in the presence of increasing amounts of either MAb AP33 or MAb 2/69a (Fig. 4B to D). E2 binding by peptide I-Ig fractions obtained from both patients was inhibited, in a dose-dependent manner, by MAb AP33 but was unaffected by MAb 2/69a (Fig. 4B and D). Similarly, the binding of peptide II-Ig was inhibited by MAb 2/69a but was unaffected by MAb AP33 (Fig. 4C and E). Together, these data showed that the binding of antibodies to the region of E2 encompassing aa 412 to 423 was unaffected by antibodies that target the region encompassing aa 434 to 446 and vice versa.
Fig 4.

Antibodies that target the region of E2 encompassing aa 434 to 446 do not compete with antibodies that target the region of E2 encompassing aa 412 to 423 for E1E2 binding. (A) 1A76 peptide I-Ig (■), 3A64 peptide I-Ig (●), 1A76 peptide II-Ig (□), 3A64 peptide II-Ig (○), MAb AP33 (▲), and MAb 2/69a (△) were tested for their abilities to bind to GNA-captured E1E2 protein from strain H77c. Data are presented as the differences in optical densities between the antibody sample and a negative control in the absence of target E1E2 protein. (B to E) To test for cross-competition between antibody groups, the binding of GNA-captured E1E2 to 3A64 peptide I-Ig (B), 3A64 peptide II-Ig (C), 1A76 peptide I-Ig (D), or 1A76 peptide II-Ig (E) was determined in the presence of competing MAb AP33 (▲) or MAb 2/69a (△). Data are presented as the percent binding of an uninhibited control. The mean percentages of antibody binding obtained for the highest concentrations of test and control antibodies were compared by using a t test. P values of <0.01 are indicated by double asterisks.
Peptide I-Ig and peptide II-Ig neutralize H77c HCVpp infectivity but differ in their abilities to neutralize JFH-1 HCVpp.
We next determined the abilities of the peptide I-Ig and peptide II-Ig fractions and MAbs AP33 and 2/69a to neutralize HCVpp-expressing strain H77c (genotype 1a) and JFH1 (genotype 2a) E1E2 glycoproteins. A dose-dependent neutralization of H77 HCVpp was observed for all of the Ig fractions and MAbs tested (Fig. 5A). Differences in neutralization potency were evident, with estimated 50% inhibitory concentrations (IC50s) ranging from <0.1 to >8 μg ml−1 for the different peptide I-Ig and peptide II-Ig fractions and monoclonal antibodies. MAb AP33 was the most potently neutralizing, while MAb 2/69a was the least neutralizing. When tested with HCVpp supplemented with E1E2 of the JFH-1 strain, a much lower neutralization potency was observed at the concentrations tested (Fig. 5B). A dose-dependent inhibition of entry was achieved by using the 1A76 peptide I-Ig and the 3A64 peptide II-Ig preparations as well as MAb AP33. In contrast, 1A76 peptide II-Ig, 3A64 peptide I-Ig, and MAb 2/69a did not neutralize JFH-1 HCVpp. This finding suggests that although the antibodies isolated from patients recognize the same peptide sequence, they recognize different epitopes, resulting in different neutralizing phenotypes.
Fig 5.

Human and murine antibodies that target regions of E2 encompassing aa 412 to 423 and aa 434 to 446 neutralize HCVpp entry but differ in their neutralization breadths. Dilutions of 1A76 peptide I-Ig (■), 3A64 peptide I-Ig (●), 1A76 peptide II-Ig (□), 3A64 peptide II-Ig (○), MAb AP33 (▲), or MAb 2/69a (△) were used to neutralize the infectivity of H77c HCVpp (A) or JFH-1 HCVpp (B). Two different negative controls were used for the neutralization assays (♢): a normal human serum sample mock purified using the magnetic bead process was used as a negative control in the Ig neutralization assays, whereas HIV-1-specific monoclonal antibody 2F5 was used as a negative control in the MAb neutralization assays. Although neutralization assays were performed at the same time, for clarity, they are plotted on two panels corresponding to each peptide Ig and a third panel for the monoclonal antibodies. The same negative-control NHS data set is included in both Ig panels. The mean infectivities observed at the highest concentrations of each test antibody and the negative-control antibody were compared by using a t test. P values are indicated as follows: ns, P > 0.05; *, P < 0.01; ***, P < 0.001; ****, P < 0.0001. The mean luminescences observed for the uninhibited H77c HCVpp and JFH-1 HCVpp were 3,830 relative light units (RLU) and 3,819 RLU, and those for the no-env controls were 165 RLU and 138 RLU, respectively.
Peptide I-Ig and peptide II-Ig potently neutralize H77c chimeric HCVcc infectivity but differ in their abilities to neutralize wild-type JFH-1 HCVcc.
To assess whether the Ig fractions were capable of neutralizing the entry of authentic HCVcc virions, neutralization assays were performed by using cell culture-grown JFH-1 virus and a JFH-1 chimera possessing the structural proteins derived from clone H77c (Fig. 6). H77/JFH-1 was neutralized by peptide I-Ig and peptide II-Ig isolated from patients 1A76 and 3A64 as well as by MAb AP33 and MAb 2/69a. Again, the neutralization of wild-type JFH-1 HCVcc was more variable (Fig. 6B). Dose-dependent neutralization, which differed in potency, was observed for 1A76 peptide I-Ig, 3A64 peptide I-Ig, 3A64 peptide II-Ig, and MAb AP33. In contrast, both 1A76 peptide II-Ig and MAb 2/69a failed to neutralize JFH-1 HCVcc at the antibody concentrations tested.
Fig 6.

Human and murine antibodies that target regions of E2 encompassing aa 412 to 423 and aa 434 to 446 neutralize HCVcc infection. Dilutions of 1A76 peptide I-Ig (■), 3A64 peptide I-Ig (●), 1A76 peptide II-Ig (□), 3A64 peptide II-Ig (○), MAb AP33 (▲), or MAb 2/69a (△) were used to neutralize the infectivity of H77c HCVcc (A) or JFH-1 HCVcc (B). As for the HCVpp assays, two different negative controls were used in these assays (♢): a normal human serum sample mock purified using the magnetic bead process was used as a negative control in the Ig neutralization assays, whereas HIV-1-specific monoclonal antibody 2F5 was used as a negative control in the MAb neutralization assays. Although neutralization assays were performed at the same time, for clarity, they are plotted on two panels corresponding to each peptide Ig and a third panel for the monoclonal antibodies. The same negative-control NHS data set is included in both Ig panels. The mean infectivities observed at the highest concentrations of each test antibody and the negative-control antibody were compared by using a t test. P values are indicated as follows: ns, P > 0.05; *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
Antibodies that target regions of E2 encompassing aa 412 to 423 and 434 to 446 show additive neutralization.
Having shown that antibodies that target the region of E2 encompassing aa 434 to 446 bind E2 independently of antibodies that target the region encompassing aa 412 to 423 and that both groups have a neutralizing phenotype, we assessed whether or not antibodies to these distinct regions could mediate additive neutralization. To assess this, increasing doses of peptide II-Ig were mixed with autologous peptide I-Ig at or near its estimated IC50 and then used to neutralize H77c HCVpp (Fig. 7A) or JFH-1/H77c HCVcc (Fig. 7B). Dose-dependent additive neutralization was observed for antibodies derived from both patients 1A76 and 3A64, with maximal combined neutralization reaching 93%. Combinations of MAbs AP33 and 2/69a resulted in the same additive neutralization, demonstrating that both human and murine antibodies share this property.
Fig 7.

Peptide I-Ig and peptide II-Ig display additive neutralization of HCVpp and HCVcc infection. Increasing concentrations of 1A76 peptide II-Ig (□), 3A64 peptide II-Ig (○), or MAb 2/69a (△) were supplemented into neutralization reaction mixtures using fixed concentrations (approximate IC50s) of 1A76 peptide I-Ig, 3A64 peptide I-Ig, or MAb AP33 and either HCVpp supplemented with H77c E1E2 (A) or an HCVcc chimera possessing the structural proteins of H77c (B). A fixed concentration of HIV-1-specific antibody 2F5 (▾) was used as a control antibody. The residual infectivity, expressed as a percentage of that observed in the absence of antibody, is plotted against the concentration of the supplemented antibody. The neutralization obtained with peptide I-Ig or MAb AP33 in the absence of the supplementary antibody is indicated by the dashed lines. Dose-dependent additive neutralization was consistently observed for all patient-isolated antibody preparations and MAb combinations. The mean infectivities observed at the highest concentrations of each test antibody and the negative-control antibody were compared by using a t test. P values are indicated as follows: ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Analysis of circulating variants in patients harboring antibodies directed to regions encompassing aa 412 to 423 and aa 434 to 446.
To determine if the presence of antibodies to regions encompassing aa 412 to 423 and aa 434 to 446 resulted in the selection of variants, we attempted to recover and sequence the corresponding E1E2 coding regions; only the serum sample obtained from patient 3A64 was successfully amplified (Fig. 8A). The region encompassing aa 412 to 423 of the majority sequence was identical to that of H77c, whereas the regions corresponding to peptide II harbored W437F, L438I, Y442S, Q443Y, and H444Y substitutions. All substitutions, except Y442S, are commonly found in genotype 3a sequences in the GenBank database (Fig. 1).
Fig 8.

Epitope mapping by enrichment of random peptide phage display libraries reveals the presence of distinct yet overlapping epitopes within regions of E2 encompassing aa 412 to 423 and aa 434 to 446. Affinity-purified Ig from patients 3A64 (A) and 1A76 (B) and MAb 2/69a (C) were used to enrich phages from a 12-mer random peptide library. Following three to four rounds of biopanning, individual phage clones were isolated, amplified, and tested for reactivity to the selecting antibody, and the amino acid sequence of the peptide insert was determined by DNA sequencing. The resulting sequences were aligned to the corresponding region of the H77c E2 amino acid sequence. The phage clone identification is presented, and individual residues aligning to the H77c sequence are shaded. Numbers in parentheses indicate the number of times each peptide sequence was recovered. The reactivity of each phage to the selecting antibody in a phage capture enzyme immunoassay is shown alongside each peptide sequence and is shown as the relative optical density compared to that of capture by an antibody preparation from an HCV-negative donor (NHS-Ig). +++, >4-fold greater than the optical density obtained for the negative control NHS-IgG; ++ >3-fold greater; +, >1.5-fold greater; −, <1.5-fold greater. In addition, the consensuses of the RNA sequences of the viruses infecting patient 3A64 were deduced by reverse transcription-PCR (RT-PCR) and sequencing (A). This sequence is presented below the alignment for the peptides selected by antibodies isolated from patient 3A64.
Peptide mapping of epitopes recognized by peptide I-Ig, peptide II-Ig, and MAb 2/69a reveals the presence of multiple epitopes.
We have previously enriched random peptide display libraries to map the residues critical for MAb AP33 (56). Therefore, we adopted the same approach to determine residues involved in the binding of peptide I-Ig, peptide II-Ig, and MAb 2/69a. Following 3 to 4 rounds of biopanning, the binding of individual phage clones to the target Ig/MAb was assessed, and their peptide inserts were determined by DNA sequencing. Six of the seven phage clones isolated using 3A64 peptide I-Ig were strongly reactive to the selecting peptide I-Ig. Clone 6 had a lower level of reactivity, although this level of reactivity was still more than twice that observed for the negative control normal human serum (NHS) IgG (Fig. 8A). An alignment of the deduced amino acid sequences of the peptide inserts identified key residues that appeared to be critical for binding (Fig. 8A). One peptide sequence was identified in 7 independent clones, and residues that aligned to those present in the corresponding H77c sequence were L413, I414, N415, T416, G418, S419, W420, and I422. Analysis of the all of the peptide sequences showed that the most frequently observed amino acid residues aligned to the sequence 416TxGxW420.
A similar analysis using 1A76 peptide I-Ig showed that three of four clones isolated were well recognized by the target Ig fraction in the phage binding immunoassay (Fig. 8B). The most prevalent phage clone sequence was present in 50% of the clones selected. This sequence possessed residues that mapped to positions L413, N415, and W420. The remaining clones were typified by the presence of a tryptophan, corresponding to residue W420, and either L413 or I414. The clone that was nonreactive in the phage immunoassay (clone 18) contained a tryptophan but did not contain any other amino acids that aligned to the E2 sequence.
Similar analyses of the antibodies that target the E2 region corresponding to peptide II were also able to identify key residues involved in binding. Seven clones enriched by 3A64 peptide II-Ig were selected and analyzed, and of these clones, two showed specific reactivity to the selecting antibody in the phage immunoassay (Fig. 8A). Sequence analysis of these clones revealed that the most frequent canonical binding sequence was GWLxG, corresponding to E2 residues G436, W437, L438, and G440. All phage-displayed peptides enriched by peptide II-Ig isolated from sera of patient 1A76 also demonstrated specific binding in the phage enzyme immunoassay, and analyses of the peptide sequence revealed that these antibodies recognized an overlapping yet distinct epitope consisting of G436, W437, and L441. Finally, the residues involved in recognition by MAb 2/69a were defined (Fig. 8C). Key residues were identified as G440, Y443, and K446. Together, the peptide mapping data demonstrate that antibodies directed to each of these peptides recognize overlapping yet distinct epitopes.
DISCUSSION
Studies of both sera and human monoclonal antibodies have shown that cross-neutralizing antibodies arise during chronic HCV infection (4, 19, 28, 31, 34, 37, 39, 41, 44, 45, 57). Several mechanisms have been proposed to explain how HCV persists. One mechanism proposed recently was the induction of nonneutralizing antibodies that target epitopes within the E2 region encompassing amino acid residues 434 to 446, which reportedly interfere with antibodies that target part of the E2-CD81 binding site. In particular, these antibodies targeted broadly neutralizing epitopes located in a highly conserved region of E2 corresponding to amino acids 412 to 423 (65, 66). This finding was contrary to previous reports that murine monoclonal antibodies to the region encompassing aa 434 to 446 neutralized E2-CD81 binding (9) and autologous H77c HCVpp infectivity (27). In order to clarify this, we studied the prevalences and neutralizing phenotypes of antibodies that target these two regions from sera from two chronically infected individuals. Crucially, our data, derived by using affinity-purified human Ig fractions, showed that human antibodies that recognize both of these regions neutralized HCVpp and HCVcc. We did not observe any interference between these two groups of antibodies. The depletion of antibodies that target the region encompassing aa 412 to 423 and aa 434 to 446 with saturating amounts of peptide had a minimal effect on the overall neutralizing titer (data not shown), confirming our previously reported observation that antibody specificities to these epitopes contribute only a small part of the total polyclonal neutralizing response in chronically infected subjects (55).
Analysis of sera obtained from more than 500 individuals infected with a broad range of HCV genotypes revealed that the overall prevalence of peptide I-targeting antibodies was less than 3%, in accordance with our previously reported estimate (55). In contrast, Zhang et al. reported previously that two of nine chronically infected patients harbored antibodies specific for a slightly extended (aa 412 to 425) peptide I (66). It is possible that extending the peptide increased the detection of antibodies with different specificities; however, this is not supported by previously reported mapping experiments, which suggested that the additional residues had no impact on antibody recognition (65, 66). Similar to the findings reported previously by Zhang et al., we confirmed that antibodies that target the region encompassing aa 412 to 423 coexisted with those that target the region encompassing aa 434 to 446.
Random peptide phage display epitope mapping experiments showed that both regions contained overlapping, yet distinct, epitopes. Phage binding enzyme immunoassays enabled the identification of specific binding mimotopes, thereby increasing our confidence that the canonical sequences identified key residues involved in binding. This is important, as the biopanning process can also enrich for irrelevant peptides (40). Some of our selected phage-displayed peptides did not demonstrate specific binding to their selecting antibody despite possessing sequence motifs that would implicate specific enrichment during selection. It is possible that the affinity of the interaction between these phage-displayed peptides and the target antibody falls below the sensitivity of our assays. Further experiments are under way to investigate this possibility.
Analysis of the peptide sequences of phages enriched using peptide I-Ig from both patients 1A76 and 3A64 revealed a consistent preservation of tryptophan, corresponding to W420, which we had previously shown to be essential for recognition by murine MAbs AP33 and 3/11 (56) and human MAb e137 (47). This was also implicated in the binding of HCV immunoglobulin (HCVIG) pools to an equivalent peptide antigen (66). While some critical residues were common, phage mapping experiments showed that the exact epitopes targeted by the patients' antibodies differed. Global conformation can also affect the exposure of epitopes within this region (48). Both of these factors are likely to contribute to the differences in the neutralization breadths and potencies of antibodies that target this conserved region of E2. In contrast, the region encompassing aa 434 to 446 contains both variable and conserved residues, both of which were targeted by peptide II-Ig and MAb 2/69a. This variability impacts the neutralization breadths and potencies observed for the various antibodies specific for the region encompassing aa 434 to 446. Finally, we have recently shown that different E1E2s exhibit inherent differences in the overall sensitivity to antibody neutralization, which is independent of genotype. In particular, we have shown that H77c E1E2 is one of the most easily neutralized (57).
It is possible that different epitope specificities might explain the contrast in neutralizing antibody phenotypes identified here compared to the interfering phenotype reported previously by Zhang et al. However, peptide mapping of epitopes recognized by peptide II-Ig fractions and MAb 2/69a revealed that they collectively targeted epitopes spanning almost the entire region encompassing aa 434 to 446. Furthermore, in their mapping experiments, Zhang et al. showed that the majority of the reactivity of the interfering antibodies in HCVIG was targeted to residues 440 to 445, which is encompassed within the proposed 2/69a epitope. Together, these findings would argue against there being both neutralizing and interfering antibodies against this region.
Antibody-based immune selection is known to play a role in the evolution of intrahost HCV populations (12, 17, 36). As the region of E2 under investigation tolerates genetic diversity at specific amino acid positions, we examined whether the presence of these antibodies resulted in the selection of novel variants in these patients. PCR amplification of the full-length E1E2 coding region was possible only for sera of patient 3A64. Interestingly, the E1E2 coding sequence of the virus present in this patient contained several mutations compared to the H77c sequence, some of which were predicted antibody contact residues. It was demonstrated recently that amino acid substitutions at positions 443 and 444 may be involved in escape from antibody neutralization (17). The phenotype of this variant virus is currently being assessed.
We have previously shown that residues within the region encompassing aa 412 to 423 are involved in CD81 binding (46). Similarly, mutational analysis of a fragment of the region of aa 434 to 446, encompassing the sequence 436GWLAGLFY443, implicated a role for this region in CD81 binding (15). Therefore, it is likely that antibodies that target both of these regions neutralize HCV entry by directly blocking the interaction between E2 and the cellular receptor CD81. Importantly, the demonstration that some peptide II-Ig was capable of cross-neutralization and could be used in combination with antibodies that target the region encompassing aa 412 to 423 could inform the development of future antibody therapies. In conclusion, this study has shown that human antibodies that target the region encompassing aa 434 to 446, which were previously thought to reduce the neutralizing potency of broadly neutralizing antibodies that target epitopes located within the region encompassing aa 412 to 423, are not inhibitory but are instead capable of neutralizing HCVpp and HCVcc entry. It is difficult to explain this discrepancy, but our findings are consistent with previously reported data demonstrating the inhibition of CD81 binding and with the limited HCVpp neutralization data obtained for murine MAbs that target this region.
ACKNOWLEDGMENTS
We thank François-Loïc Cosset for the provision of the HCVpp packaging and reporter plasmids, Jens Bukh for the H77c E1E2 plasmid, Takaji Wakita for JFH-1, and Charles Rice for Huh7.5 cells, H77c-chimeric JFH1cc, and MAb 9E10. Arvind Patel (via Genentech Inc.) provided monoclonal antibody AP33.
This work was supported by the Medical Research Council (grant G0801169), the European Union (grant MRTN-CT-2006-035599), and the Nottingham Digestive Diseases Centre Biomedical Research Unit.
Footnotes
Published ahead of print 14 December 2011
REFERENCES
- 1. Alter MJ, et al. 1992. The natural history of community-acquired hepatitis C in the United States. The Sentinel Counties Chronic Non-A, Non-B Hepatitis Study Team. N. Engl. J. Med. 327:1899–1905 [DOI] [PubMed] [Google Scholar]
- 2. Anonymous 1999. Global surveillance and control of hepatitis C. J. Viral Hepat. 6:35–47 [PubMed] [Google Scholar]
- 3. Barth H, et al. 2005. Scavenger receptor class B type I and hepatitis C virus infection of primary Tupaia hepatocytes. J. Virol. 79:5774–5785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bartosch B, et al. 2003. In vitro assay for neutralizing antibody to hepatitis C virus: evidence for broadly conserved neutralization epitopes. Proc. Natl. Acad. Sci. U. S. A. 100:14199–14204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197:633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bartosch B, et al. 2003. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 278:41624–41630 [DOI] [PubMed] [Google Scholar]
- 7. Brimacombe CL, et al. 2011. Neutralizing antibody-resistant hepatitis C virus cell-to-cell transmission. J. Virol. 85:596–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bukh J, Miller RH, Purcell RH. 1995. Genetic heterogeneity of hepatitis C virus: quasispecies and genotypes. Semin. Liver Dis. 15:41–63 [DOI] [PubMed] [Google Scholar]
- 9. Clayton RF, et al. 2002. Analysis of antigenicity and topology of E2 glycoprotein present on recombinant hepatitis C virus-like particles. J. Virol. 76:7672–7682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davis GL, et al. 2005. A randomized, open-label study to evaluate the safety and pharmacokinetics of human hepatitis C immune globulin (Civacir) in liver transplant recipients. Liver Transpl. 11:941–949 [DOI] [PubMed] [Google Scholar]
- 11. Dietzschold B, et al. 1990. Structural and immunological characterization of a linear virus-neutralizing epitope of the rabies virus glycoprotein and its possible use in a synthetic vaccine. J. Virol. 64:3804–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dowd KA, Netski DM, Wang X-H, Cox AL, Ray SC. 2009. Selection pressure from neutralizing antibodies drives sequence evolution during acute infection with hepatitis C virus. Gastroenterology 136:2377–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dreux M, et al. 2007. The exchangeable apolipoprotein ApoC-I promotes membrane fusion of hepatitis C virus. J. Biol. Chem. 282:32357–32369 [DOI] [PubMed] [Google Scholar]
- 14. Dreux M, et al. 2006. High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J. Biol. Chem. 281:18285–18295 [DOI] [PubMed] [Google Scholar]
- 15. Drummer HE, Boo I, Maerz AL, Poumbourios P. 2006. A conserved Gly436-Trp-Leu-Ala-Gly-Leu-Phe-Tyr motif in hepatitis C virus glycoprotein E2 is a determinant of CD81 binding and viral entry. J. Virol. 80:7844–7853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Evans MJ, et al. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805 [DOI] [PubMed] [Google Scholar]
- 17. Fafi-Kremer S, et al. 2010. Viral entry and escape from antibody-mediated neutralization influence hepatitis C virus reinfection in liver transplantation. J. Exp. Med. 207:2019–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Falkowska E, Kajumo F, Garcia E, Reinus J, Dragic T. 2007. Hepatitis C virus envelope glycoprotein E2 glycans modulate entry, CD81 binding, and neutralization. J. Virol. 81:8072–8079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Farci P, et al. 1994. Prevention of hepatitis C virus infection in chimpanzees after antibody-mediated in vitro neutralization. Proc. Natl. Acad. Sci. U. S. A. 91:7792–7796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feray C, et al. 1998. Incidence of hepatitis C in patients receiving different preparations of hepatitis B immunoglobulins after liver transplantation. Ann. Intern. Med. 128:810–816 [DOI] [PubMed] [Google Scholar]
- 21. Flint M, et al. 1999. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol. 73:6235–6244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Folgori A, et al. 2006. A T-cell HCV vaccine eliciting effective immunity against heterologous virus challenge in chimpanzees. Nat. Med. 12:190–197 [DOI] [PubMed] [Google Scholar]
- 23. Grove J, et al. 2008. Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J. Virol. 82:12020–12029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gurusamy KS, Tsochatzis E, Xirouchakis E, Burroughs AK, Davidson BR. 2010. Antiviral therapy for recurrent liver graft infection with hepatitis C virus. Cochrane Database Syst. Rev. 2010(1):CD006803 doi:10.1002/14651858.CD006803.pub3 [DOI] [PubMed] [Google Scholar]
- 25. Helle F, Dubuisson J. 2008. Hepatitis C virus entry into host cells. Cell. Mol. Life Sci. 65:100–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Helle F, et al. 2007. The neutralizing activity of anti-hepatitis C virus antibodies is modulated by specific glycans on the E2 envelope protein. J. Virol. 81:8101–8111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hsu M, et al. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U. S. A. 100:7271–7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Johansson DX, et al. 2007. Human combinatorial libraries yield rare antibodies that broadly neutralize hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 104:16269–16274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Judith MG, et al. 2009. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49:364–377 [DOI] [PubMed] [Google Scholar]
- 30. Kato T, et al. 2001. Sequence analysis of hepatitis C virus isolated from a fulminant hepatitis patient. J. Med. Virol. 64:334–339 [DOI] [PubMed] [Google Scholar]
- 31. Keck Z-Y, et al. 2008. Definition of a conserved immunodominant domain on hepatitis C virus E2 glycoprotein by neutralizing human monoclonal antibodies. J. Virol. 82:6061–6066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuo A, Terrault NA. 2006. Management of hepatitis C in liver transplant recipients. Am. J. Transplant. 6:449–458 [DOI] [PubMed] [Google Scholar]
- 33. Lavillette D, et al. 2005. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology 41:265–274 [DOI] [PubMed] [Google Scholar]
- 34. Law M, et al. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14:25–27 [DOI] [PubMed] [Google Scholar]
- 35. Lindenbach BD, et al. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626 [DOI] [PubMed] [Google Scholar]
- 36. Liu L, et al. 2010. Acceleration of hepatitis C virus envelope evolution in humans is consistent with progressive humoral immune selection during the transition from acute to chronic infection. J. Virol. 84:5067–5077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Logvinoff C, et al. 2004. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 101:10149–10154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Makris M, et al. 1996. The natural history of chronic hepatitis C in haemophiliacs. Br. J. Haematol. 94:746–752 [DOI] [PubMed] [Google Scholar]
- 39. Mancini N, et al. 2009. Hepatitis C virus (HCV) infection may elicit neutralizing antibodies targeting epitopes conserved in all viral genotypes. PLoS One 4:e8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Menendez A, Scott JK. 2005. The nature of target-unrelated peptides recovered in the screening of phage-displayed random peptide libraries with antibodies. Anal. Biochem. 336:145–157 [DOI] [PubMed] [Google Scholar]
- 41. Meunier J-C, et al. 2005. Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc. Natl. Acad. Sci. U. S. A. 102:4560–4565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. 1982. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 42:3858–3863 [PubMed] [Google Scholar]
- 43. Owsianka A, Clayton RF, Loomis-Price LD, McKeating JA, Patel AH. 2001. Functional analysis of hepatitis C virus E2 glycoproteins and virus-like particles reveals structural dissimilarities between different forms of E2. J. Gen. Virol. 82:1877–1883 [DOI] [PubMed] [Google Scholar]
- 44. Owsianka A, et al. 2005. Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glycoprotein. J. Virol. 79:11095–11104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Owsianka AM, et al. 2008. Broadly neutralizing human monoclonal antibodies to the hepatitis C virus E2 glycoprotein. J. Gen. Virol. 89:653–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Owsianka AM, et al. 2006. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J. Virol. 80:8695–8704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Perotti M, et al. 2008. Identification of a broadly cross-reacting and neutralizing human monoclonal antibody directed against the hepatitis C virus E2 protein. J. Virol. 82:1047–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pestka JM, et al. 2007. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc. Natl. Acad. Sci. U. S. A. 104:6025–6030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48a. Ploss A, et al. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27:493–497 [Google Scholar]
- 50. Samuel D, et al. 1993. Liver transplantation in European patients with the hepatitis B surface antigen. N. Engl. J. Med. 329:1842–1847 [DOI] [PubMed] [Google Scholar]
- 51. Schiano TD, et al. 2006. Monoclonal antibody HCV-Ab(XTL)68 in patients undergoing liver transplantation for HCV: results of a phase 2 randomized study. Liver Transpl. 12:1381–1389 [DOI] [PubMed] [Google Scholar]
- 52. Shimizu YK, et al. 1994. Neutralizing antibodies against hepatitis C virus and the emergence of neutralization escape mutant viruses. J. Virol. 68:1494–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Simmonds P, et al. 2005. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42:962–973 [DOI] [PubMed] [Google Scholar]
- 54. Tao W, et al. 2009. A single point mutation in E2 enhances hepatitis C virus infectivity and alters lipoprotein association of viral particles. Virology 395:67–76 [DOI] [PubMed] [Google Scholar]
- 55. Tarr AW, et al. 2007. Determination of the human antibody response to the epitope defined by the hepatitis C virus-neutralizing monoclonal antibody AP33. J. Gen. Virol. 88:2991–3001 [DOI] [PubMed] [Google Scholar]
- 56. Tarr AW, et al. 2006. Characterization of the hepatitis C virus E2 epitope defined by the broadly neutralizing monoclonal antibody AP33. Hepatology 43:592–601 [DOI] [PubMed] [Google Scholar]
- 57. Tarr AW, et al. 2011. Hepatitis C patient-derived glycoproteins exhibit marked differences in susceptibility to serum neutralizing antibodies: genetic subtype defines antigenic but not neutralization serotype. J. Virol. 85:4246–4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Todo S, et al. 1991. Orthotopic liver transplantation for patients with hepatitis B virus-related liver disease. Hepatology 13:619–626 [PMC free article] [PubMed] [Google Scholar]
- 60. Voisset C, et al. 2005. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J. Biol. Chem. 280:7793–7799 [DOI] [PubMed] [Google Scholar]
- 61. von Hahn T, et al. 2007. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology 132:667–678 [DOI] [PubMed] [Google Scholar]
- 62. Wakita T, et al. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Witteveldt J, et al. 2009. CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J. Gen. Virol. 90:48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang J, et al. 2004. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 78:1448–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang P, et al. 2007. Hepatitis C virus epitope-specific neutralizing antibodies in Igs prepared from human plasma. Proc. Natl. Acad. Sci. U. S. A. 104:8449–8454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang P, et al. 2009. Depletion of interfering antibodies in chronic hepatitis C patients and vaccinated chimpanzees reveals broad cross-genotype neutralizing activity. Proc. Natl. Acad. Sci. U. S. A. 106:7537–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zheng A, et al. 2007. Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J. Virol. 81:12465–12471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zibert A, Kraas W, Meisel H, Jung G, Roggendorf M. 1997. Epitope mapping of antibodies directed against hypervariable region 1 in acute self-limiting and chronic infections due to hepatitis C virus. J. Virol. 71:4123–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]