

Abstract
Vesicular stomatitis virus (VSV) is a promising oncolytic agent against a variety of cancers. However, it has never been tested in any pancreatic cancer model. Pancreatic ductal adenocarcinoma (PDA) is the most common and aggressive form of pancreatic cancer. In this study, the oncolytic potentials of several VSV variants were analyzed in a panel of 13 clinically relevant human PDA cell lines and compared to conditionally replicative adenoviruses (CRAds), Sendai virus and respiratory syncytial virus. VSV variants showed oncolytic abilities superior to those of other viruses, and some cell lines that exhibited resistance to other viruses were successfully killed by VSV. However, PDA cells were highly heterogeneous in their susceptibility to virus-induced oncolysis, and several cell lines were resistant to all tested viruses. Resistant cells showed low levels of very early VSV RNA synthesis, indicating possible defects at initial stages of infection. In addition, unlike permissive PDA cell lines, most of the resistant cell lines were able to both produce and respond to interferon, suggesting that intact type I interferon responses contributed to their resistance phenotype. Four cell lines that varied in their permissiveness to VSV-ΔM51 and CRAd dl1520 were tested in mice, and the in vivo results closely mimicked those in vitro. While our results demonstrate that VSV is a promising oncolytic agent against PDA, further studies are needed to better understand the molecular mechanisms of resistance of some PDAs to oncolytic virotherapy.
INTRODUCTION
Oncolytic virus (OV) therapy is an anticancer approach that utilizes replication-competent viruses to specifically kill tumor cells (9, 59, 69). Such selectivity is possible because many tumors are characterized by defective innate immune responses or tumor-related abnormalities in the regulation of mRNA translation or certain cellular signaling pathways, facilitating selective replication of viruses in cancer cells. For example, many cancer cells have defective type I interferon (IFN) responses, which provides growth advantages to tumor cells; however, it also makes them more susceptible to viral infections (51, 66). As a result, OVs can infect, replicate within, and kill tumor cells. Successful virus replication in cancer cells leads to the release of newly formed infectious virus particles that go on to infect neighboring tumor cells.
Vesicular stomatitis virus (VSV) is a promising OV and has demonstrated preclinical success against a variety of malignancies, including prostate cancer (1, 11, 49), breast cancer (3, 20, 53, 63), melanoma (20, 24), colorectal cancer (16, 32, 64), liver cancer (4–5, 75), glioblastoma (10, 56, 73), and other cancers (6). As a result, at least two VSV OVs have been considered for clinical trials by the NIH Recombinant-DNA Advisory Committee (10). However, VSV's oncolytic potential has never been studied in any pancreatic cancer models. About 95% of pancreatic cancers are pancreatic ductal adenocarcinomas (PDAs), which are highly invasive, with aggressive local growth and rapid metastasis to surrounding tissues (65). PDA is considered one of the most lethal abdominal malignancies, with annual deaths closely matching the annual incidence of the disease (19, 45), resulting in a 5-year survival rate of only 8 to 20%. Several cancer therapies proven successful in other tumor types have shown little efficacy in treating PDA. Chemotherapy is the primary treatment available; however, patients exhibit little improvement or develop chemoresistance (65). Therefore, development of new treatment strategies for patients suffering from PDA is of utmost importance.
OV therapy with several viruses, including adenoviruses (28, 33, 41), herpesviruses (17, 36, 52, 61, 71), and reoviruses (18, 29–30), has recently shown promise in several PDA tumor models. However, there are several advantages of using VSV as an anticancer therapy. VSV is the prototypic nonsegmented negative-strand (NNS) RNA virus (order Mononegavirales, family Rhabdoviridae), and its basic biology and interactions with host immune responses have been extensively studied (47). This knowledge has led to the development of rationally designed VSV vectors for use in vaccines, gene therapy, and OV therapy (6, 70). While VSV is very sensitive to IFN-mediated antiviral responses (and therefore unable to productively infect healthy cells), it can specifically infect and kill tumor cells, the majority of which are believed to be defective in type I IFN production and responses (6, 44). Also, the mechanisms of VSV-mediated killing by apoptosis have been established (22). In addition to tumor specificity, VSV has several important advantages as an OV: (i) replication occurs in the cytoplasm of host cells with no risk of host cell transformation, (ii) cellular uptake in many mammalian cell types occurs rapidly and there is no cell cycle dependency, (iii) the genome is easily manipulated with the possibility for strong and adjustable levels of foreign gene expression to enhance oncolysis and specificity, and (iv) there is no preexisting immunity against VSV in humans (6). While VSV is not considered a significant human pathogen, it can cause neurotoxicity in mice, nonhuman primates, and even humans (58). However, several VSV mutants have been generated that are not neurotropic but retain their oncolytic activity (2, 37, 73). In this study, we focused on two such VSV mutants, VSV-ΔM51-GFP and VSV-p1-GFP (73). VSV-p1-GFP has the green fluorescent protein (GFP) open reading frame (ORF) inserted at position 1 of the viral genome, resulting in slower viral replication kinetics, reducing VSV-p1-GFP's ability to evade innate immune responses (73). VSV-ΔM51-GFP has a deletion at amino acid position 51 of the matrix (M) protein, as well as the GFP ORF inserted in position 5 of the viral genome (73). Both attenuated VSV recombinants have shown a desirable phenotype characterized by retention of their oncolytic activities but lack of neurotoxicity in vivo (2, 73).
In our study, the oncolytic potentials of VSV variants were analyzed in a panel of 13 clinically relevant human PDA cell lines and compared to conditionally replicative adenoviruses (CRAds), Sendai virus (SeV) and respiratory syncytial virus (RSV). VSV showed oncolytic abilities superior to those of all other viruses tested and was effective in killing the majority of tested PDA cell lines. However, we identified some PDA cell lines that showed general resistance to oncolysis by all tested viruses. These results were confirmed for several PDA cell lines in vivo in nude mice. We also conducted an initial analysis of PDA resistance to virus-induced cell death. Our in vitro and in vivo results demonstrate that VSV has good potential as an OV against PDA, while further studies are needed to better understand the molecular mechanisms of resistance of some PDA cell lines to virotherapy.
MATERIALS AND METHODS
Cell lines.
The human PDA cell lines used in this study were CFPAC-1 (ATCC CRL-1918), Hs766T (ATCC HTB-134), Capan-2 (ATCC HTB-80), T3M4 (54), AsPC-1 (ATCC CRL-1682), HPAF-II (ATCC CRL-1997), Suit2 (34), HPAC (ATCC CRL-2119), BxPC-3 (ATCC CRL-1687), MIA PaCa2 (ATCC CRL-1420), SU.86.86 (ATCC CRL-1837), Capan-1 (ATCC HTB-79), and Panc-1 (ATCC CRL-1469). In addition, the immortal human pancreatic duct epithelial (HPDE) cell line (21) was used in this study and maintained in Keratinocyte-SFM (Gibco). This cell line, which was generated by introduction of the E6 and E7 genes of human papillomavirus 16 into normal adult pancreas epithelium, retains a genotype similar to that of pancreatic duct epithelium and is nontumorigenic in nude mice (21). The mouse breast cancer cell line 4T1 (ATCC CRL-2539), baby hamster kidney BHK-21 fibroblasts (ATCC CCL-10), the human cervix adenocarcinoma HeLa cell line (ATCC CCL-2), African green monkey kidney Vero cells (ATCC CCL-81), and human epidermoid cancer Hep-2 cells (ATCC CCL-23) were used to grow viruses and/or as controls for viral replication. CFPAC-1, Suit2, HPAC, MIA PaCa2, Capan-1, Panc-1, 4T1, and Vero cells were all maintained in Dulbecco's modified Eagle's medium (DMEM) (Cellgro). Capan-2, T3M4, AsPC-1, BxPC-3, and SU.86.86 cells were maintained in RPMI 1640 (HyClone). HPAF-II, Hs766T, BHK-21, A549, and HeLa cells were maintained in modified Eagle's medium (MEM) (Cellgro). All cell lines were supplemented with 9% fetal bovine serum (Gibco). For all experiments, PDA cell lines were passaged no more than 10 times.
Viruses.
The following viruses were used in this study: recombinant wild-type VSV (Indiana serotype) (VSV-wt) (42), VSV-p1-GFP, VSV-ΔM51-GFP (p5), CRAd-dl1520 (ONYX-015), CRAd-hTERT (Adv-TERTp-E1A), SeV-GFP, and RSV-GFP. VSV-p1-GFP has the GFP ORF inserted at position 1 of the viral genome (73). VSV-ΔM51-GFP has a deletion at amino acid position 51 of the M protein, as well as the GFP ORF inserted in position 5 of the viral genome (73). Both attenuated VSV recombinants have been shown to retain their oncolytic activity while lacking neurotoxicity in vivo (2, 73). CRAd-dl1520 is attenuated by deletion of a large part of the coding sequence for the E1b55k viral gene product and selectively replicates in and kills cancer cells (8, 12). CRAd-hTERT is a human telomerase reverse transcriptase (hTERT) promoter-dependent CRAd that selectively replicates in and kill cells with active hTERT (85 to 90% of tumor cells) (31). SeV-GFP (SeV-GFP-Fmut) has the GFP ORF at position 1 of the viral genome and a mutation in the cleavage site of the fusion (F) protein, allowing F activation and production of infectious virus particles in cells without acetylated trypsin in the medium through a ubiquitous furin-like protease (72). RSV-GFP has the GFP ORF at position 1 of the viral genome (26). All VSV variants were grown in BHK-21 cells, SeV-GFP was grown in Vero cells, CRAds were grown in HeLa cells, and RSV-GFP was grown in Hep-2 cells. For animal experiments, VSV-ΔM51-GFP was dialyzed (Slide-A-Lyzer; Pierce) in 2 liters of chilled dialysis buffer (25 mM Tris, pH 7.4, 140 mM NaCl, 5 mM KCl, 0.6 mM Na2HPO4, 0.5 mM MgCl2, 0.9 mM CaCl2, and 5% [wt/vol] sucrose) for 2 h at 4°C and then for 4 h at 4°C in fresh dialysis buffer. CRAd-dl1520 was dialyzed in 10 mM Tris, pH 8, 135 mM NaCl, 1 mM MgCl2, and 50% (vol/vol) glycerol three times for 1 h each time at 4°C. The dialyzed viruses were tested for infectivity on A549 cells.
Cell viability assay.
Cells were seeded in 96-well plates so that they reached 80% confluence at 24 h and then virus infected at a multiplicity of infection (MOI) of 1 or 0.01 CIU (cell infectious units) per cell (based on VSV titration on 4T1 cells) or mock infected in MegaVir HyQSFM4 serum-free medium (SFM) (HyClone). One hour postinfection (p.i.), the virus was aspirated and the cells were incubated in growth medium containing 5% fetal bovine serum (FBS). Cell viability was analyzed at 5 days p.i. by a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) cell viability assay (Biotium). To determine the kinetics of virus-associated cytopathogenicity, cells were seeded in 96-well plates so that they reached 50% confluence at 24 h. The cells were then mock infected or infected with VSV-ΔM51-GFP at a low (0.001 CIU/cell), intermediate (0.1 CIU/cell), or high (1 CIU/cell) MOI. At 1 h p.i., the virus was aspirated and the cells were overlaid with growth medium containing 5% FBS. MTT cell viability assays were performed at 1, 16, 24, 48, and 72 h p.i.
Permissiveness of cells to virus infection.
Cells were incubated with serial dilutions of VSV-wt, VSV-p1-GFP, VSV-ΔM51-GFP, SeV-GFP, CRAd-dl1520, or CRAd-hTERT in SFM for 1 h. At 1 h p.i., the virus was aspirated and growth medium containing 5% FBS was added to each well. The infectious foci of VSV-ΔM51-GFP, VSV-p1-GFP, and SeV-GFP were analyzed by fluorescence microscopy at 24 and 48 h p.i. The infectious foci of CRAd-dl1520 and CRAd-hTERT were analyzed by immunocytochemistry (ICC) at 5 days p.i. Briefly, cells were washed with phosphate-buffered saline (PBS) and fixed in 3% paraformaldehyde (PFA) (Sigma) for 10 min, followed by permeabilization for 2 min on ice with a solution containing 20 mM HEPES (pH 7.5), 300 mM sucrose, 50 mM NaCl, 3 mM MgCl2, and 0.5% Triton X-100. The cells were then blocked with 5% bovine serum albumin (BSA) (Sigma) in PBS for 20 min and incubated with anti-adenovirus hexon primary antibodies (1:600; U.S. Biologicals; catalog number A0880-14) for 1.5 h. The cells were washed, incubated with peroxidase-conjugated goat anti-mouse IgG antibodies (1:300; Jackson ImmunoResearch) for 1.5 h, and detected by addition of the peroxidase substrate 3,3′-diaminobenzidine tetrahydrochloride hydrate (DAB) (Amresco). The infectious foci of VSV-wt were also analyzed by ICC as described above but using 1:100 rabbit polyclonal anti-VSV antibodies (raised against VSV virions) and anti-rabbit secondary antibodies. The cells were infected with serial dilutions of VSV-wt in triplicate, and infectious foci were analyzed by ICC at 48 h p.i.
One-step virus growth kinetics.
Selected PDA cells were seeded in 96-well plates to reach confluence at 24 h. They were infected in duplicate with VSV-wt, VSV-ΔM51-GFP, or VSV-p1-GFP at an MOI of 10 CIU/cell based on the reference cell line 4T1. At 1 h p.i., the virus was aspirated, and the cells were washed twice with PBS (to prevent carryover of virions) and overlaid with growth medium containing 5% FBS. At 1, 24, 50, and 72 h p.i., supernatant was collected from the wells and flash frozen at −80°C. Virus titers were later determined by plaque assay analysis. Briefly, BHK-21 cells were incubated with serial dilutions of the samples for 1 h. The virus was aspirated, and the cells were overlaid with an SFM-2% Bacto Agar mixture to limit virus spread. Infectious foci were counted by light and fluorescence microscopy at 16 h p.i.
Type I interferon sensitivity and production.
Cells were seeded in 96-well plates so that they reached 80% confluence at 24 h. For type I interferon sensitivity, the cells were treated with 5,000 U/ml alpha interferon (IFN-α) (Calbiochem; catalog number 407294) in SFM or with SFM only. Twenty-four hours posttreatment, the cells were infected with serial dilutions of VSV-ΔM51-GFP, and infectious foci were analyzed 16 h p.i. by fluorescence microscopy. Treatments and infections were performed in duplicate. For type I interferon production, cells were infected with VSV-ΔM51-GFP at an MOI of 10 CIU/cell or mock treated with SFM only. One hour p.i., the virus was aspirated and the cells were incubated in SFM. Eighteen hours p.i., supernatant was harvested and analyzed by enzyme-linked immunosorbent assay (ELISA) for production of human IFN-β (PBL; catalog number 41410-1) or human IFN-α (multisubtype; PBL; catalog number 41105-1) according to the manufacturer's instructions (PBL InterferonSource). Infections were performed in triplicate.
Western blotting.
Cellular lysates were prepared by mock infecting cells or infecting them with VSV-ΔM51-GFP at an MOI of 1 or 10 CIU/cell. One hour p.i., the virus was aspirated, and the cells were extensively washed and incubated in growth medium containing 5% FBS. Cells were harvested at 16 h p.i. and lysed in lysis buffer containing 1% Triton X-100, 20 mM HEPES, 0.15 M NaCl, 2 mM EDTA and supplemented with protease inhibitor cocktail (2×; Roche). The total protein concentration was determined by Bradford assay. Three micrograms (for VSV detection) or 30 μg (for GFP detection) of total protein was separated by electrophoresis on 10% or 12% SDS-PAGE gels, respectively, and electroblotted to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked using 5% nonfat powdered milk in TBS-T (0.5 M NaCl, 20 mM Tris [pH 7.5], 0.1%Tween 20), which was also used for antibody dilutions. The membranes were incubated with 1:10,000 rabbit polyclonal anti-VSV antibodies (raised against VSV virions) or 1:3,000 mouse anti-GFP clone 9F9.F9 (Rockland). Detection was done with 1:5,000 goat anti-rabbit or 1:5,000 goat anti-mouse horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) using the Enhanced Chemiluminescence Plus (ECL+) protein detection system (GE Healthcare). The membranes were reprobed with mouse anti-actin clone C4 (50) to verify sample loading. Image capture and densitometry analyses were performed using VisionWorksLS v6.8 software (UVP).
Northern blotting.
The pVSVFL(+)g.1 plasmid, which carries a complete cDNA copy of the VSV (Indiana strain) antigenome (42), was used as a template for addition of a SP6 promoter to the 3′ end of a 279-bp fragment of N by PCR using the following primers: 5′-ATCCAGTGGAATACCCGGCAGATT-3′ and 5′-ATTTAGGTGACACTATAGAAGTGCTCGTCAGATTCAAGCTCAGGCTG-3′. A probe for detection of N mRNA and VSV antigenomic RNA was synthesized from the PCR product by in vitro transcription in the presence of [32P]UTP using the Maxiscript T7 kit (Ambion). Cells were mock treated or treated with 100 μg/ml cycloheximide for 30 min prior to mock infection or infection with VSV-ΔM51-GFP at an MOI of 10, and treatment was continued with cycloheximide. At 4 h p.i., cells were collected and total RNA was extracted using the Quick-RNA MiniPrep kit (Zymo Research). For each sample, 1 μg of RNA was separated on a 1.2% agarose-formaldehyde gel containing ethidium bromide for confirmation of RNA loading by visualization of rRNA. The RNA was transferred to a nylon membrane and incubated with probe overnight at 58°C. Bands were detected using a phosphorimager and quantitated using Image Quant 5.2 (Molecular Dynamics).
Surface expression of CAR.
Single-cell suspensions were obtained by detaching cells using cell scrapers without trypsin to rule out a potential proteolytic effect of trypsin on surface proteins. The cells then were incubated with Fc block at a concentration of 0.5 μg/ml at room temperature for 30 min. The cells were stained for cellular coxsackievirus and adenovirus receptor (CAR) using anti-CAR antibody (clone RmCB; Millipore) for 30 min (or mock treated), washed, and subsequently stained with secondary antibody, goat anti-mouse IgG-fluorescein isothiocyanate (FITC) (Santa Cruz; 0.5 μg/ml) for 30 min. Expression of CAR was determined by flow cytometry (Beckman Coulter). Analysis was conducted using FlowJo (Treestar, Ashland, OR).
Animal experiments.
Mice were handled and maintained under veterinary supervision in accordance with institutional guidelines and under a University of North Carolina at Charlotte Institutional Animal Care and Use Committee (IACUC)-approved protocol. Six- to 8-week-old male athymic nude mice (Hsd:Athymic Nude-Foxn1nu; Harlan Laboratories, Inc., Fredrick MD) were subcutaneously injected with 1 of 4 human PDA cell lines. All cell lines used in animal experiments tested negative for an extended panel of pathogens (MIA PaCa2, SU.86.86, and Panc-1 were tested by Charles River Laboratories, and HPAF-II was tested by Bioreliance). Based on the preceding titration experiments (data not shown), mice were injected with 5 × 106 MIA PaCa2, 5 × 106 Panc-1, 3 × 106 HPAF-II, or 3 × 106 SU.86.86 cells (in 100 μl of PBS) in the right flank (n = 18 per group). Two additional untreated age-matched mice were used in the experiment to compare body weights with those of the treated experimental mice. The mice were palpated starting at 9 days after tumor injection. Tumors were established by day 13, and the mice were randomly divided into 3 groups (n = 6 per group). One group served as a control and received one intratumoral (i.t.) administration of 50 μl PBS only. The other two groups were administered VSV-ΔM51-GFP or CRAd-dl1520 i.t. once at a dose of 5 × 107 CIU in 50 μl PBS. The dose was determined based on CIU established on A549 cells for both viruses. Tumor size was monitored by caliper measurements every other day, and body weight was measured once weekly. Tumor weight was calculated according to the following formula: grams = [(length in cm) × (width in cm)2]/2. Upon sacrifice, tumor and brain tissues were harvested and tested for the presence of VSV-ΔM51-GFP.
RESULTS
Susceptibility of PDA cell lines to viral oncolysis.
The susceptibility of human PDA cells to virus-mediated oncolysis was tested in a panel of 13 clinically relevant PDA cell lines derived from primary PDA tumors or PDA metastasis to the liver or lymph nodes. In addition to the PDA cell line, the immortal HPDE cell line, which retains a genotype similar to that of pancreatic duct epithelium and is nontumorigenic in nude mice (21), was employed as a “benign” control cell line to determine virus specificity for PDA cells. In addition to VSV-wt, we tested two VSV variants, VSV-ΔM51-GFP and VSV-p1-GFP (Fig. 1), with a particular focus on VSV-ΔM51-GFP (73). Several previous studies showed that VSV mutants with a deletion or mutation of methionine at position 51 (ΔM51) of the M protein exhibited good oncolytic potential but lacked undesirable neurotoxicity (2, 13, 25, 67, 73, 75). A similar phenotype was recently demonstrated for VSV-p1-GFP (73). To evaluate the relative efficacy of VSV as an OV, we compared VSV variants to four other viruses: SeV-GFP, RSV-GFP, CRAd-dl1520, and CRAd-hTERT (Fig. 1). SeV-GFP and RSV-GFP are also NNS RNA viruses shown to have oncolytic potential (14, 15, 38–40, 76), while CRAds have shown some success in several PDA cell lines in vitro and in vivo (28, 33, 41), although they have not been tested in most of the PDA cell lines used in this study. The inclusion of additional viruses would also help to discriminate between virus-specific and general resistance phenotypes if any PDA cell lines were identified as nonpermissive to VSV.
Fig 1.

Viruses used in this study. VSV-p1-GFP has the GFP ORF inserted in position 1 of the viral genome, resulting in attenuation of the virus. VSV-ΔM51-GFP has a deletion at amino acid position 51 of the M protein, reducing its ability to suppress host immunity. In addition, VSV-ΔM51-GFP has the GFP ORF inserted in position 5 of the viral genome. SeV-GFP has the GFP ORF inserted at position 1 of the viral genome and a mutation in the cleavage site of the F protein, allowing F activation and production of infectious virus particles in cells without trypsin addition. RSV-GFP has the GFP ORF inserted at position 1 of the viral genome. CRAd-dl1520 is attenuated by deletion of a large part of the coding sequence for the E1b55k viral gene product. CRAd-hTERT is an hTERT-dependent CRAd.
To analyze the ability of viruses to kill cancer cells, PDA cell lines were infected at either a low MOI (0.01 CIU/cell) or a higher MOI (1.0 CIU/cell), and at 5 days p.i., an MTT cell viability assay was performed. The MOI values for each virus-cell line combination are relative and were calculated based on titration of all VSV variants and SeV on 4T1 cells and of RSV and CRAds on HeLa cells. These two reference cell lines (4T1 and HeLa) were selected based on their abilities to support robust replication of the viruses used in this study. Therefore, for each MOI, the same amount of virus stock was added to each cell line. VSV-wt, VSV-ΔM51-GFP, and VSV-p1-GFP all caused significant death in the majority of cell lines at both high (Fig. 2A) and low (Fig. 2B) MOIs compared to mock-infected cells. In general, at the higher MOI, VSVs and CRAds caused more significant cell death than SeV-GFP and RSV-GFP (Fig. 2A). At the lower MOI, VSVs caused more significant cell death than all other viruses, including CRAds (Fig. 2B).
Fig 2.

PDA cell viability following infection with viruses. PDA cell lines and HPDE cells were seeded in 96-well plates so that they reached 80% confluence at 24 h. The cells were infected with the indicated viruses at an MOI of of 1 (A) or 0.01 (B) CIU/cell or mock infected. Cell viability was analyzed at 5 days p.i. by an MTT cell viability assay and is expressed as a ratio of virus-treated to mock-treated cells for each time point. All MTT assays were done in triplicate, and the data represent the means and standard deviations. Cell lines are grouped arbitrarily based on their susceptibilities to virus-induced oncolysis.
Several PDA cell lines showed various degrees of resistance to oncolysis by VSVs, with HPAF-II, Hs766T, and BxPC-3 displaying the strongest resistance. Interestingly, we observed a substantial difference in the susceptibilities of HPAF-II, Hs766T, and benign HPDE cells to oncolysis with different VSV variants. These cell lines were effectively killed by VSV-wt (both MOIs) and VSV-p1-GFP (HPAF-II at the high MOI only) at 5 days p.i. but were resistant to VSV-ΔM51-GFP, even at an MOI of 1. Importantly, all three PDA cell lines were also among the most resistant to other tested viruses, suggesting that general antiviral mechanisms may contribute to their phenotype (see below).
To analyze the kinetics of PDA cell death following VSV-ΔM51-GFP (Fig. 3) or VSV-wt (data not shown) infection, cells were infected at an MOI of 0.001, 0.1, or 1 CIU/cell (Fig. 3), and cell viability was analyzed at different time points. The majority of cell lines had significantly decreased viability after infection with VSV-ΔM51-GFP at any tested MOI. Consistent with the data presented in Fig. 2, HPAF-II, Hs766T, and BxPC-3 cells were most resistant to VSV-mediated cell death in the presence of any amount of VSV-ΔM51-GFP. CFPAC-1, HPAC, and benign HPDE cells were resistant to VSV-ΔM51-GFP only when infected at the lowest MOI (0.001).
Fig 3.

Kinetics of cytopathogenicity of VSV-ΔM51-GFP in PDA cells. Cells were seeded in 96-well plates so that they reached 50% confluence at 24 h. The cells were then mock infected or virus infected at low (0.001 CIU/cell), intermediate (0.1 CIU/cell), or high (1 CIU/cell) MOI. MTT cell viability assays were performed at 1, 16, 24, 48, and 72 h p.i. Cell viability is expressed as the percentage of mock-infected cells at 1 h p.i. All MTT assays were done in triplicate, and the data represent means and standard deviations.
Permissiveness of PDA cell lines to viral infection.
The failure of OVs to kill cancer cells can be explained by their inability to infect and/or replicate in these cells, although cellular defects in apoptosis may also be responsible for the defect in virus-mediated oncolysis. To determine whether variations in viral oncolysis observed between different PDA cell lines were due to different levels of permissiveness of these cell lines to virus infection, monolayer cultures of PDA cells were infected with serial virus dilutions. To test whether the differences between cell line levels of permissiveness to virus infection were specific for VSVs or general (e.g., if they have intact antiviral responses), we examined all viruses (Fig. 1) except RSV. The infectious foci of VSV-ΔM51-GFP, VSV-p1-GFP, and SeV-GFP were analyzed by fluorescence microscopy at 24 (VSV) or 48 (SeV) h p.i. The number and size of viral plaques produced by VSV-wt, CRAd-dl1520, and CRAd-hTERT were analyzed by ICC as described in Materials and Methods. The virus permissiveness shown in Fig. 4 is expressed as the ratio of the virus titer on the pancreatic cell line under study to the titer on a reference cell line (4T1 or HeLa), and higher numbers indicate greater permissiveness.
Fig 4.

Permissiveness of PDA cell lines to different viruses. PDA cell lines and HPDE cells were incubated with serial dilutions of viruses. The infectious foci of VSV-ΔM51-GFP, VSV-p1-GFP (24 h p.i.), and SeV-GFP (48 h p.i.) were analyzed by fluorescence microscopy. The infectious foci of VSV-wt, CRAd-dl1520, and CRAd-hTERT were analyzed by ICC as described in Materials and Methods. Virus permissiveness (relative yield) is expressed as the log10 of the ratio of the virus titer on the pancreatic cell line under study to the titer on a reference cell line (4T1 for VSV and SeV; HeLa for CRAds). The following titers were observed on reference cell lines: VSV-wt, 1.6 × 109 CIU/ml on 4T1; VSV-ΔM51-GFP, 3.3 × 108 CIU/ml on 4T1; VSV-p1-GFP, 3 × 107 CIU/ml on 4T1; SeV-GFP, 1.5 × 107 CIU/ml on 4T1; CRAd-hTERT, 1.5 × 107 CIU/ml on HeLa; and CRAd-dl1520, 4 × 108 CIU/ml on HeLa. A relative yield of 0 indicates that the PDA cell line and a reference cell line are equally permissive to the virus, while higher numbers indicate greater permissiveness for the PDA cell line. The area of infectious foci was analyzed using Image J software (NIH): small, area of <10 (surface area units); medium, area of 10 to 30; large, area of >30; nd, not done.
The degree of curvature in Fig. 4 indicates that the adenoviruses have less variability among PDA cells than VSV and SeV. Interestingly, while BxPC-3 and Hs766T were resistant to all tested viruses, HPAF-II showed an intermediate permissiveness to infection by both adenoviruses (Fig. 4 and 5 [for CRAd-dl1520]), although this PDA cell line was resistant to virus-mediated oncolysis by either CRAd (Fig. 2). As shown in Fig. 4, the majority of cell lines were highly permissive to VSV-ΔM51-GFP infection, with a relative ratio greater than or close to 1 (log10 = 0) (AsPC-1, SU.86.86, Capan-1, Panc-1, MIA PaCa2, Suit2, and Capan-2). In these cell lines, we observed rapid spread of VSV-ΔM51-GFP forming large infectious foci (filled circles in Fig. 4; large GFP foci in Fig. 5). Cell lines that were less permissive to VSV-ΔM51-GFP infection include benign HPDE cells (6.6 times less, with very small foci), as well as T3M4 (2.2 times less than 4T1), CFPAC-1 (3.8 times less), and HPAC (10 times less), all of which also formed smaller infectious foci at 16 h p.i. BxPC-3, HPAF-II, and Hs766T cells appeared highly resistant to VSV-ΔM51-GFP infection, with relative susceptibilities much less than that of 4T1 (62, 971, and 25,385 times less, respectively), and infectious foci were much smaller than those of all the other cell lines tested (Fig. 5). VSV-ΔM51-GFP was also analyzed at 5 days p.i., when the majority of cell lines highly permissive to VSV-ΔM51-GFP infection were no longer viable and had detached from the culture plastic. However, HPAF-II, BxPC-3, and Hs766T cells remained attached to the plastic with decreased GFP intensity, again indicating that VSV-ΔM51-GFP infection is restricted in these cell lines (data not shown).
Fig 5.

Permissiveness of selected PDA cell lines to virus infection. Representative PDA cell lines (not all are shown) were incubated with serial dilutions of VSV-ΔM51-GFP or CRAd-dl1520. The infectious foci of VSV-ΔM51-GFP were analyzed by fluorescence microscopy at 24 h p.i. The infectious foci of CRAd-dl1520 were analyzed by ICC at 5 days p.i, as described in Materials and Methods.
SU.86.86 showed a very intriguing phenotype, being highly permissive to VSVs and SeV but resistant to both CRAd-dl1520 and CRAd-hTERT. To test whether this cell line may lack CAR, required for adenovirus attachment (which would explain the phenotype) (41), we analyzed all PDA cell lines for CAR expression by flow cytometry and found that SU86.86 was the only cell line completely lacking CAR (Fig. 6), while all other cell lines (including HPAF-II, Hs766T, and BXPC-3, displaying a general resistance phenotype) had varying but detectable levels of CAR (data not shown), which is in agreement with our data (Fig. 4 and 5), indicating that these cell lines (unlike SU86.86) have reasonably good susceptibility to both adenoviruses and also suggesting that they are not defective in CAR expression. Although other factors may also contribute to the resistance of SU86.86 to CRAds, the lack of CAR expression alone might be sufficient to cause the phenotype.
Fig 6.

Surface expression of CAR. Single-cell suspensions of HeLa (positive control) or SU.86.86 (obtained without trypsin) cells were analyzed for adenovirus CAR using anti-CAR antibody and secondary IgG-FITC antibody (solid lines) or secondary IgG-FITC antibody only (gray areas). Expression of CAR was determined by flow cytometry (Beckman Coulter) and analyzed using FlowJo (Treestar, Ashland, OR) as described in Materials and Methods.
To examine if reduced permissiveness to VSV-ΔM51-GFP also resulted in a decrease in new viral protein synthesis, lysates were prepared from uninfected cells and from cells infected with VSV-ΔM51-GFP at MOIs of 1 and 10 CIU/cell and harvested at 16 h p.i. Equal amounts of total protein were then examined by Western blotting for both VSV proteins and GFP expression. The expression levels of viral proteins within the different cell lines were in agreement with GFP expression (Fig. 7). Protein expression (GFP level measurements are shown in Fig. 7) was also generally consistent with cell line permissiveness and oncolysis, especially when protein accumulation levels were compared after lower-MOI infection. Viral protein expression was strongly reduced in BxPC-3, HPAF-II, Hs766T, and benign HPDE cells, which are the most “nonpermissive,” and all demonstrated small focus sizes when infected with VSV-ΔM51-GFP (Fig. 4). Viral protein expression was also reduced in CFPAC-1 and HPAC cells, which had reduced permissiveness and medium focus sizes.
Fig 7.

Analysis of viral protein accumulation in cells at 16 h p.i. Cells were mock infected or infected with VSV-ΔM51-GFP at an MOI of 1 or 10 CIU/cell. The cells were harvested at 16 h p.i., and the cell lysates were analyzed by Western blotting for VSV proteins, GFP, or actin.
To directly examine the growth potential of VSVs in resistant cell lines, we tested all 3 VSVs in the majority of PDA cell lines (and in benign HPDE cells) using a standard one-step growth kinetics assay (Fig. 8). In general, our data show that, while all tested cell lines were able to support productive replication of VSVs, the lowest production was observed in benign HPDE cells and in most PDA cell lines displaying a resistant phenotype. Also, most cells showed very similar growth kinetics for all 3 viruses, while HPAF-II supported a significantly lower level of VSV-ΔM51-GFP production than other VSVs. This result may explain, at least partially, why HPAF-II cells were particularly resistant to VSV-ΔM51-GFP (Fig. 2A). BxPC-3 cells showed a surprisingly high level of new particle production when infected at an MOI of 10. However, it is important to note that an MOI of 10, used for one-step growth kinetics, is never attainable during oncolytic treatment in vivo. The experiments on virus-mediated cell death shown in Fig. 2 and 3 were conducted at more realistic MOIs of 0.001 to 1 (maximum).
Fig 8.

One-step growth kinetics of VSVs in PDA cell lines. PDA cells were infected with VSV-wt, VSV-ΔM51-GFP, or VSV-p1-GFP at an MOI of 10 CIU/cell, which was calculated based on the reference cell line 4T1. At 1 h p.i., the virus was aspirated and the cells were washed and overlaid with 5% growth medium. At 1, 24, 50, and 72 h p.i., the supernatant was collected, and virus titers were determined by plaque assay on BHK-21 cells. All infections were done in duplicate, and the data represent means ± standard deviations.
Timing and cellular factors of resistance of PDA cell lines to VSV-ΔM51-GFP.
To analyze why PDA cells differ in their permissiveness to VSV-ΔM51-GFP, we looked at the early stages of virus infection and at cellular characteristics that could explain the observed differences.
Antigenome and VSV N mRNA synthesis was determined by Northern blotting of total RNA isolated at 4 h p.i. from cells that were untreated or treated with cycloheximide and infected with VSV-ΔM51-GFP at an MOI of 10 (Fig. 9 and Table 1). Cycloheximide blocks new protein synthesis and thereby viral genome synthesis and secondary transcription. Expression of both VSV N mRNA and antigenomic RNA was strongly reduced in BxPC-3, HPAC, HPAF-II, and Hs766T cells and somewhat reduced in CFPAC-1 cells, consistent with the reduced viral protein synthesis and permissiveness to VSV-ΔM51-GFP infection seen in these cell lines. Interestingly, RNA synthesis in benign HPDE cells was quite robust despite low protein synthesis 16 h p.i. (Fig. 7) and reduced permissiveness in the cell line, suggesting a block at a later stage of viral infection. In all cases where secondary transcription was reduced, primary transcription was reduced proportionately (Table 1). This suggests that in cell lines with lower mRNA synthesis, viral genome release into the cytoplasm was inhibited, and that for genomes that were released, early infection proceeded normally.
Fig 9.

Early viral RNA levels in infected cells. Cells were mock treated or treated with 100 μg/ml cycloheximide (CHX) for 30 min prior to mock infection or infection with VSV-ΔM51-GFP at an MOI of 10, and treatment was continued with CHX. At 4 h p.i., cells were collected, and total RNA was extracted and analyzed by Northern blotting for VSV antigenome RNA (top) or N mRNA (bottom).
Table 1.
Early viral RNA synthesis in cells infected with VSV-ΔM51-GFP
| Cell line | Primary TXNa | Total TXNb | Total TXN/primary TXN | Antigenome RNAc | Total TXN/antigenome RNA |
|---|---|---|---|---|---|
| AsPC-1 | 668 | 29,232 | 44 | 1,276 | 23 |
| BxPC-3 | 37 | 1,517 | 41 | 40 | 38 |
| CFPAC | 172 | 10,911 | 63 | 230 | 47 |
| Capan-1 | 1,728 | 31,394 | 18 | 2,205d | 14 |
| Capan-2 | 249 | 14,807 | 59 | 1,401d | 11 |
| HPAC | 56 | 1,549 | 28 | 60d | 26 |
| HPAF-II | 27 | 899 | 34 | 18d | 49 |
| Hs766T | 181 | 3,891 | 22 | 86 | 45 |
| MIA PaCa2 | 1,520 | 31,434 | 21 | 1,857 | 17 |
| Panc-1 | 1,126 | 34,698 | 31 | 1,401 | 25 |
| Su.86.86 | 5,162 | 46,195 | 9 | 1,428 | 32 |
| Suit2 | 854d | 41,203d | 48 | 2,988d | 14 |
| T3M4 | 378 | 14,363 | 38 | 1,051 | 14 |
| HPDE | 1,682 | 32,759 | 19 | 1,803 | 18 |
VSV N mRNA transcription (TXN) level 4 h p.i. in the presence of cycloheximide.
VSV N mRNA transcription level 4 h p.i. in the absence of cycloheximide.
VSV antigenome RNA synthesis level 4 h p.i. in the absence of cycloheximide.
The values are for RNA bands detected using a phosphorimager and quantitated using Image Quant software and are the averages of two independent repeats, except as indicated.
VSV is sensitive to type I IFN responses. However, many different tumor types are known to lack these responses, allowing VSV to productively infect cancer cells while sparing healthy cells (7, 44). Here, we wanted to test the hypothesis that the resistance of some PDA cell lines to VSV (and other viruses) was a result of their intact IFN responses. To determine if PDA cell lines were sensitive to type I IFN, all cells were mock treated or treated with 5,000 U/ml IFN-α for 24 h prior to infection with serial dilutions of VSV-ΔM51-GFP. A ratio of mock-treated to IFN-α-treated cell titers was determined for each PDA cell line (Fig. 10). We observed that certain cell lines did not significantly suppress VSV-ΔM51-GFP infection in response to IFN-α. VSV-ΔM51-GFP titers were no more than 26-fold reduced following IFN treatment in Panc-1, SU.86.86, MIA PaCa2, and HPAC cells, while Capan-2, Hs766T, T3M4, and benign HPDE cells showed an intermediate sensitivity to IFN-α. HPAC cells displayed an interesting phenotype, with comparable titers with or without IFN treatment; however, IFN-treated HPAC cells required an additional day for visible foci to appear. Surprisingly, several PDA cancer cell lines (Capan-1, AsPC-1, HPAF-II, BxPC-3, Suit2, and CFPAC-1) were highly responsive to IFN-α. Among these IFN-sensitive cells are AsPC-1, Capan-1, and Suit2, which support robust infection by VSV-ΔM51-GFP in the absence of IFN-α pretreatment.
Fig 10.

Type I interferon sensitivities of PDA cell lines. PDA cell lines and HPDE cells were either treated with 5,000 U/ml IFN-α in SFM or mock treated with SFM only. Twenty-four hours posttreatment, the cells were infected with serial dilutions of VSV-ΔM51-GFP, and infectious foci were analyzed 16 h p.i. by fluorescence microscopy to calculate the virus titer under these conditions. Treatments and infections were performed in duplicate, and average values are shown. For HPAC cells pretreated with IFN-α, virus-driven GFP signal was delayed by 24 h p.i.
To further study the role of IFN in the resistance of PDA cells to VSV, we examined the abilities of PDA cell lines to produce IFN-α and/or IFN-β in response to VSV-ΔM51-GFP infection (MOI, 10 CIU per cell) at 18 h p.i. As expected, significant amounts of IFN-β were produced by benign HPDE cells, which are expected to retain normal antiviral responses (Fig. 11). Importantly, all three cell lines (HPAF-II, HPAC, and Hs766T) producing significant amounts of IFN-β at 18 h p.i. were among the most resistant cell lines (Fig. 11). As illustrated in Table 2, except for BxPC-3, all PDA cell lines highly resistant to VSV showed an HPDE-like phenotype characterized by both the production of IFN-β and sensitivity to IFN treatment. In addition, our data experimentally explain the phenotypes AsPC-1, Suit2, and Capan-1, which are sensitive to IFN but support robust virus infection without added IFN, as they all are defective in IFN production. Interestingly, we were unable to detect any significant production of IFN-α in response to virus infection by any tested cell line at 18 h p.i. (data not shown); however, it is produced later than IFN-β. Future experiments will analyze PDA cells for production of various IFNs at different time points after infection.
Fig 11.
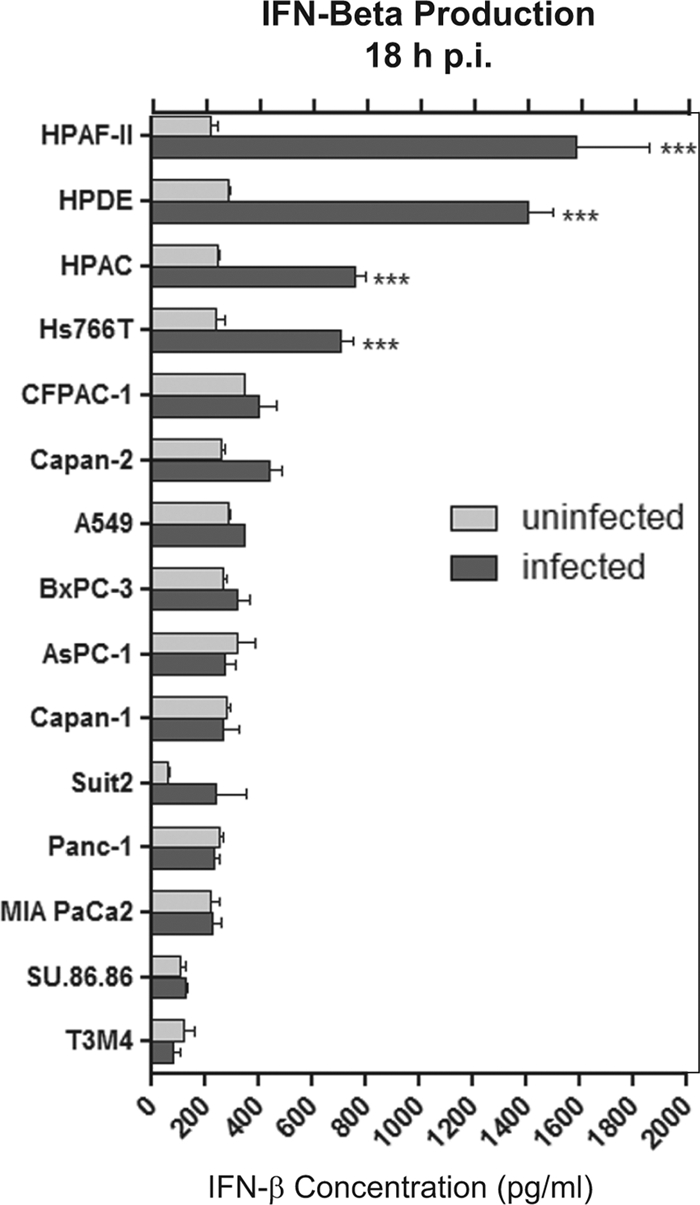
Type I interferon production by PDA cell lines. Cells were infected with VSV-ΔM51-GFP at an MOI of 10 CIU/cell or mock treated with SFM only. One hour p.i., the virus was aspirated and the supernatant was harvested and analyzed by ELISA for production of human IFN-β. Infections were performed in triplicate, and the data represent the means and standard deviations. Comparison of groups was done by using 2-way analysis of variance (ANOVA), followed by the Bonferroni posttest for multiple comparisons (***, P < 0.001).
Table 2.
Correlation between IFN sensitivity, production, and resistance of PDA cells to VSV
| Cell line | IFN sensitivity (24 h p.i.)a | IFN-β production (18 h p.i.)a | In vitro resistance to VSV-ΔM51-GFPa |
|---|---|---|---|
| AsPC-1 | +++ | − | − |
| Su.86.86 | − | − | − |
| Capan-1 | +++ | − | − |
| Panc-1 | − | − | − |
| MIA PaCa2 | − | − | − |
| Suit2 | ++ | − | − |
| Capan-2 | + | − | − |
| T3M4 | + | − | − |
| CFPAC-1 | ++ | − | ++ |
| HPDE | + | +++ | +++ |
| HPAC | − | +++ | ++ |
| BxPC-3 | +++ | − | +++ |
| HPAF-II | +++ | +++ | +++ |
| Hs766T | + | +++ | +++ |
+++, high levels of IFN sensitivity, IFN production, or virus resistance; ++, intermediate levels of IFN sensitivity, IFN production, or virus resistance; +, low levels of IFN sensitivity, IFN production, or virus resistance; −, no detectible levels of IFN sensitivity, IFN production, or virus resistance.
Together, our data show surprising diversity among PDA cells in regard to their ability to produce and respond to type I IFN. Moreover, we demonstrate that a combination of IFN sensitivity and IFN-β production may be used to predict the responsiveness of most PDA cells to oncolytic treatment.
Efficacy of VSV-ΔM51-GFP and CRAd-dl1520 in nude mice bearing human PDA tumors.
To test the efficacy of VSV-ΔM51-GFP in vivo and to determine the relevance of our in vitro results to an in vivo situation, we chose four cell lines for in vivo testing based on our in vitro virus permissiveness and oncolysis experiments. MIA PaCa2 and Panc-1 cells are highly permissive to both VSV-ΔM51-GFP and CRAd-dl1520, SU.86.86 is highly permissive to VSV-ΔM51-GFP but not CRAd-dl1520, and HPAF-II has limited permissiveness to both VSV-ΔM51-GFP and CRAd-dl1520 (Fig. 2 to 5). These human pancreatic cancer cell lines were injected subcutaneously into the right flank of male nude mice (n = 18 per cell line). Once the mice developed palpable tumors (5 to 7 mm) they were divided into three equal groups (n = 6). A control group received an i.t. injection of PBS, one group received an i.t. injection of 5 × 107 CIU VSV-ΔM51-GFP, and one group received an i.t. injection of 5 × 107 CIU CRAd-dl1520. The mice were monitored daily for signs of distress, and tumor size was measured every other day for 14 days. VSV-ΔM51-GFP and CRAd-dl1520 had the greatest therapeutic effect in mice bearing Panc-1 and MIA PaCa2 tumors (Fig. 12). VSV-ΔM51-GFP seemed to stabilize SU.86.86 tumor growth compared to treatment of SU.86.86 tumors with CRAd-dl1520 and PBS, which had no effect on tumor growth (Fig. 12). SU.86.86 grew more rapidly than all other cell lines in vivo, and several tumors became ulcerated over the course of the experiment (Fig. 12). While mice bearing SU.86.86 tumors showed no signs of distress at any point during the experiment, several were euthanized at an earlier time point due to large tumor size (day 21 instead of day 25). Tumor growth continued in the presence or absence of VSV-ΔM51-GFP and CRAd-dl1520 for mice bearing HPAF-II tumors (Fig. 12). In general, our in vivo experiments closely mimicked our in vitro results. Fourteen days after injection with VSV-ΔM51-GFP, CRAd-dl1520, or PBS, all mice were euthanized, the tumors were harvested, and wet weights and presence of virus were determined.
Fig 12.

Efficacy of VSV-ΔM51-GFP and CRAd-dl1520 in nude mice bearing human PDA tumors. Six- to 8-week-old male athymic nude mice were subcutaneously injected with MIA PaCa2, Panc-1, HPAF-II, or Su.86.86 cells in the right flank (n = 18 per group). Tumors were established by day 13, and the mice were randomly divided into 3 groups (n = 6 per group). One group served as a control and received one i.t. administration of 50 μl PBS only. The other two groups were treated i.t. once with VSV-ΔM51-GFP or CRAd-dl1520 at a dose of 5 × 107 CIU in 50 μl PBS. Tumor size was monitored by caliper measurements, and tumor weight was calculated according to the following formula: grams = (length in centimeters × width2)/2. Comparison of groups was done by using 2-way ANOVA, followed by the Bonferroni posttest for multiple comparisons (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
It has been demonstrated that VSV-wt can cause encephalitis in mice; however, VSV-ΔM51-GFP is a nonneurotropic OV (73). In agreement with this, animals infected with VSV-ΔM51-GFP showed no signs of encephalitis or distress over the course of the experiment. Nevertheless, brain tissues of VSV-ΔM51-GFP-infected animals were analyzed for the presence of virus by standard plaque assay on BHK-21 cells, and no VSV-ΔM51-GFP was detected. Interestingly, despite the robust oncolytic effect achieved for animals bearing Panc-1 and MIA PaCa2 following i.t. infection with VSV-ΔM51-GFP, when a similar analysis was conducted on tumor samples, only two samples (one SU.86.86 and one MIA PaCa2 sample) had detectable VSV-ΔM51-GFP present at 14 days p.i. (data not shown).
DISCUSSION
In this study, for the first time, we evaluated VSV as an OV against pancreatic cancer cells. VSV variants showed oncolytic abilities superior to those of other viruses and were effective against the majority of the 13 tested human PDA cell lines. We also identified several cell lines highly resistant to oncolytic virotherapy by VSV and/or other tested viruses.
Among VSV variants, we focused primarily on VSV-ΔM51-GFP because several previous studies showed that VSV variants with a ΔM51 mutation were effective OVs with no neurotoxicity in animals (2, 13, 25, 67, 73, 75). To evaluate the relative efficacy of VSV as an OV, we initially compared VSV variants to four other viruses. We chose CRAd-dl1520 (also known as “ONYX-15”) as a relevant control for further in vitro and in vivo experiments, as this DNA virus is unrelated to VSV, has been tested in several clinical trials, and has shown some success in previous PDA studies (12, 35). It is important to point out that although our in vitro data suggest a possible use of CRAds for PDA treatment, any viable strategy for treatment of patients using CRAds remains to be determined due to some of their reported limitations, including their dependence on CAR expression in target cells and their quick elimination from the bloodstream by the liver and inactivation by binding to blood cells and other components of the immune system, as well as their limited spread throughout the tumor (41).
Our in vitro experiments indicated great variability in permissiveness of PDA cell lines to all viruses. Overall, VSV variants were the most effective, but even for VSVs, some cell lines, including HPAF-II, Hs766T, and CFPAC-1, were less effectively killed by VSV-ΔM51-GFP than by VSV-wt and VSV-p1-GFP. There are two major hypotheses explaining varying susceptibilities of PDA cell lines to oncolysis by a particular virus in vitro. First, PDA cells may differ in their susceptibility to virus infection and/or their ability to support virus replication. This may happen because PDA cells may lack key lack key cellular factors (e.g., receptors) required for successful virus infection or because resistant cells have intact antiviral responses preventing successful virus spread. Alternatively, some PDA cells may have defective apoptotic pathways, so that even if a virus can successfully infect and replicate in these cells, they are not efficiently killed by apoptosis.
The oncolytic potential of viruses is generally contingent on their ability to infect and replicate in the cells. In our study, PDA cell permissiveness to all viruses closely mirrored our cell death analysis, with several cell lines (HPAF-II, Hs766T, and BxPC-3) showing various degrees of resistance to all tested viruses. The six least permissive cell lines were all defective in cell killing for at least some of the MOIs tested. Five of these cell lines, BxPC-3, HPAF-II, HPAC, Hs766T, and CFPAC, showed low levels of early (4 h p.i.) viral RNA synthesis (including primary transcription of viral genomes) when infected with VSV-ΔM51-GFP compared to the more permissive cell lines, indicating a possible defect at a very early stage in infection, such as attachment, entry, or endosomal escape. Experiments are under way in these PDA cell lines to further define the affected steps in viral infection and the responsible cellular mechanisms. In contrast to VSV-resistant PDA cell lines, in benign HPDE cells (also resistant to VSV), early viral mRNA and genome synthesis equaled that found in many permissive cell lines, but viral protein synthesis at 16 h p.i and virion production were sharply reduced, suggesting a defect at later stages of viral infection. This phenotype is expected for benign cells with intact innate antiviral responses.
To address differences in permissiveness to VSV in PDA cell lines, we also looked at their abilities to produce and respond to type I IFN. In general, many tumor cells are defective in producing type I IFNs but may remain sensitive to type I IFN, which could be produced by infected benign cells that surround the tumor. Still other tumor cells may retain the ability to produce their own IFN (51, 66). Responsiveness of cancer cells to IFN could be an important factor in predicting their behavior in vivo, where VSV infection would induce IFN production in surrounding healthy tissues, thus limiting oncolytic potential toward cancer cells sensitive to IFN. Our data showed surprising diversity among PDA cells in regard to their ability to produce and respond to type I IFN (Table 2). With the exception of BxPC-3, all VSV-resistant PDA cell lines were characterized by both the production of IFN-β and sensitivity to IFN treatment. The same phenotype was shown by benign HPDE cells, which are expected to retain normal antiviral responses. The VSV-resistant phenotype of BxPC-3 in vitro (it is sensitive to IFN but does not produce IFN-β) could be due to an IFN-independent block of virus infection. Interestingly, we identified some PDA cell lines (AsPC-1, Suit2, and Capan-1) that are responsive to IFN but highly susceptible to infection in vitro (without added IFN), as they all are defective in IFN production. High heterogeneity in response to type I IFN has been reported in several other cancer types, including mesothelioma (60), melanomas (46, 74), lymphomas (68), bladder cancers (48), and renal cancers (57), and likely in other types (67). Our data suggest that a combination of IFN sensitivity and IFN-β production may be used to predict the responsiveness of most PDA cells to oncolytic treatment.
Together, our data suggest that VSV-resistant cell lines have more than one “defect” responsible for their virus-resistant phenotype. If their resistance was solely dependent on their intact IFN pathways, we would expect them to have a phenotype similar to that of benign HPDE cells. HPDE cells do not have any defects in early steps of VSV infection (demonstrated by “normal” RNA synthesis, including primary transcription of the viral genome at 4 h p.i.), but robust type I IFN responses inhibit subsequent virus replication, resulting in very low protein accumulation at 16 h p.i. However, unlike HPDE cells, all PDA cell lines highly resistant to VSV also showed defective early viral RNA synthesis, suggesting that they have some defects inhibiting early steps of VSV infection (e.g., attachment or entry).
Most of our data show a correlation between the permissibility of PDA cells to VSV infection and its oncolytic potential. However, if cells are successfully infected at a high MOI (one-step infection), they are able to successfully produce new viral particles. BxPC-3 cells showed surprisingly high production of new particles when infected at an MOI of 10. Interestingly, it is also the only one of the most resistant cell lines that did not produce significant amounts of IFN-β (Fig. 11 and Table 2). At the same time, BxPC-3 cells were characterized by deficient RNA synthesis at 4 h p.i., suggesting that BxPC-3 cells have some defects in virus attachment/internalization or another early step in VSV infection. It also showed a low level of viral (and GFP) protein synthesis when BxPC-3 cells were infected at a lower MOI of 1 (Fig. 7, compare AsPC1 and BxPC-3 at MOIs of 1 and 10). It is important to note that infection at an MOI of 10, used in Fig. 8 for one-step growth kinetics, is never attainable during oncolytic treatment in vivo. The experiments on virus-mediated cell death shown in Fig. 2 and 3 were conducted at more realistic MOIs between 0.001 and 1.
Previous studies have shown that many cancer cells are able to inhibit apoptosis to allow prolonged proliferation (27). As VSV has been shown to cause cell death by apoptosis via either the intrinsic or extrinsic pathway or both (10, 22, 23, 62), cell lines with decreased expression or activation of certain apoptotic proteins have the potential to limit/delay cell death following VSV infection. Furthermore, differences in oncolytic potential between VSV variants could be due to differences in their mechanisms of cell death induction. It has been demonstrated that VSV-wt induces apoptosis via the mitochondrial pathway due to wt M protein inhibition of gene expression, while VSV-ΔM51-GFP, with a mutant M protein, induces apoptosis primarily via the death receptor pathway (23). While we cannot fully address these possibilities at this point, our preliminary experiments show significant increases in caspase 3 cleavage following VSV-ΔM51-GFP infection in all cell lines except Hs766T and HPAC at 17 h p.i. (data not shown). More studies are needed to determine whether a reduced level of apoptotic response or the delayed induction of apoptosis in some of these cell lines plays a role in restricting VSV oncolysis. These defects could also (in addition to intact IFN pathways) explain why cell lines resistant to VSV are also resistant to other, unrelated viruses.
Based on our in vitro studies, we chose 4 cell lines with varying permissiveness to VSV-ΔM51-GFP and CRAd-dl1520 to determine if our in vitro studies are relevant in vivo. We observed in vitro that MIA PaCa2 and Panc-1 are highly permissive to both VSV-ΔM51-GFP and CRAd-dl1520, SU.86.86 is highly permissive to VSV-ΔM51-GFP but not CRAd-dl1520, and HPAF-II has limited permissiveness to both. The induced tumors in nude mice showed the same permissiveness pattern observed in vitro, indicating in vitro testing can be used to identify cancers resistant to a particular virus. It is important to emphasize that the ability of a virus to kill cancer cells in vitro, or even in vivo (in nude mice), does not guarantee its efficacy in cancer patients due to complex tumor microenvironments and compromised immune responses (9). However, our data clearly show that if cells are resistant to viral oncolysis in vitro, it is highly unlikely that they could be effectively eliminated in vivo, suggesting the importance of in vitro pretesting (when possible) in identifying virus-resistant cancers.
There are several important characteristics of VSV that in combination make it a more attractive candidate for PDA treatment than other tested viruses: (i) there are few, if any, restrictions to VSV attachment and entry, as it is believed not to be dependent on any host receptor in target cells; (ii) there is no preexisting immunity against VSV in humans; (iii) VSV is not considered a significant human pathogen, and several VSV mutants, including VSV-ΔM51-GFP and VSV-p1-GFP, are not neurotropic but retain their oncolytic activity; (iv) cellular uptake in many mammalian cell types occurs very rapidly, and there is no cell cycle dependency; (vi) our comparative analysis here demonstrated that VSV variants showed oncolytic abilities superior to those of other viruses, and some cell lines that exhibited resistance to other viruses were successfully killed by VSV.
There are several potential options for virus-resistant cancer cells. Prescreening cells against an array of different OVs could identify the best option for treating a particular tumor. For example, VSV-ΔM51-GFP is more suitable than CRAds for treating PDAs similar to SU.86.86 cells, which showed a complete lack of the CAR expression required for adenovirus attachment (data not shown). In the cases where cells are less permissive to VSV-ΔM51-GFP than VSV-wt or VSV-p1-GFP (HPAF-II and Hs766T), the use of VSV-p1-GFP might be a better option, especially because this virus is also nonneurotoxic in vivo. Combination therapies have also demonstrated some success. Virotherapy in combination with chemotherapy can enhance the oncolytic effect compared to either treatment alone (55). Treating tumors with more than one OV (combined virotherapy) could also potentially lead to enhanced oncolysis (43). Importantly, understanding the mechanisms and identifying potential biomarkers of resistance is critical for the development of prescreening approaches and individualized oncolytic virotherapy against PDA.
ACKNOWLEDGMENTS
We are grateful to the following laboratories for kindly providing reagents for this project: Jack Rose (Yale University) for VSV-ΔM51-GFP and VSV-p1-GFP viruses, Savio L. C. Woo (Mount Sinai Medical Center) for Ad5-hTERT-E1 (CRAd-hTERT) virus, Wolfgang Neubert (Max Planck Institute of Biochemistry) for SeV-GFP-Fmut (SeV-GFP) virus, Mark Peeples (Ohio State University College of Medicine) for RSV-GFP virus, David McConky (M. D. Anderson Cancer Center) for CFPAC-1 and Hs766T cells, Randall Kimple (UNC-Chapel Hill) for Capan-2 and T3M4 cells, Timothy Wang (Columbia University) for AsPC-1 cells, Andrei Ivanov (University of Rochester Medical School) for HPAF-II cells, Michael Hollingsworth (University of Nebraska Medical Center) for Suit2 cells, Emmanuel Zervos (Tampa General Hospital) for HPAC cells, and Sanjay Srivastava (Texas Tech University Health Sciences Center) for BxPC-3 cells. We thank Sue Moyer (University of Florida College of Medicine) for providing a range of reagents for this project and Eric Hastie, Nirav Shah, Amritha Kidiyoor, Carlos Molestina, Jennifer Curry, and Virat Patel for technical assistance.
Footnotes
Published ahead of print 11 January 2012
REFERENCES
- 1. Ahmed M, Cramer SD, Lyles DS. 2004. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology 330:34–49 [DOI] [PubMed] [Google Scholar]
- 2. Ahmed M, Marino TR, Puckett S, Kock ND, Lyles DS. 2008. Immune response in the absence of neurovirulence in mice infected with M protein mutant vesicular stomatitis virus. J. Virol. 82:9273–9277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ahmed M, Puckett S, Lyles DS. 2010. Susceptibility of breast cancer cells to an oncolytic matrix (M) protein mutant of vesicular stomatitis virus. Cancer Gene Ther. 17:883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Altomonte J, et al. 2009. Enhanced oncolytic potency of vesicular stomatitis virus through vector-mediated inhibition of NK and NKT cells. Cancer Gene Ther. 16:266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ausubel LJ, et al. 2011. Current good manufacturing practice production of an oncolytic recombinant vesicular stomatitis viral vector for cancer treatment. Hum. Gene Ther. 22:489–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barber GN. 2004. Vesicular stomatitis virus as an oncolytic vector. Viral Immunol. 17:516–527 [DOI] [PubMed] [Google Scholar]
- 7. Barber GN. 2005. VSV-tumor selective replication and protein translation. Oncogene 24:7710–7719 [DOI] [PubMed] [Google Scholar]
- 8. Bischoff JR, et al. 1996. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 274:373–376 [DOI] [PubMed] [Google Scholar]
- 9. Breitbach CJ, Reid T, Burke J, Bell JC, Kirn DH. 2010. Navigating the clinical development landscape for oncolytic viruses and other cancer therapeutics: no shortcuts on the road to approval. Cytokine Growth Factor Rev. 21:85–89 [DOI] [PubMed] [Google Scholar]
- 10. Cary ZD, Willingham MC, Lyles DS. 2011. Oncolytic vesicular stomatitis virus induces apoptosis in U87 glioblastoma cells by a type II death receptor mechanism and induces cell death and tumor clearance in vivo. J. Virol. 85:5708–5717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang G, et al. 2010. Enhanced oncolytic activity of vesicular stomatitis virus encoding SV5-F protein against prostate cancer. J. Urol. 183:1611–1618 [DOI] [PubMed] [Google Scholar]
- 12. Crompton AM, Kirn DH. 2007. From ONYX-015 to armed vaccinia viruses: the education and evolution of oncolytic virus development. Curr. Cancer Drug Targets 7:133–139 [DOI] [PubMed] [Google Scholar]
- 13. Ebert O, Harbaran S, Shinozaki K, Woo SL. 2005. Systemic therapy of experimental breast cancer metastases by mutant vesicular stomatitis virus in immune-competent mice. Cancer Gene Ther. 12:350–358 [DOI] [PubMed] [Google Scholar]
- 14. Echchgadda I, et al. 2011. Oncolytic targeting of androgen-sensitive prostate tumor by the respiratory syncytial virus (RSV): consequences of deficient interferon-dependent antiviral defense. BMC Cancer 28:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Echchgadda I, et al. 2009. Anticancer oncolytic activity of respiratory syncytial virus. Cancer Gene Ther. 16:923–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Edge RE, et al. 2008. A let-7 MicroRNA-sensitive vesicular stomatitis virus demonstrates tumor-specific replication. Mol. Ther. 16:1437–1443 [DOI] [PubMed] [Google Scholar]
- 17. Eisenberg DP, et al. 2010. Hyperthermia potentiates oncolytic herpes viral killing of pancreatic cancer through a heat shock protein pathway. Surgery 148:325–334 [DOI] [PubMed] [Google Scholar]
- 18. Etoh T, et al. 2003. Oncolytic viral therapy for human pancreatic cancer cells by reovirus. Clin. Cancer Res. 9:1218–1223 [PubMed] [Google Scholar]
- 19. Farrow B, Albo D, Berger DH. 2008. The role of the tumor microenvironment in the progression of pancreatic cancer. J. Surg. Res. 149:319–328 [DOI] [PubMed] [Google Scholar]
- 20. Fernandez M, Porosnicu M, Markovic D, Barber GN. 2002. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J. Virol. 76:895–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Furukawa T, et al. 1996. Long-term culture and immortalization of epithelial cells from normal adult human pancreatic ducts transfected by the E6E7 gene of human papilloma virus 16. Am. J. Pathol. 148:1763–1770 [PMC free article] [PubMed] [Google Scholar]
- 22. Gaddy DF, Lyles DS. 2007. Oncolytic vesicular stomatitis virus induces apoptosis via signaling through PKR, Fas, and Daxx. J. Virol. 81:2792–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gaddy DF, Lyles DS. 2005. Vesicular stomatitis viruses expressing wild-type or mutant M proteins activate apoptosis through distinct pathways. J. Virol. 79:4170–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Galivo F, et al. 2010. Interference of CD40L-mediated tumor immunotherapy by oncolytic VSV. Hum. Gene Ther. 21:439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goel A, et al. 2007. Radioiodide imaging and radiovirotherapy of multiple myeloma using VSV(Delta51)-NIS, an attenuated vesicular stomatitis virus encoding the sodium iodide symporter gene. Blood 110:2342–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hallak LK, Collins PL, Knudson W, Peeples ME. 2000. Iduronic acid-containing glycosaminoglycans on target cells are required for efficient respiratory syncytial virus infection. Virology 271:264–275 [DOI] [PubMed] [Google Scholar]
- 27. Hamacher R, Schmid RM, Saur D, Schneider G. 2008. Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol. Cancer 7:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. He X, et al. 2009. E1B-55kD-deleted oncolytic adenovirus armed with canstatin gene yields an enhanced anti-tumor efficacy on pancreatic cancer. Cancer Lett. 285:89–98 [DOI] [PubMed] [Google Scholar]
- 29. Himeno Y, et al. 2005. Efficacy of oncolytic reovirus against liver metastasis from pancreatic cancer in immunocompetent models. Int. J. Oncol. 27:901–906 [PubMed] [Google Scholar]
- 30. Hirano S, et al. 2009. Reovirus inhibits the peritoneal dissemination of pancreatic cancer cells in an immunocompetent animal model. Oncol. Rep. 21:1381–1384 [DOI] [PubMed] [Google Scholar]
- 31. Huang TG, Savontaus MJ, Shinozaki K, Sauter BV, Woo SL. 2003. Telomerase-dependent oncolytic adenovirus for cancer treatment. Gene Ther. 10:1241–1247 [DOI] [PubMed] [Google Scholar]
- 32. Huang TG, Ebert O, Shinozaki K, Garcia-Sastre A, Woo SL. 2003. Oncolysis of hepatic metastasis of colorectal cancer by recombinant vesicular stomatitis virus in immune-competent mice. Mol. Ther. 8:434–440 [DOI] [PubMed] [Google Scholar]
- 33. Huch M, José GAA, González JR, Alemany R, Fillat C. 2009. Urokinase-type plasminogen activator receptor transcriptionally controlled adenoviruses eradicate pancreatic tumors and liver metastasis in mouse models. Neoplasia 11:518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Iwamura T, Katsuki T, Ide K. 1987. Establishment and characterization of a human pancreatic cancer cell line (SUIT-2) producing carcinoembryonic antigen and carbohydrate antigen 19-9. Jpn. J. Cancer Res. 78:54–62 [PubMed] [Google Scholar]
- 35. Kasuya H, Takeda S, Nomoto S, Nakao A. 2005. The potential of oncolytic virus therapy for pancreatic cancer. Cancer Gene Ther. 12:725–736 [DOI] [PubMed] [Google Scholar]
- 36. Kasuya H, et al. 2007. Suitability of a US3-inactivated HSV mutant (L1BR1) as an oncolytic virus for pancreatic cancer therapy. Cancer Gene Ther. 14:533–542 [DOI] [PubMed] [Google Scholar]
- 37. Kelly EJ, Nace R, Barber GN, Russell SJ. 2010. Attenuation of vesicular stomatitis virus encephalitis through microRNA targeting. J. Virol. 84:1550–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kinoh H, Inoue M. 2008. New cancer therapy using genetically-engineered oncolytic Sendai virus vector. Front. Biosci. 13:2327–2334 [DOI] [PubMed] [Google Scholar]
- 39. Kinoh H, et al. 2004. Generation of a recombinant Sendai virus that is selectively activated and lyses human tumor cells expressing matrix metalloproteinases. Gene Ther. 11:1137–1145 [DOI] [PubMed] [Google Scholar]
- 40. Komaru A, et al. 2009. Sustained and NK/CD4+ T cell-dependent efficient prevention of lung metastasis induced by dendritic cells harboring recombinant Sendai virus. J. Immunol. 183:4211–4219 [DOI] [PubMed] [Google Scholar]
- 41. Kuhlmann KF, Gouma DJ, Wesseling JG. 2008. Adenoviral gene therapy for pancreatic cancer: where do we stand? Dig. Surg. 25:278–292 [DOI] [PubMed] [Google Scholar]
- 42. Lawson ND, Stillman EA, Whitt MA, Rose JK. 1995. Recombinant vesicular stomatitis viruses from DNA. Proc. Natl. Acad. Sci. U. S. A. 92:4477–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Le Boeuf F, et al. 2010. Synergistic interaction between oncolytic viruses augments tumor killing. Mol. Ther. 18:888–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lichty BD, Power AT, Stojdl DF, Bell JC. 2004. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol. Med. 10:210–216 [DOI] [PubMed] [Google Scholar]
- 45. Lindsay TH, et al. 2005. Pancreatic cancer pain and its correlation with changes in tumor vasculature, macrophage infiltration, neuronal innervation, body weight and disease progression. Pain 119:233–246 [DOI] [PubMed] [Google Scholar]
- 46. Linge C, Gewert D, Rossmann C, Bishop JA, Crowe JS. 1995. Interferon system defects in human malignant melanoma. Cancer Res. 55:4099–4104 [PubMed] [Google Scholar]
- 47. Lyles D, Rupprecht CE. 2007. Rhabdoviridae, p 1363–1408 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 48. Matin SF, et al. 2001. Impaired alpha-interferon signaling in transitional cell carcinoma: lack of p48 expression in 5637 cells. Cancer Res. 61:2261–2266 [PubMed] [Google Scholar]
- 49. Moussavi M, et al. 2010. Oncolysis of prostate cancers induced by vesicular stomatitis virus in PTEN knockout mice. Cancer Res. 70:1367–1376 [DOI] [PubMed] [Google Scholar]
- 50. Moyer SA, Baker SC, Lessard JL. 1986. Tubulin: a factor necessary for the synthesis of both Sendai virus and vesicular stomatitis virus RNAs. Proc. Natl. Acad. Sci. U. S. A. 83:5405–5409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Naik S, Russell SJ. 2009. Engineering oncolytic viruses to exploit tumor specific defects in innate immune signaling pathways. Expert Opin. Biol. Ther. 9:1163–1176 [DOI] [PubMed] [Google Scholar]
- 52. Nakao A, et al. 2007. Clinical experiment of mutant herpes simplex virus HF10 therapy for cancer. Curr. Cancer Drug Targets 7:169–174 [DOI] [PubMed] [Google Scholar]
- 53. Obuchi M, Fernandez M, Barber GN. 2003. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J. Virol. 77:8843–8856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Okabe T, Yamaguchi N, Ohsawa N. 1983. Establishment and characterization of a carcinoembryonic antigen (CEA)-producing cell line from a human carcinoma of the exocrine pancreas. Cancer 51:662–668 [DOI] [PubMed] [Google Scholar]
- 55. Ottolino-Perry K, Diallo JS, Lichty BD, Bell JC, McCart JA. 2010. Intelligent design: combination therapy with oncolytic viruses. Mol. Ther. 18:251–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ozduman K, Wollmann G, Piepmeier JM, van den Pol AN. 2008. Systemic vesicular stomatitis virus selectively destroys multifocal glioma and metastatic carcinoma in brain. J. Neurosci. 28:1882–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pfeffer LM, et al. 1996. Human renal cancers resistant to IFN′s antiproliferative action exhibit sensitivity to IFN′s gene-inducing and antiviral actions. J. Urol. 156:1867–1871 [PubMed] [Google Scholar]
- 58. Quiroz E, Moreno N, Peralta PH, Tesh RB. 1988. A human case of encephalitis associated with vesicular stomatitis virus (Indiana serotype) infection. Am. J. Trop. Med. Hyg. 39:312–314 [DOI] [PubMed] [Google Scholar]
- 59. Russell SJ, Peng KW. 2007. Viruses as anticancer drugs. Trends Pharmacol. Sci. 28:326–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Saloura V, et al. 2010. Evaluation of an attenuated vesicular stomatitis virus vector expressing interferon-beta for use in malignant pleural mesothelioma: heterogeneity in interferon responsiveness defines potential efficacy. Hum. Gene Ther. 21:51–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sarinella F, Calistri A, Sette P, Palu G, Parolin C. 2006. Oncolysis of pancreatic tumour cells by a gamma 34.5-deleted HSV-1 does not rely upon Ras-activation, but on the PI 3-kinase pathway. Gene Ther. 13:1080–1087 [DOI] [PubMed] [Google Scholar]
- 62. Sharif-Askari E, et al. 2007. Bax-dependent mitochondrial membrane permeabilization enhances IRF3-mediated innate immune response during VSV infection. Virology 365:20–33 [DOI] [PubMed] [Google Scholar]
- 63. Shi W, et al. 2009. Antitumor and antimetastatic activities of vesicular stomatitis virus matrix protein in a murine model of breast cancer. J. Mol. Med. 87:493–506 [DOI] [PubMed] [Google Scholar]
- 64. Shinozaki K, Ebert O, Woo SL. 2005. Treatment of multi-focal colorectal carcinoma metastatic to the liver of immune-competent and syngeneic rats by hepatic artery infusion of oncolytic vesicular stomatitis virus. Int. J. Cancer 114:659–664 [DOI] [PubMed] [Google Scholar]
- 65. Stathis A, Moore MJ. 2010. Advanced pancreatic carcinoma: current treatment and future challenges. Nat. Rev. Clin. Oncol. 7:163–172 [DOI] [PubMed] [Google Scholar]
- 66. Stojdl DF, et al. 2000. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 6:821–825 [DOI] [PubMed] [Google Scholar]
- 67. Stojdl DF, et al. 2003. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4:263–275 [DOI] [PubMed] [Google Scholar]
- 68. Sun WH, et al. 1998. Interferon-alpha resistance in a cutaneous T-cell lymphoma cell line is associated with lack of STAT1 expression. Blood 91:570–576 [PubMed] [Google Scholar]
- 69. Vähä-Koskela MJ, Heikkilä JE, Hinkkanen AE. 2007. Oncolytic viruses in cancer therapy. Cancer Lett. 254:178–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. von Messling V, Cattaneo R. 2004. Toward novel vaccines and therapies based on negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 283:281–312 [DOI] [PubMed] [Google Scholar]
- 71. Watanabe I, et al. 2008. Effects of tumor selective replication-competent herpes viruses in combination with gemcitabine on pancreatic cancer. Cancer Chemother. Pharmacol. 61:875–882 [DOI] [PubMed] [Google Scholar]
- 72. Wiegand MA, Bossow S, Schlecht S, Neubert WJ. 2007. De novo synthesis of N and P proteins as a key step in Sendai virus gene expression. J. Virol. 81:13835–13844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wollmann G, Simon RVI, Rose JK, van den Pol AN. 2010. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J. Virol. 84:1563–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wong LH, et al. 1997. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3gamma. J. Biol. Chem. 272:28779–28785 [DOI] [PubMed] [Google Scholar]
- 75. Wu L, et al. 2008. rVSV(M Delta 51)-M3 is an effective and safe oncolytic virus for cancer therapy. Hum. Gene Ther. 19:635–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yonemitsu Y, Kinoh UYH, Hasegawa M. 2008. Immunostimulatory virotherapy using recombinant Sendai virus as a new cancer therapeutic regimen. Front. Biosci. 13:4953–4959 [DOI] [PubMed] [Google Scholar]