

Abstract
While work with viral fusion proteins has demonstrated that the transmembrane domain (TMD) can affect protein folding, stability, and membrane fusion promotion, the mechanism(s) remains poorly understood. TMDs could play a role in fusion promotion through direct TMD-TMD interactions, and we have recently shown that isolated TMDs from three paramyxovirus fusion (F) proteins interact as trimers using sedimentation equilibrium (SE) analysis (E. C. Smith, et al., submitted for publication). Immediately N-terminal to the TMD is heptad repeat B (HRB), which plays critical roles in fusion. Interestingly, addition of HRB decreased the stability of the trimeric TMD-TMD interactions. This result, combined with previous findings that HRB forms a trimeric coiled coil in the prefusion form of the whole protein though HRB peptides fail to stably associate in isolation, suggests that the trimeric TMD-TMD interactions work in concert with elements in the F ectodomain head to stabilize a weak HRB interaction. Thus, changes in TMD-TMD interactions could be important in regulating F triggering and refolding. Alanine insertions between the TMD and HRB demonstrated that spacing between these two regions is important for protein stability while not affecting TMD-TMD interactions. Additional mutagenesis of the C-terminal end of the TMD suggests that β-branched residues within the TMD play a role in membrane fusion, potentially through modulation of TMD-TMD interactions. Our results support a model whereby the C-terminal end of the Hendra virus F TMD is an important regulator of TMD-TMD interactions and show that these interactions help hold HRB in place prior to the triggering of membrane fusion.
INTRODUCTION
Membrane fusion is a complex biological phenomenon requiring the juxtaposition and deformation of two membranes prior to their eventual merger into one continuous bilayer. Despite the various types of membrane fusion events, all require the presence of one or more specialized proteins to catalyze this energy-intensive process. Enveloped viruses generally express one or more membrane glycoproteins which are critical in promoting virus-cell membrane fusion (26, 65, 77). Paramyxoviruses, including measles, respiratory syncytial virus (RSV), and the zoonotic Hendra virus, typically express two surface glycoproteins: an attachment protein (G, HN, or H), which is responsible for receptor binding and cellular attachment, and a fusion (F) protein, which is responsible for driving fusion between the viral and cellular membranes (32). Virus-cell fusion ultimately culminates in the deposition of the viral genome into the host cell and thus constitutes a pivotal step in the virus life cycle.
Paramyxovirus F proteins are type I integral membrane proteins which are cotranslationally folded as trimers with extensive monomer-monomer contacts (80). All F proteins are initially synthesized as inactive (F0) precursors that must be proteolytically processed by intracellular (22, 55, 57, 58) or extracellular (4, 5) proteases to form the disulfide-linked fusogenically active heterodimer (F1+F2). Hendra virus F cleavage is unique among paramyxoviruses in that F is initially surface expressed as an uncleaved, fusion-inactive (F0) form, endocytosed, cleaved by the endosomal/lysosomal protease cathepsin L, and subsequently retrafficked to the cell surface (47, 58, 59). This fusogenically active form resides on the virus or cell surface in a metastable state which must then be triggered to undergo conformational changes intimately linked to membrane fusion. Like other class I viral fusion proteins, paramyxovirus F proteins share common structural features (Fig. 1A) such as a single-pass transmembrane domain (TMD), two heptad repeat regions (heptad repeat A [HRA] and HRB), and a hydrophobic fusion peptide (FP), all suggestive of a conserved mechanism of membrane fusion (70, 77). Crystal structures of both the prefusion (80) and postfusion (14, 79) forms of paramyxovirus F proteins exist and, along with numerous studies, these provide a structural model for how the conserved domains drive membrane fusion (17). Once triggered, the hydrophobic FP is inserted into the target cell membrane, causing extension and formation of the HRA coiled coil (2, 11, 77). Subsequent unfolding and refolding of HRB around the HRA coiled coil results in the formation of an extremely thermostable six-helix bundle which is critical for membrane fusion (3, 8, 10, 13, 41, 76).
Fig 1.

Schematic of Hendra virus F and centrifugation constructs. (A) A schematic of the cleaved F1+F2 form of Hendra virus F. FP, fusion peptide; HRA and HRB, heptad repeat regions; TMD, transmembrane domain; CT, cytoplasmic/intraviral tail. (B) Diagram of SN-TMD and SN-HRB-TMD centrifugation constructs.
Despite existing structural data, the potential roles of the TMD in protein folding, prefusion stability, and membrane fusion are poorly understood as this domain is not present in any of the available crystal structures. Studies with class I and III fusion proteins, including influenza virus hemagglutinin (HA) (1, 28, 42, 52), HIV gp120 (50, 56, 63, 67, 68, 75, 78), and vesicular stomatitis virus (VSV) G protein (16, 53), have demonstrated that the TMD is critical for fusion promotion beyond a role as a membrane anchor. Additionally, the importance of both TMDs for a flavivirus class II fusion protein has recently been reported (21). Replacing the TMD of influenza virus HA with a glycophosphatidylinositol (GPI) anchor abolished aqueous content mixing (28, 42), while a GPI-anchored VSV G protein was completely fusion incompetent (53), suggesting that a proteinaceous TMD is essential for fusion protein-promoted fusion. However, little is known about the sequence dependency of TMDs required for fusion promotion. Substitution of viral fusion protein TMDs with TMDs from diverse viral or nonviral proteins can often result in a fusion-competent protein (30, 50, 53, 78) though replacement of the TMD of Newcastle disease virus (NDV) F with the TMD of either Sendai virus (SeV) F, measles virus (MV) F, or VSV G resulted in fusion-defective F proteins (24). Additionally, the presence or absence of certain TMD residues can act to modulate fusion efficiency. Examples include central glycine motifs (i.e., GXXXG, where X is any amino acid) (16, 48, 49) and a leucine zipper-like (or heptad repeat-like) arrangement of leucine/isoleucine residues (25, 29) though the absence of these motifs does not necessarily preclude fusion competency.
GXXXG and leucine zipper motifs have been implicated in promotion of TMD-TMD interactions, and we have recently demonstrated that these interactions are important in the maintenance of prefusion stability and membrane fusion of the Hendra virus F protein (E. C. Smith et al., submitted for publication). Sedimentation equilibrium (SE) analytical ultracentrifugation analysis of isolated TMDs from three paramyxoviruses fused to staphylococcal nuclease (SN) found that the TMDs interact in a monomer-trimer equilibrium and that mutations which significantly increase or decrease the strength of these interactions have dramatic affects on F protein stability and fusion promotion (Smith et al., submitted). This study highlighted the importance of TMD-TMD interactions in F protein folding, but the sequences that drive these interactions and how they aid in fusion promotion remain unknown. During fusion, the stability of these trimeric TMD-TMD interactions could be modulated by nearby regions within F, such as HRB, which is N-terminal to the TMD. Data presented here demonstrate that the addition of HRB to the isolated Hendra virus F TMD destabilizes the trimeric TMD-TMD interactions, suggesting that these interactions, along with interactions in the head domain of F, likely are important in stabilizing the HRB coiled coil prior to fusion as both of these regions could act to clamp HRB together. Additionally, we generated mutant Hendra virus F proteins harboring alanine insertions at either the N or C terminus of the TMD and examined their effect on protein stability and fusion promotion. Though increasing the length of the TMD moderately affected protein stability overall, N-terminal insertions resulted in a dramatic reduction of the surface-expressed prefusion cleaved F1 form, likely by destabilizing the F1 form, while C-terminal insertions had greater effects on membrane fusion. SE analysis of isolated SN-TMD constructs corresponding to the alanine insertion mutants demonstrated that C-terminal insertions significantly disrupted TMD-TMD interactions while N-terminal insertions had no effect. Closer examination of the C-terminal end of the Hendra virus F TMD revealed that mutation of C-terminal residues with β-branched side chains leads to hypofusogenic proteins, suggesting that TMD β-branched residues play a role in membrane fusion. Our results support a model in which TMD-TMD interactions are critical for HRB stability and demonstrate that the C-terminal end of the Hendra virus F TMD acts as an important regulator of TMD-TMD interactions. Furthermore, our data show that the absolute spacing between the TMD and HRB is essential for maintenance of prefusion stability and implicate β-branched residues within the TMD in membrane fusion promotion.
MATERIALS AND METHODS
Cell lines and culture.
All cell lines were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco/Invitrogen, Carlsbad, CA) supplemented with 1% penicillin-streptomycin (P-S) and 10% fetal bovine serum (FBS). BSR cells (generously provided by Karl-Klaus Conzelman, Pettenkofer Institut) (7) were selected for expression of the T7 polymerase every third passage by the addition of G418 sulfate (Gibco/Invitrogen, Carlsbad, CA) to the growth medium.
Plasmids and site-directed mutagenesis.
The Hendra virus F and G genes were provided by Lin-Fa Wang (Australian Animal Health Laboratory) and were transiently expressed using the pCAGGS vector (51). Mutant Hendra virus F constructs in pGEM-4z were created using a QuikChange site-directed mutagenesis kit (Agilent Technologies/Stratagene, Santa Clara, CA), digested with SalI, and subcloned into XhoI-digested pCAGGS. Staphylococcal nuclease fused to the transmembrane domain (TMD) of glycophorin A (GPA) in the pET-11a expression vector was generously provided by Karen Fleming (The Johns Hopkins University). Analytical ultracentrifugation constructs containing either the wild-type or mutant Hendra virus F TMD and HRB were cloned into pET-11a (replacing the TMD of GPA) using XmaI and BamHI sites at the 5′ and 3′ ends, respectively. Constructs containing only the TMD were cloned into pET-11a using XmaI and XhoI sites at the 5′ and 3′ ends, respectively.
Recombinant protein expression and purification.
Wild-type Hendra virus F TMD centrifugation constructs corresponding to the sequence VNPSLISM*LSMIILYVLSIAALCIGLITFISF**VIVE KK (where * and ** denote positions for insertion of N- and C-terminal alanine residues, respectively) were expressed as C-terminal fusions with staphylococcal nuclease. The sequences for the construct with two alanine insertions in the C terminus (C2), C3, the construct with one alanine insertion in the N terminus (N1), and N2 were identical except for the alanine insertions indicated in Fig. 3A. For the TMD-HRB constructs, HRB (VYTDKVDISSQISSMNQSLQQSKDYIKEAQKILDT) was inserted between SN and the TMD, resulting in an SN-HRB-TMD fusion construct, where HRB and the TMD were in phase as in the wild-type full-length protein. All constructs were transformed into Rosetta-Gami cells (EMD Chemicals, Gibbstown, NJ) and grown at 37°C in 2× yeast extract-tryptone (YT) medium under the selection of 0.015 mg/ml kanamycin, 0.0125 mg/ml tetracycline, 0.05 mg/ml streptomycin, 0.034 mg/ml chloramphenicol, and 0.1 mg/ml ampicillin. The cultures were grown to an optical density at 600 nm (OD600) of 0.6 to 0.8, induced for protein expression by the addition of 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG; Sigma, St. Louis, MO) for 4 h, and then pelleted at 6,000 × g for 10 min. Cell pellets were resuspend in a 1:20 culture volume of lysis buffer (50 mM HEPES, 2 mM EDTA, and 1 mM phenylmethylsulfonyl fluoride [PMSF] added just prior to use, at pH 8.0), subjected to three freeze-thaw cycles, and incubated with 0.1 mg/ml hen egg white lysozyme for 30 min on ice. The cell lysate was then sonicated (three 20-s pulses), 5 mM CaCl2 was added, and the solution was incubated for 30 min on ice. Insoluble protein was pelleted at 12,000 × g for 10 min and single-salt extracted with 25 ml of lysis buffer containing 1 mM PMSF and 1 M ammonium acetate (NH4OAc) (73). The lysate was then subjected to centrifugation twice more at 12,000 × g for 10 min, with the pellet first resuspended in lysis buffer containing 1 mM PMSF and 2% (vol/vol) polyethylene glycol 400 dodecyl ether (Thesit; Sigma, St. Louis, MO) and then in lysis buffer containing 1 mM PMSF and 1 M NH4OAc with 2% (vol/vol) Thesit. The solution was then clarified by centrifugation at 15,000 × g for 30 min and dialyzed (molecular-weight cutoff [MWCO] of 6,000 to 8,000) overnight at 4°C against 0.1 M NH4OAc and 0.2% (vol/vol) Thesit in lysis buffer. Recombinant protein was then purified by fast protein liquid chromatography (FPLC) using cation exchange chromatography and a 1-ml HiTrap SP FF column (GE Healthcare, Piscataway, NJ) (31), followed by a washing step with lysis buffer containing 0.1 M NH4OAc and 0.2% (vol/vol) Thesit (wash buffer I) and elution with wash buffer I containing 1 M NaCl. Fractions containing recombinant protein were then combined, diluted to 200 mM NaCl, and FPLC purified again, followed by a washing step with an aqueous solution of 200 mM NaCl, 20 mM Na2HPO4/NaH2PO4 at pH 7.0, 29% D2O, and the Zwittergent detergent 3-(N,N-dimethylmyristyl-ammonio) propane sulfonate (C14SB; Sigma/Fluka, St. Louis, MO) (wash buffer II) (9). Recombinant protein was eluted using wash buffer II containing 1 M NaCl and dialyzed using Slide-A-Lyzer Mini Dialysis Units (10,000 MWCO; Pierce, Rockford, IL) against wash buffer II overnight at 4°C. If needed, purified protein was concentrated using Amicon Ultra centrifugal filter units (100,000 MWCO; Millipore, Billerica, MA).
Fig 3.

N- and C-terminal TMD insertions differentially affect protein expression and fusion. (A) Sequence of the predicted Hendra virus F TMD and alanine insertion mutants, where L494 is the predicted start of the TMD. (B) Biotinylation of cell surface-expressed wild-type (WT) and mutant Hendra virus F proteins labeled with [trans-35S]cysteine-methionine. Surface proteins were biotinylated and immunoprecipitated, and the total and surface protein populations were separated by streptavidin pulldown, followed by 15% SDS-PAGE and visualization using autoradiography. A representative gel of three independent experiments is shown. (C) Quantification of cleavage time points for wild-type or mutant Hendra virus F proteins. The proteins were radiolabeled with [trans-35S]cysteine-methionine, immunoprecipitated, analyzed by 15% SDS-PAGE, and visualized using autoradiography. Error bars represent the mean ± the standard error of the mean for three independent experiments. (D) Quantification of cell surface expression levels of F1 and membrane fusion levels of wild-type and mutant F proteins. Vero cells were transfected with wild-type or mutant Hendra virus F and wild-type Hendra virus G along with a luciferase construct, overlaid with BSR cells, and assayed for luciferase activity. Error bars represent the mean ± the standard error of the mean for three independent experiments.
Analytical ultracentrifugation and data analysis.
Analytical ultracentrifugation sedimentation equilibrium experiments were performed at three different protein concentrations and two rotor speeds (20,000 and 30,000 rpm) using a Beckman XL-A analytical ultracentrifuge equipped with an An-60 Ti rotor. Data were collected at 25°C, and the radial absorbance was monitored at 280 nm. Attainment of equilibrium was determined by comparing radial scans at different times, and once radial scans taken 6 h apart were indistinguishable, the system was considered to be at equilibrium. Typically, equilibration times of ≥24 h were sufficient to reach equilibrium. To negate the weight contribution of TMD-associated detergent, density matching of the solution to the density of C14SB was performed as previously described using D2O (9). For data analysis, the partial specific volumes of each recombinant protein were estimated using SEDNTERP (D. B. Hayes, T. Laue, and J. Philo, Boston Biomedical Research Institute, Watertown, MA [http://www.rasmb.bbri.org]). Data analysis was performed using KaleidaGraph (Synergy Software, Reading, PA) software and the following equation:
where A represents the total absorbance of the solution at radial position r, while αm,0 and αd/t,0 represent the monomer (m) and dimer/trimer (d/t) absorbance, respectively, at the reference radius, r0. SEDNTERP was used to calculate the estimated molecular mass (Mm) and partial specific volume (v̄) of the monomer in solution, and the molecular mass for a dimer or trimer (Md/t) is a multiple of Mm. R is the universal gas constant, T is the absolute temperature, ρ is the solvent density, ω is the angular velocity, and ζ is the baseline offset. The best-fit model at each protein concentration and speed was chosen by examining residual distribution around zero, the correlation constant (R), and chi-square (χ2) values.
Biotinylation of cell surface proteins.
Subconfluent monolayers of Vero cells in 60-mm dishes were transiently transfected with 3 μg of either wild-type or mutant Hendra virus F in pCAGGS using Lipofectamine Plus (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. At 18 h posttransfection cells were starved for 45 min with DMEM deficient in cysteine-methionine and labeled for 3 h with 100 μCi/ml trans-35S-labeled cysteine-methionine (MP Biomedicals, Irvine, CA or PerkinElmer, Waltham, MA). After labeling, cells were washed three times with ice-cold phosphate-buffered saline (PBS; pH 8.0), and cell surface proteins were biotinylated using 1 mg/ml EZ-Link sulfo-N-hydroxysuccinimide-biotin (Sulfo-NHS-biotin; Pierce, Rockford, IL), diluted in PBS, at 4°C for 35 min (with rocking), followed by 15 min at room temperature. The cells were then washed three times with ice-cold PBS (pH 8.0) and lysed in radio immunoprecipitation assay (RIPA) buffer (100 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1% deoxycholic acid) containing 0.3 M NaCl. Cellular lysates were centrifuged at 136,500 × g for 15 min at 4°C, and 8 μl of Hendra virus F peptide antibody was added to the supernatant. The supernatant was then incubated for 3 h or overnight at 4°C, followed by incubation with 30 μl of protein A-Sepharose beads (GE Healthcare, Piscataway, NJ) for 30 min. Immunoprecipitated proteins were washed twice with RIPA buffer containing 0.30 M NaCl, twice with RIPA buffer containing 0.15 M NaCl, and once with SDS wash II buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.4, 2.5 mM EDTA) and boiled away from the beads in 10% SDS. Ten percent of the protein was removed for analysis (total population), while the remaining 90% was diluted in biotinylation dilution buffer (20 mM Tris [pH 8], 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.2% bovine serum albumin) and incubated with immobilized streptavidin beads (Pierce, Rockford, IL) at 4°C for 1 h, with rocking. Samples were washed as described above, analyzed via 15% SDS-PAGE, and visualized using a Typhoon imaging system (GE Healthcare, Piscataway, NJ). Band densitometry was performed using ImageQuant, version 5.2 (GE Healthcare, Piscataway, NJ), and results are expressed as percentages of wild-type levels.
Cleavage time point immunoprecipitation.
Vero cells in six-well plates were transiently transfected with 2 μg of wild-type or mutant F in pCAGGS using Lipofectamine Plus (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. At 18 h posttransfection, cells were starved and labeled as described above except with a 30-min label. The cells were either immediately lysed in RIPA buffer or chased with DMEM for 2, 4, 6, or 24 h and then lysed. Lysates were spun, incubated with 4 μl of Hendra virus F peptide antibody, pulled down with protein A-conjugated Sepharose beads, and washed as described above. Protein was analyzed via 15% SDS-PAGE and visualized using a Typhoon imaging system (GE Healthcare, Piscataway, NJ). Band densitometry was performed as described above, and percent cleavage was defined as [F1/(F1+F0)].
Luciferase reporter gene assay.
Vero cells in six-well plates were transiently transfected with 0.8 μg of luciferase under the control of the T7 promoter, 0.9 μg of wild-type Hendra virus G in pCAGGS, and 0.3 μg of either wild-type or mutant Hendra virus F in pCAGGS using Lipofectamine Plus (Invitrogen, Carlsbad, CA). At 18 h posttransfection, cells were washed with PBS and overlaid with BSR cells (which stably express the T7 polymerase) at an approximate 1:1 ratio at 37°C for 3 h. The cells were then lysed and assayed for luciferase activity using a luciferase assay system (Promega, Madison, WI) according to the manufacturer's instructions. Activity was measured using an Lmax luminometer (Molecular Devices, Sunnyvale, CA). Values were normalized to samples containing wild-type Hendra virus F and G, with the wild-type levels set at 100% after subtraction of the values for the Hendra virus G alone.
RESULTS
Addition of HRB destabilizes Hendra virus F TMD-TMD interactions.
Isolated TMDs of three paramyxovirus F proteins, parainfluenza virus 5 (PIV5) F, human metapneumovirus (HMPV) F, and Hendra virus F, have recently been shown to interact in a monomer-trimer equilibrium using SE analysis (Smith et al., submitted). While TMD-TMD interactions were shown to be important in maintenance of F prefusion stability, how these interactions affect membrane fusion remains unknown. One possibility is that the stability of these TMD-TMD interactions is modulated by nearby regions of the F protein. HRB, which is located directly N-terminal to the TMD, is a coiled coil in the prefusion structure, suggesting that it might help stabilize these TMD-TMD interactions (80). Alternatively, TMD interactions may stabilize HRB, and changes in TMD-TMD interactions could be important in both F triggering and in subsequent conformational rearrangement. To examine the potential influence of HRB on TMD-TMD interactions, fusion proteins containing either the isolated TMD or HRB and the TMD fused to staphylococcal nuclease (SN) were made, resulting in SN-TMD or SN-HRB-TMD constructs, respectively (Fig. 1B). This system has been used extensively to characterize TMD-TMD interactions of other membrane proteins, including glycophorin A (19, 20, 37, 38, 74), synaptobrevin and syntaxin (31), and the ErB receptor (71). SN has been shown to be exclusively monomeric under many experimental conditions (20). Expressing the TMD or HRB-TMD as SN chimeras increases the molar extinction coefficient of the complex and allows for centrifugation at lower speeds, thus reducing pressure-specific effects on the system (20). The chimeric proteins were grown in the Escherichia coli strain Rosetta-Gami (EMD Chemicals, Gibbstown, NJ), purified using a 1-ml HiTrap SP cation exchange column (GE Healthcare, Piscataway, NJ) (31), and exchanged into the detergent 3-(N,N-dimethylmyristyl-ammonio) propane sulfonate (C14SB) prior to centrifugation (9). Previous work with wild-type Hendra virus F SN-TMD constructs analyzed in another detergent, pentaethylene glycol monooctyl ether (C8E5), demonstrated that the observed trimeric TMD-TMD interactions occurred independently of the detergent utilized (Smith et al., submitted); thus, only C14SB was used in this report. Three concentrations of both the SN-TMD and SN-HRB-TMD constructs were brought to sedimentation equilibrium in a Beckman XL-A analytical ultracentrifuge, and this was performed at two different rotor speeds (20,000 and 30,000 rpm) at 25°C. Equilibrium was considered to be obtained once radial scans taken 6 h apart were indistinguishable, and equilibrium was typically reached within 24 h. Radial absorbance data were collected at 280 nm, and equilibrium scans were analyzed for best fit using KaleidaGraph software by fitting data to the equation given in Materials and Methods. Best-fit models for each protein concentration and speed were determined by examining the distribution of residual values around zero and selecting fits which also returned high correlation constants (R) and low chi-square values (χ2).
The Hendra virus F SN-TMD (residues 485 to 520) construct was best fit as a monomer-trimer equilibrium at all concentrations at 20,000 and 30,000 rpm (Fig. 2A and C). Each of the six data sets has residuals evenly distributed around zero (Fig. 2A and data not shown), indicating that the monomer-trimer fit was in good agreement with the molecular-weight distribution present in the system. A monomer-dimer fit resulted in lower correlation constants (R) and higher chi-square (χ2) values, as did the inclusion of additional terms (i.e., monomer-dimer-trimer), indicating that a monomer-trimer best fit the data. Furthermore, monomer–N-mer fits, which allow for implicit curve fitting, returned stoichiometries very close to a monomer-trimer fit (Fig. 2C) at both speeds and all protein concentrations, also ruling out a monomer-dimer equilibrium model. Surprisingly, inclusion of HRB between SN and the TMD resulted in the data being best described by a monomer-dimer equilibrium (Fig. 2B and C), with monomer–N-mer fits very close to the monomer-dimer model (Fig. 2C). Fitting the same data set to the monomer-trimer equilibrium observed for the wild-type TMD alone resulted in significantly lower R and higher χ2 values. Since addition of HRB had a destabilizing effect, these data suggest that TMD-TMD interactions, along with the F ectodomain at the N-terminal end of HRB, may hold the HRB trimer in place and, thus, could act in regulating the unfolding and refolding of HRB around HRA, a step essential for membrane fusion.
Fig 2.

Inclusion of HRB disrupts TMD-TMD interactions. (A) SE analysis of wild-type Hendra virus F TMDs fused to SN and fit to a monomer-trimer equilibrium. The residuals and curve for one concentration of the 20,000 (20K)-rpm and 30,000 (30K)-rpm rotor speeds are shown. (B) The experiment is the same as described for panel A except that the SN-HRB-TMD construct is fit to a monomer-dimer equilibrium. (C) Summary of fit statistics, including monomer–N-mer fits (± standard error), for additional rotor speeds and protein concentrations. WT, wild type.
Spacing between HRB and the TMD is important for F1 stability.
A previous report examined how spacing between HRB and the TMD of PIV5 F affects proteins stability and membrane fusion (82). In this study, Zhou et al. made insertions, deletions, or substitutions to the 7-amino-acid linker region between the PIV5 F HRB and TMD. While all mutant PIV5 F proteins were expressed at nearly wild-type levels, F proteins with insertions between the two domains were fusion incompetent, demonstrating that the spacing between these two domains is crucial for biological activity (82). To examine the role of HRB-TMD spacing for the Hendra virus F protein, one, two, or three alanine residues were inserted at the N terminus of the putative TMD boundary (Fig. 3A). Prior to an examination of the fusogenicity of these mutant F proteins, cell surface expression (CSE) was monitored by surface biotinylation. Vero cells were transiently transfected with either wild-type or mutant Hendra virus F, starved, and metabolically labeled, followed by immunoprecipitation and separation of the biotinylated F fraction using streptavidin-conjugated beads. In contrast to PIV5 F, insertion of two or three alanine residues progressively and dramatically reduced CSE of the fusion-competent F1 form (Fig. 3B, lanes 6 to 8) compared to wild-type (lane 2). Reduced CSE of F1 could be due to protein misfolding, premature triggering of the F1 form due to decreased protein stability, or problems with protein trafficking. To help distinguish between these possibilities, pulse-chase analysis using a 30-min pulse was performed to examine the kinetics of F expression and processing (Fig. 3C). Insertion of one alanine had only a small effect on F1 processing (Fig. 3C, empty square), while insertion of two (empty diamond) or three (empty triangle) alanine residues led to moderate or severe reductions in F1 cleavage efficiency, respectively. As the initial slopes for each graph are similar, we hypothesize that the folding equilibrium for the N2 and N3 mutants was shifted such that a larger percentage of F misfolds (as more alanine residues are inserted) but the population of the F protein which folds correctly is properly trafficked and cleaved. Thus, these data suggest that, unlike insertions in PIV5 F, insertions between Hendra virus F HRB and the TMD dramatically affect protein expression and stability.
As the above alanine insertions could also lengthen the TMD, which could be responsible for the observed phenotype, identical insertions at the C-terminal end of the TMD were made (Fig. 3A). Though the C-terminal alanine insertions reduced cell surface levels of F1 by up to 50% (Fig. 3B, lanes 3 to 5) compared to the wild-type level (lane 2), this reduction was not nearly as dramatic as that observed with the N-terminal insertions. Furthermore, all of the C-terminal insertion mutants were cleaved with wild-type kinetics (Fig. 3C, filled shapes), suggesting that these mutations did not significantly affect the conformation of the Hendra virus F ectodomain. Together, these data demonstrate that while increasing the overall length of the TMD leads to moderate reductions in F protein expression, progressive N-terminal insertions severely reduce the stability of the cleaved, prefusion F1 conformation.
C-terminal alanine insertions within the TMD reduce cell-cell fusion.
Since the TMDs of various viral fusion proteins have been shown to be critical in membrane fusion (reviewed in references 36 and 66), a luciferase reporter gene assay was performed for both the C- and N-terminal mutants to examine cell-cell fusion. Vero cells were transiently transfected with a luciferase construct under the control of the T7 promoter, wild-type Hendra virus G, and wild-type or mutant Hendra virus F. The next day, cells were overlaid with BSR cells, which stably express the T7 polymerase, and luciferase activity was measured. As expected based on F1 cell surface expression, two or three N-terminal alanine insertions almost completely abolished cell-cell fusion, while the insertion of one alanine resulted in fusion levels barely above background (Fig. 3D, black bars). Unexpected, however, was the dramatic reduction in fusion observed for the C-terminal insertion mutants. Progressive C-terminal alanine insertions resulted in significant decreases in cell-cell fusion, and the insertion of three alanine residues (C3) completely abolished fusion activity. In all cases, observed cell-cell fusion levels were inconsistent with levels expected based on F1 cell surface expression (Fig. 3D, gray bars) (69). Combined, these data suggest that increasing the length of the TMD through C-terminal insertions results in hypofusogenic proteins.
N- and C-terminal insertions differentially affect TMD-TMD interactions.
Mechanistically, the effects of both the N- and C-terminal alanine insertions could be explained through alterations in TMD-TMD interactions. Recent work from our laboratory demonstrated that mutations within the TMD which alter TMD-TMD associations affect the stability of the prefusion, cleaved F1 form (Smith et al., submitted). To examine this possibility, SN constructs fused to the TMD of four of the N- or C-terminal mutants (N1 and N2 or C2 and C3) were made, and oligomerization was examined using SE analysis as described above. Neither the N1 (Fig. 4C) nor N2 (Fig. 4B and C) SN-TMD construct deviated from the monomer-trimer model observed with the wild-type TMD. All data sets from both mutants were best described as demonstrating monomer-trimer equilibrium, and fitting to a monomer-dimer model decreased R values and increased χ2 values. Furthermore, monomer–N-mer fits returned stoichiometries very close to those of a monomer-trimer (Fig. 4C), strongly indicating that alanine insertions at the N-terminal end of the Hendra virus F TMD do not affect TMD-TMD trimerization. These data support the concept that spacing between HRB and the TMD is vital in maintaining prefusion F1 stability while not directly affecting TMD-TMD interactions. Surprisingly, analysis of the C3 SN-TMD construct shows that the insertion of three alanine residues at the C-terminal end of the TMD decreases trimeric interactions as these data are best fit by a monomer-dimer equilibrium (Fig. 4A and C) and are very similar to the observed monomer–N-mer fits (Fig. 4C). The C2 SN-TMD construct is best fit by a monomer-trimer equilibrium at lower rotor speeds, while at high speeds, the data are best fit by a monomer-dimer equilibrium (Fig. 4C). A speed-dependent shift in best-fit models observed for C2 is likely due to the decreased stability of the TMD-TMD interactions at higher rotor speeds, which has been previously described for this system (Smith et al., submitted), and suggests that C2 TMD-TMD interactions are likely weakened upon the insertion of two alanine residues. Ultimately, these data support a model in which the C-terminal portion of the Hendra virus F TMD is an important regulator of TMD-TMD interactions and in which disruption of these interactions decreases membrane fusion promotion.
Fig 4.

C-terminal insertions disrupt TMD-TMD interactions. (A) SE analysis of the C3 SN-TMD construct showing the residuals and curve at 20,000 and 30,000 rpm for one concentration fit to a monomer-dimer model. (B) SE analysis of the N2 SN-TMD construct fit to a monomer-trimer model. (C) Additional SN-TMD constructs and fit statistics, including monomer–N-mer fits (± standard error), for different rotor speeds and protein concentrations.
Mutation of nonpolar β-branched residues within the Hendra virus F TMD reduces fusion.
The observed decreases in cell-cell fusion and TMD-TMD interactions suggested that residues within the C terminus of the TMD are likely important for fusion promotion through modulation of TMD-TMD interactions. To test this hypothesis, alanine scanning mutagenesis was performed on the last 12 amino acids of the Hendra virus F TMD (Fig. 5A). Surface biotinylation was performed as described above, and all of the mutant F proteins were surface expressed at wild-type levels with the exception of the mutant with LI replaced by AA (LI mutant) (Fig. 5B), which was reduced about 50% compared to the wild type. Both the F1 and F0 forms of LI had a small population of lower apparent molecular weight species, which could be indicative of heterogeneous glycosylation (Fig. 5B, lane 4). Immunoprecipitation of the Hendra virus F LI mutant followed by treatment with peptide N-glycosidase F (PNGase F) (12), an enzyme which removes the entire N-glycan, failed to completely resolve the species into a tighter band (data not shown), suggesting that additional alterations to the protein may be present. For the remaining mutant F proteins, both F0 and F1 had CSE levels almost identical to wild-type levels, indicating that these specific sequences are not required for protein expression and folding (Fig. 5B and C). As these mutations could also affect fusion promotion, a luciferase reporter gene analysis was performed as described above. Surprisingly, despite wild-type F1 CSE levels, the majority of the mutant F proteins (IG, TF, FV, and IV mutants) were impaired for fusion by as much as 60%, while cell-cell fusion levels for the LI and IS mutants were at levels consistent with F1 CSE of those proteins (Fig. 5C). As many mutations resulted in hypofusogenic phenotypes, these data suggest that the C-terminal region of the Hendra virus F TMD is critical for membrane fusion.
Fig 5.
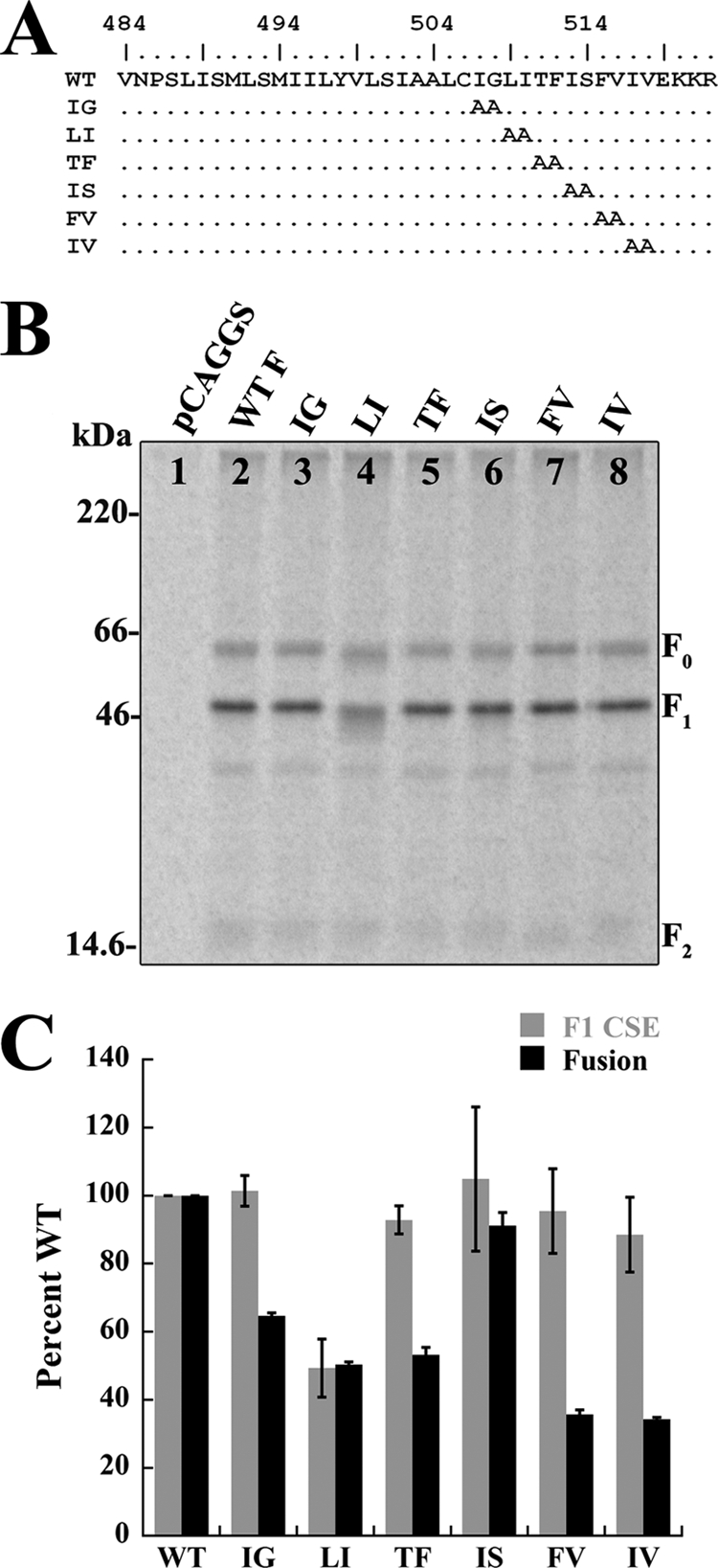
β-Branched residues within the C-terminal end of the TMD are important for fusion. (A) Sequence of the Hendra virus F TMD and alanine substitution mutants, where L492 is the predicted start of the TMD. Mutants are named by abbreviations for the residues replaced with alanines. (B) Biotinylation of cell surface-expressed wild-type and mutant F proteins. The proteins were labeled with [trans-35S]cysteine-methionine, biotinylated, and immunoprecipitated, and the total and surface protein populations were separated by streptavidin pulldown, followed by 15% SDS-PAGE and visualization using autoradiography. (C) Quantification of F1 cell surface expression levels and cell-cell fusion levels for wild-type and mutant F proteins. Cells were transfected with wild-type or mutant Hendra virus F and wild-type Hendra virus G along with a luciferase construct, overlaid with BSR cells, and assayed for luciferase activity. Error bars represent the mean ± the standard error of the mean for three independent experiments.
DISCUSSION
Transmembrane domains of viral fusion proteins have been hypothesized to serve important functions in membrane fusion promotion, especially in driving the hemifusion-to-fusion transition (1, 28, 42, 44–46, 52). One of the ways in which TMDs have been proposed to aid in membrane fusion promotion is through TMD-TMD interactions, and recent work with isolated TMDs of PIVF 5, HMPV F, and Hendra virus F demonstrated that these domains interact in a monomer-trimer equilibrium (Smith et al., submitted). What remain unknown, however, are the sequence requirements for these TMD-TMD interactions and how they could mechanistically modulate F-mediated fusion. Results from this study suggest that TMD-TMD interactions could be an important regulator of HRB refolding and that spacing between these two domains is essential in maintaining stability of the prefusion F1 form. Furthermore, our data demonstrate that the C-terminal end of the Hendra virus F TMD is important in TMD-TMD interactions. Finally, mutational studies suggest that β-branched residues near the C terminus of the TMD are important in membrane fusion.
An attractive model for how TMD-TMD interactions could regulate membrane fusion is through potential stabilization of the HRB coiled coil prior to F protein triggering. As the prefusion structure of PIV5 F showed the HRB and the trimeric GCN4t region as a continuous coiled coil (80), we first determined if inclusion of HRB between SN and the Hendra virus F TMD stabilized the trimeric TMD-TMD interactions (Fig. 2A and C). Surprisingly, inclusion of HRB resulted in a poorer monomer-trimer fit than observed for the wild-type Hendra virus F TMD alone and, instead, a monomer-dimer model was the best fit (Fig. 2B and C), demonstrating that addition of HRB has a destabilizing effect. These data suggest that HRB may require additional elements within the F protein, such as the TMD and portions of the head domain of F, to stabilize a coiled-coil conformation. Furthermore, the SE analysis data suggest that TMD-TMD interactions could play a role in holding the unstable HRB coiled-coil together, and evidence from other groups supports this hypothesis. Isolated HRB peptides, unlike HRA peptides, fail to form a coiled-coil structure in solution (27), and the prefusion structure of PIV5 F was obtained only when the TMD was replaced with the trimeric GCN4t region (80). It could be that the “spring-like” coiled-coil structure of HRB is dependent on both the ectodomain and the TMD to clamp it together, a model which our data would support (Fig. 6, step a). Modulation of TMD-TMD interactions following triggering could cause this clamp to be released (Fig. 6, steps b and c) and facilitate subsequent HRB refolding, a step required for six-helix bundle formation (Fig. 6, steps d and e). While specific interactions with homotypic attachment proteins are generally thought to trigger paramyxovirus F proteins (32, 70), other domains, such as the FP region in Hendra virus F (69) and the HRB linker in HMPV (64) and PIV5 F (62), also seem to play a role in triggering.
Fig 6.

A model for TMD-TMD interactions as potential regulators of membrane fusion. In the prefusion cleaved form of F (step a), HRB forms a coiled coil and the TMDs interact as trimers. Triggering of F (step b) causes changes in TMD-TMD interactions and local unfolding mediated by the α-helix destabilizing β-branched residues (*). Unfolding of the TMDs (steps c and d) causes unfolding of HRB, extension of HRA, and insertion of the FP into the target membrane. Step e shows the postfusion six-helix bundle conformation of the F protein with the TMDs and FPs on the same end of the molecule. Green, TMD; red, HRB; blue, HRA, yellow, FP.
Such a mechanism for regulation of membrane fusion may require a certain distance between both HRB and the TMD as studies with PIV5 F demonstrated that spacing between these two domains is important for fusion promotion (82). Additionally, studies with SNARE proteins have also demonstrated that increased spacing between the SNARE TMD and the conserved coiled-coil domain significantly reduced SNARE-mediated fusion (43). In our work, progressive alanine insertions between HRB and the TMD in Hendra virus F caused the F1 form of the protein to become increasingly destabilized and less efficiently expressed on the cell surface (Fig. 3B and D). Several explanations could be given for this phenotype, including disruption of the TMD-TMD helix-helix interacting face or lengthening of the TMD. It is unlikely that such a decrease in F1 expression/stability was due to disruption of the helix-helix interacting face as insertion of three alanine residues (N3), which should insert one complete helical turn and thus restore the helix-helix interacting face, failed to restore expression levels. Additionally, while control mutations lengthening the TMD did affect protein expression and stability (Fig. 3D), this effect was not nearly as pronounced, given that the F1 forms of C1, C2, and C3 remained at 45 to 80% of wild-type expression levels. Lastly, our SE analysis data for N1 and N2 resulted in a monomer-trimer best-fit model and suggest that TMD-TMD interactions were unaffected by N-terminal alanine insertions (Fig. 4B and C). Thus, our data support a model in which spacing between HRB and the TMD F is important for stability and triggering of the prefusion, cleaved form of Hendra virus F, likely affecting the ability of TMD-TMD interactions to hold HRB together. Increasing the spacing between HRB and the TMD could result in a less stable fold of the C-terminal portion of HRB, resulting in premature triggering following F protein cleavage.
Beyond increasing the spacing between HRB and the TMD, the N-terminal insertions also increased the length of the TMD. While our control C-terminal insertions (Fig. 3A) demonstrated that increasing the length of the TMD had moderate effects on F1 expression, these mutants also had serious fusion defects (Fig. 3D). Even though F1 is expressed at 40% of wild-type levels, C2 and C3 promoted much lower levels of membrane fusion than expected based on surface expression levels (Fig. 3D) (69). Our subsequent SE analysis of C2 and C3 constructs demonstrated that C-terminal insertions affected TMD-TMD interactions and that the isolated C3 TMD was not best fit by a monomer-trimer but, rather, by a monomer-dimer equilibrium (Fig. 4A). Since alanine residues were inserted near the C terminus (and not directly at the end), these insertions could be disrupting structural determinants required for proper maintenance of TMD-TMD interactions. It is unlikely that this is caused solely by disruption of the helical face as the presence of three alanine residues failed to restore membrane fusion. Ultimately, our data suggest that elements within the C terminus of the Hendra virus F TMD are important for TMD-TMD interactions and subsequently membrane fusion promotion.
The sequence requirements which determine TMD-TMD interactions in both viral and nonviral systems remain poorly understood though several lines of evidence point toward central glycine motifs (16, 48, 49, 68), polar residues (18, 23, 81), and/or heptad repeat-like arrangements of leucine and isoleucine (25, 29). A recent study of various paramyxovirus F proteins demonstrated that swapping the TMD of NDV F with that of SeV F, MV F, or VSV G resulted in F proteins defective for membrane fusion, suggesting that the actual sequences of paramyxovirus TMDs are important for fusion (24). Additionally, patches of hydrophobic residues at the N and C termini of the PIV5 F TMD seem to be important for F-mediated membrane fusion, especially β-branched residues at the N terminus, further suggesting a sequence dependence of paramyxovirus F protein TMDs for fusion (6). Alanine substitution of the last 12 amino acids within the Hendra virus F TMD (Fig. 5A) revealed that this region is tolerant to alanine substitution, particularly for protein expression and processing (Fig. 5B and C). Interesting, however, was the hypofusogenic phenotype associated with the Hendra virus F protein FV and IV mutants, which immediately precede the highly charged EKKR region (Fig. 5A). Combining these data with the data from the C-terminal insertions (which immediately preceded the VIV sequence) suggests that β-branched residues in the sequence FVIV could be important modulators of F protein-promoted membrane fusion. Recent work with the baboon reovirus p15 TMD (15) and model peptides (34, 35, 60, 61) suggests that β-branched residues are important in membrane fusion in a variety of systems. Nonpolar β-branched residues (valine and isoleucine) are traditionally thought of as being disruptive of α-helical structures; however, studies of synthetic peptides in detergent micelles demonstrated that these residues are incorporated into hydrophobic α-helices as easily as leucine (39, 40). Additionally, as β-branched residues are thought to add conformational plasticity to membrane α-helices (33, 54, 72), these residues could be important for driving lipid disordering and later steps of membrane fusion. Thus, inclusion of these residues within viral TMDs may both facilitate promotion of α-helical structure and impart a degree of structural flexibility which alanine or residues of similar α-helical propensity could not. Consequently, mutation of the VIV sequence to alanine in the context of the FV and IV mutants might not disrupt the initial α-helical structure of the Hendra virus F TMD within a membrane but could either stabilize TMD-TMD interactions or abolish the conformational flexibility needed for efficient membrane fusion. Future SE analysis of the scanning alanine mutants, especially the FV and IV mutants, will be needed to delineate the potential role(s) of β-branched residues in modulating TMD-TMD interactions. Beyond this, further experiments will be needed to firmly establish the mechanistic role(s) of paramyxovirus TMDs in membrane fusion, but this work presents an exciting model by which TMD-TMD interactions affect not only protein stability but also fusogenicity. Furthermore, these data also support a role for TMD-TMD interactions and TMD-HRB spacing in Hendra virus F triggering.
ACKNOWLEDGMENTS
We thank Lin-Fa Wang (Australian Animal Health Laboratory) for providing the Hendra virus F and G plasmids, Karen Fleming (The Johns Hopkins University) for providing the SN pET-11a constructs, Stacy Smith for helping to express and purify some of the recombinant proteins, and Anna Smith for helping to generate some of the Hendra virus F mutant constructs. We also acknowledge members of the Dutch laboratory for critically reviewing the manuscript.
This work was supported by NIAID/NIH grant R01AI051517 to R.E.D., NIH grant R01GM070662 to M.G.F., and NIH grant 2P20 RR020171 from the National Center for Research Resources to R.E.D., M.G.F., and T.P.C.
Footnotes
Published ahead of print 11 January 2012
REFERENCES
- 1. Armstrong RT, Kushnir AS, White JM. 2000. The transmembrane domain of influenza hemagglutinin exhibits a stringent length requirement to support the hemifusion to fusion transition. J. Cell Biol. 151:425–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Asano K, Asano A. 1985. Why is a specific amino acid sequence of F glycoprotein required for the membrane fusion reaction between envelope of HVJ (Sendai virus) and target cell membranes? Biochem. Int. 10:115–122 [PubMed] [Google Scholar]
- 3. Baker KA, Dutch RE, Lamb RA, Jardetzky TS. 1999. Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell 3:309–319 [DOI] [PubMed] [Google Scholar]
- 4. Biacchesi S, et al. 2006. Modification of the trypsin-dependent cleavage activation site of the human metapneumovirus fusion protein to be trypsin independent does not increase replication or spread in rodents or nonhuman primates. J. Virol. 80:5798–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biacchesi S, et al. 2004. Recombinant human Metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: deletion of G yields a promising vaccine candidate. J. Virol. 78:12877–12887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bissonnette ML, Donald JE, DeGrado WF, Jardetzky TS, Lamb RA. 2009. Functional analysis of the transmembrane domain in paramyxovirus F protein-mediated membrane fusion. J. Mol. Biol. 386:14–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buchholz UJ, Finke S, Conzelmann KK. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73:251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bullough PA, Hughson FM, Skehel JJ, Wiley DC. 1994. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371:37–43 [DOI] [PubMed] [Google Scholar]
- 9. Burgess NK, Stanley AM, Fleming KG. 2008. Determination of membrane protein molecular weights and association equilibrium constants using sedimentation equilibrium and sedimentation velocity. Methods Cell Biol. 84:181–211 [DOI] [PubMed] [Google Scholar]
- 10. Buzon V, et al. 2010. Crystal Structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions. PLoS Pathog. 6:e1000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carr CM, Kim PS. 1993. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 73:823–832 [DOI] [PubMed] [Google Scholar]
- 12. Carter JR, Pager CT, Fowler SD, Dutch RE. 2005. Role of n-linked glycosylation of the Hendra virus fusion protein. J. Virol. 79:7922–7925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273 [DOI] [PubMed] [Google Scholar]
- 14. Chen L, et al. 2001. The structure of the fusion glycoprotein of Newcastle disease virus suggests a novel paradigm for the molecular mechanism of membrane fusion. Structure 9:255–266 [DOI] [PubMed] [Google Scholar]
- 15. Clancy EK, Duncan R. 2011. Helix-destabilizing, β-branched, and polar residues in the baboon reovirus p15 transmembrane domain influence the modularity of FAST proteins. J. Virol. 85:4707–4719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cleverley DZ, Lenard J. 1998. The transmembrane domain in viral fusion: essential role for a conserved glycine residue in vesicular stomatitis virus G protein. Proc. Natl. Acad. Sci. U. S. A. 95:3425–3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Colman PM, Lawrence MC. 2003. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 4:309–319 [DOI] [PubMed] [Google Scholar]
- 18. Dawson JP, Weinger JS, Engelman DM. 2002. Motifs of serine and threonine can drive association of transmembrane helices. J. Mol. Biol. 316:799–805 [DOI] [PubMed] [Google Scholar]
- 19. Doura AK, Fleming KG. 2004. Complex interactions at the helix-helix interface stabilize the glycophorin a transmembrane dimer. J. Mol. Biol. 343:1487–1497 [DOI] [PubMed] [Google Scholar]
- 20. Fleming KG, Ackerman AL, Engelman DM. 1997. The effect of point mutations on the free energy of transmembrane alpha-helix dimerization. J. Mol. Biol. 272:266–275 [DOI] [PubMed] [Google Scholar]
- 21. Fritz R, et al. 2011. The unique transmembrane hairpin of flavivirus fusion protein E is essential for membrane fusion. J. Virol. 85:4377–4385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garten W, et al. 1994. Processing of viral glycoproteins by the subtilisin-like endoprotease furin and its inhibition by specific peptidylchloroalkylketones. Biochimie 76:217–225 [DOI] [PubMed] [Google Scholar]
- 23. Gratkowski H, Lear JD, DeGrado WF. 2001. Polar side chains drive the association of model transmembrane peptides. Proc. Natl. Acad. Sci. U. S. A. 98:880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gravel KA, McGinnes LW, Reitter J, Morrison TG. 2011. The transmembrane domain sequence affects the structure and function of the Newcastle disease virus fusion protein. J. Virol. 85:3486–3497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gurezka R, Laage R, Brosig B, Langosch D. 1999. A heptad motif of leucine residues found in membrane proteins can drive self-assembly of artificial transmembrane segments. J. Biol. Chem. 274:9265–9270 [DOI] [PubMed] [Google Scholar]
- 26. Harrison SC. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Joshi SB, Dutch RE, Lamb RA. 1998. A core trimer of the paramyxovirus fusion protein: parallels to influenza virus hemagglutinin and HIV-1 gp41. Virology 248:20–34 [DOI] [PubMed] [Google Scholar]
- 28. Kemble GW, Danieli T, White JM. 1994. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell 76:383–391 [DOI] [PubMed] [Google Scholar]
- 29. Kliger Y, Shai Y. 1997. A leucine zipper-like sequence from the cytoplasmic tail of the HIV-1 envelope glycoprotein binds and perturbs lipid bilayers. Biochemistry 36:5157–5169 [DOI] [PubMed] [Google Scholar]
- 30. Kozerski C, Ponimaskin E, Schroth-Diez B, Schmidt MF, Herrman A. 2000. Modification of the cytoplasmic domain of influenza virus hemagglutinin affects enlargement of the fusion pore. J. Virol. 74:7529–7537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kroch AE, Fleming KG. 2006. Alternate interfaces may mediate homomeric and heteromeric assembly in the transmembrane domains of SNARE proteins. J. Mol. Biol. 357:184–194 [DOI] [PubMed] [Google Scholar]
- 32. Lamb RA, Parks GD. 2007. Paramyxoviridae: the viruses and their replication, p 1449–1496 In Knipe DM, et al. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 33. Langosch D, Arkin IT. 2009. Interaction and conformational dynamics of membrane-spanning protein helices. Protein Sci. 18:1343–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Langosch D, Brosig B, Pipkorn R. 2001. Peptide mimics of the vesicular stomatitis virus G-protein transmembrane segment drive membrane fusion in vitro. J. Biol. Chem. 276:32016–32021 [DOI] [PubMed] [Google Scholar]
- 35. Langosch D, et al. 2001. Peptide mimics of SNARE transmembrane segments drive membrane fusion depending on their conformational plasticity. J. Mol. Biol. 311:709–721 [DOI] [PubMed] [Google Scholar]
- 36. Langosch D, Hofmann M, Ungermann C. 2007. The role of transmembrane domains in membrane fusion. Cell. Mol. Life Sci. 64:850–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lemmon MA, et al. 1992. Glycophorin A dimerization is driven by specific interactions between transmembrane alpha-helices. J. Biol. Chem. 267:7683–7689 [PubMed] [Google Scholar]
- 38. Lemmon MA, Flanagan JM, Treutlein HR, Zhang J, Engelman DM. 1992. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry 31:12719–12725 [DOI] [PubMed] [Google Scholar]
- 39. Li SC, Deber CM. 1994. A measure of helical propensity for amino acids in membrane environments. Nat. Struct. Biol. 1:368–373 [DOI] [PubMed] [Google Scholar]
- 40. Li SC, Deber CM. 1992. Glycine and β-branched residues support and modulate peptide helicity in membrane environments. FEBS Lett. 311:217–220 [DOI] [PubMed] [Google Scholar]
- 41. Markosyan RM, Cohen FS, Melikyan GB. 2003. HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol. Biol. Cell 14:926–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Markosyan RM, Cohen FS, Melikyan GB. 2000. The lipid-anchored ectodomain of influenza virus hemagglutinin (GPI-HA) is capable of inducing nonenlarging fusion pores. Mol. Biol. Cell 11:1143–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McNew JA, Weber T, Engelman DM, Söllner TH, Rothman JE. 1999. The length of the flexible SNAREpin juxtamembrane region is a critical determinant of SNARE-dependent fusion. Mol. Cell 4:415–421 [DOI] [PubMed] [Google Scholar]
- 44. Melikyan GB, Grener SA, Ok DC, Cohen FS. 1997. Inner but not outer membrane leaflets control the transition from glycosylphosphatidylinositol-anchored influenza hemagglutinin-induced hemifusion to full fusion. J. Cell Biol. 136:995–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Melikyan GB, Markosyan RM, Roth MG, Cohen FS. 2000. A point mutation in the transmembrane domain of the hemagglutinin of influenza virus stabilizes a hemifusion intermediate that can transit to fusion. Mol. Biol. Cell 11:3765–3775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Melikyan GB, White JM, Cohen FS. 1995. GPI-anchored influenza hemagglutinin induces hemifusion to both red blood cell and planar bilayer membranes. J. Cell Biol. 131:679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Meulendyke KA, Wurth MA, McCann RO, Dutch RE. 2005. Endocytosis plays a critical role in proteolytic processing of the Hendra virus fusion protein. J. Virol. 79:12643–12649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Miyauchi K, et al. 2010. The membrane-spanning domain of gp41 plays a critical role in intracellular trafficking of the HIV envelope protein. Retrovirology. 7:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miyauchi K, et al. 2006. Mutations of conserved glycine residues within the membrane-spanning domain of human immunodeficiency virus type 1 gp41 can inhibit membrane fusion and incorporation of Env onto virions. Jpn. J. Infect. Dis. 59:77–84 [PubMed] [Google Scholar]
- 50. Miyauchi K, et al. 2005. Role of the specific amino acid sequence of the membrane-spanning domain of human immunodeficiency virus type 1 in membrane fusion. J. Virol. 79:4720–4729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants by a novel eukaryotic vector. Gene 108:193–200 [DOI] [PubMed] [Google Scholar]
- 52. Nüssler F, Clague MJ, Herrmann A. 1997. Meta-stability of the hemifusion intermediate induced by glycosylphosphatidylinositol-anchored influenza hemagglutinin. Biophys. J. 73:2280–2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Odell D, Wanas E, Yan J, Ghosh HP. 1997. Influence of membrane anchoring and cytoplasmic domains on the fusogenic activity of vesicular stomatitis virus glycoprotein G. J. Virol. 71:7996–8000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ollesch J, et al. 2008. Secondary structure and distribution of fusogenic LV-peptides in lipid membranes. Eur. Biophys. J. 37:435–445 [DOI] [PubMed] [Google Scholar]
- 55. Ortmann D, et al. 1994. Proteolytic cleavage of wild type and mutants of the F protein of human parainfluenza virus type 3 by two subtilisin-like endoproteases, furin and KEX2. J. Virol. 68:2772–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Owens R, Burke C, Rose J. 1994. Mutations in the membrane-spanning domain of the human immunodeficiency virus envelope glycoprotein that affect fusion activity. J. Virol. 68:570–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pager CT, Craft WW, Jr, Patch J, Dutch RE. 2006. A mature and fusogenic form of the Nipah virus fusion protein requires proteolytic processing by cathepsin L. Virology 346:251–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pager CT, Dutch RE. 2005. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J. Virol. 79:12714–12720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pager CT, Wurth MA, Dutch RE. 2004. Subcellular localization and calcium and pH requirements for proteolytic processing of the Hendra virus fusion protein. J. Virol. 78:9154–9163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Poschner BC, Fischer K, Herrmann JR, Hofmann MW, Langosch D. 2010. Structural features of fusogenic model transmembrane domains that differentially regulate inner and outer leaflet mixing in membrane fusion. Mol. Membr. Biol. 27:1–10 [DOI] [PubMed] [Google Scholar]
- 61. Poschner BC, Quint S, Hofmann MW, Langosch D. 2009. Sequence-specific conformational dynamics of model transmembrane domains determines their membrane fusogenic function. J. Mol. Biol. 386:733–741 [DOI] [PubMed] [Google Scholar]
- 62. Russell CJ, Kantor KL, Jardetzky TS, Lamb RA. 2003. A dual-functional paramyxovirus F protein regulatory switch segment: activation and membrane fusion. J. Cell Biol. 163:363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Salzwedel K, Johnston P, Roberts S, Dubay J, Hunter E. 1993. Expression and characterization of glycophospholipid-anchored human immunodeficiency virus type 1 envelope glycoproteins. J. Virol. 67:5279–5288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schowalter RM, Chang A, Robach JG, Buchholz UJ, Dutch RE. 2009. Low-pH triggering of human metapneumovirus fusion: essential residues and importance in entry. J. Virol. 83:1511–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schowalter RM, Smith EC, Dutch RE. 2011. Attachment and entry: viral cell fusion, p 243–260 In Agbandje-McKenna M, McKenna R. (ed), Structural virology. Royal Society of Chemistry, Cambridge, United Kingdom [Google Scholar]
- 66. Schroth-Diez B, et al. 2000. The Role of the transmembrane and of the intraviral domain of glycoproteins in membrane fusion of enveloped viruses. Biosci. Rep. 20:571–595 [DOI] [PubMed] [Google Scholar]
- 67. Shang L, Hunter E. 2010. Residues in the membrane-spanning domain core modulate conformation and fusogenicity of the HIV-1 envelope glycoprotein. Virology 404:158–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shang L, Yue L, Hunter E. 2008. Role of the membrane-spanning domain of human immunodeficiency virus type 1 envelope glycoprotein in cell-cell fusion and virus infection. J. Virol. 82:5417–5428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Smith EC, Dutch RE. 2010. Side chain packing below the fusion peptide strongly modulates triggering of the Hendra virus F protein. J. Virol. 84:10928–10932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Smith EC, Popa A, Chang A, Masante C, Dutch RE. 2009. Viral entry mechanisms: the increasing diversity of paramyxovirus entry. FEBS J. 276:7217–7227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stanley AM, Fleming KG. 2005. The transmembrane domains of ErbB receptors do not dimerize strongly in micelles. J. Mol. Biol. 347:759–772 [DOI] [PubMed] [Google Scholar]
- 72. Stelzer W, Poschner BC, Stalz H, Heck AJ, Langosch D. 2008. Sequence-specific conformational flexibility of SNARE transmembrane helices probed by hydrogen/deuterium exchange. Biophys. J. 95:1326–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sulistijo ES, Jaszewski TM, MacKenzie KR. 2003. Sequence-specific dimerization of the transmembrane domain of the “BH3-only” protein BNIP3 in membranes and detergent. J. Biol. Chem. 278:51950–51956 [DOI] [PubMed] [Google Scholar]
- 74. Treutlein HR, Lemmon MA, Engelman DM, Brunger A. 1992. The glycophorin A transmembrane domain dimer: Sequence-specific propensity for a right-handed supercoil of helixes. Biochemistry 31:12726–12732 [DOI] [PubMed] [Google Scholar]
- 75. Weiss CD, White JM. 1993. Characterization of stable Chinese hamster ovary cells expressing wild-type, secreted, and glycosylphosphatidylinositol-anchored human immunodeficiency virus type 1 envelope glycoprotein. J. Virol. 67:7060–7066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Weissenhorn W, Calder LJ, Wharton SA, Skehel JJ, Wiley DC. 1998. The central structural feature of the membrane fusion protein subunit from the Ebola virus glycoprotein is a long triple-stranded coiled coil. Proc. Natl. Acad. Sci. U. S. A. 95:6032–6036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43:189–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wilk T, Pfeiffer T, Bukovsky A, Moldenhauer G, Bosch V. 1996. Glycoprotein incorporation and HIV-1 infectivity despite exchange of the gp160 membrane-spanning domain. Virology 218:269–274 [DOI] [PubMed] [Google Scholar]
- 79. Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. U. S. A. 102:9288–9293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhou FX, Merianos HJ, Brunger AT, Engelman DM. 2001. Polar residues drive association of polyleucine transmembrane helices. Proc. Natl. Acad. Sci. U. S. A. 98:2250–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhou J, Dutch RE, Lamb RA. 1997. Proper spacing between heptad repeat B and the transmembrane domain boundary of the paramyxovirus SV5 F protein is critical for biological activity. Virology 239:327–339 [DOI] [PubMed] [Google Scholar]