

Abstract
The dynamics of the circulation and distribution of transmissible spongiform encephalopathy (TSE) agents in the blood of infected individuals remain largely unknown. This clearly limits the understanding of the role of blood in TSE pathogenesis and the development of a reliable TSE blood detection assay. Using two distinct sheep scrapie models and blood transfusion, this work demonstrates the occurrence of a very early and persistent prionemia. This ability to transmit disease by blood transfusion was correlated with the presence of infectivity in white blood cells (WBC) and peripheral blood mononucleated cells (PBMC) as detected by bioassay in mice overexpressing the ovine prion protein PrP (tg338 mice) and with the identification of abnormal PrP in WBC after using protein misfolding cyclic amplification (PMCA). Platelets and a large variety of leukocyte subpopulations also were shown to be infectious. The use of endpoint titration in tg338 mice indicated that the infectivity in WBC (per ml of blood) was 106.5-fold lower than that in 1 g of posterior brainstem sample. In both WBC and brainstem, infectivity displayed similar resistance to PK digestion. The data strongly support the concept that WBC are an accurate target for reliable TSE detection by PMCA. The presence of infectivity in short-life-span blood cellular elements raises the question of the origin of prionemia.
INTRODUCTION
Transmissible spongiform encephalopathies (TSE), or prion diseases, are fatal neurodegenerative disorders occurring in sheep (scrapie), cattle (bovine spongiform encephalopathy [BSE]), and humans (Creutzfeldt-Jakob disease [CJD]).
In 1996, a new form of TSE, named variant CJD (v-CJD), was identified in humans. Variant CJD was demonstrated to be due to the same TSE agent that causes cattle BSE, and its emergence in humans was considered to be the consequence of a dietary exposure to BSE-contaminated products (9, 13). As early as 1998, measures that intended to prevent blood-borne v-CJD transmission were implemented in a large number of countries that experienced BSE in cattle epidemics and/or v-CJD occurrence. These measures were further reinforced after the identification in the United Kingdom of at least 4 v-CJD cases in patients that received blood products prepared from donors that later developed the disease (32).
In early 2000, most of the knowledge related to the TSE agent in blood relied on the use of rodent TSE models. In these models, blood infectivity titers were reported to vary between 1 and 10 50% infectious doses (ID50) per ml of blood during the asymptomatic phase and could reach up to 100 ID50/ml during the clinical phase of the disease (8, 12). This was done by an intracerebral inoculation in a homospecific model, which also showed that about 40% of the infectivity was associated with the buffy coat, with the remainder being in the plasma (8, 21). Importantly, the buffy coat-associated infectivity could be washed off from cells by a phosphate-buffered saline (PBS) rinsing (32). Platelets were shown to have low or no infectivity in both hamsters and mice (12, 23). In the absence of information on prion distribution in blood of other species, these findings were used to develop v-CJD blood-borne transmission risk assessment (39).
A decade ago, the transmission of experimental BSE and natural scrapie were reported to occur following the transfusion of whole blood and/or buffy coat collected in asymptomatic incubating sheep (24, 28). These observations opened a new perspective for investigating the role of blood in TSE agent pathogenesis and provided an alternative model to rodents for studying the behavior and distribution of TSE agents in this tissue. In addition, the ability to collect blood volumes comparable to those used in transfusion medicine and the similarities in the pathogenesis of sheep TSE and v-CJD in humans led us to consider that the sheep model represents a pertinent source of information (29). Results recently reported for BSE-infected sheep (34) and chronic wasting disease (CWD)-infected deer (33) indicated that most blood fractions that are relevant in transfusion medicine can transmit TSE disease. In those models, blood fractions, such as platelets, that were considered of either little or no infectivity in rodents models were shown to transmit the disease.
Misfolded PrP (PrPSc) accumulation in tissues from infected individuals is the key molecular event in prion disease. Proteinase K (PK) digestion is classically used to differentiate between abnormal PrP (N-terminally truncated PrP [PrPres]) from cellular PrP, which is expressed in a large variety of cells. Whereas cellular PrP is readily degraded by PK, misfolded PrP digestion usually results in PrPres. The presence of PrPres is considered to be pathognomonic of prion diseases and correlates with the presence of infectivity. All attempts to develop a diagnosis assay based on the direct detection of PrPres in blood have failed (20). The minute amount of TSE agent in blood is the most likely explanation for this failure; however, an alternative explanation is that the TSE agent in blood is associated with PK-sensitive PrP species (16). Recent studies carried out in TSE-infected hamsters and sheep brought the proof of concept that misfolded PrP in blood can be amplified in vitro by protein misfolding cyclic amplification (PMCA) (11, 38).
These results opened new perspectives in TSE diagnosis. However, there is no clear estimation of the capacity of such an approach to ensure an early diagnosis in infected individuals, since the correlation between the detection of amplified misfolded PrP and the presence of infectivity in blood samples remains to be clarified.
In this study, the capacity of blood collected at different stages of the incubation period to transmit the disease through transfusion was investigated using both a natural and an experimental model of scrapie in sheep. In addition, the distribution of the infectivity in the blood was assessed by bioassay in transgenic mice that overexpress ovine PrP. In both models it was shown that prionemia occurs very early after infection, and the TSE agent is associated with a large variety of cellular subpopulations, including short-life-span cellular elements like platelets. Protein misfolding cyclic amplification followed by PrPres detection performed similarly to the tg338 mouse bioassay for scrapie agent detection in white blood cells (WBC). WBC infectivity cannot be washed off from the cellular surface and displayed PK resistance properties that apparently were similar to those of brain-associated infectivity.
These findings have a direct effect on our perception of the blood-associated infectivity and development of TSE blood diagnosis assays.
MATERIALS AND METHODS
Ethics statement.
All animal experiments have been performed in compliance with our institutional and national guidelines in accordance with the European Community Council Directive 86/609/EEC. The experimental protocol was approved by the INRA Toulouse/ENVT ethics committee.
Classical scrapie natural cases.
Natural classical scrapie cases included in this experiment all were VRQ/VRQ Romanov sheep born and bred in the Langlade flock. A natural scrapie epidemic has been occurring in this flock at a high incidence since 1993 (17). The dissemination of the scrapie agent in the organism of VRQ/VRQ animals naturally infected in this flock has been described in previous studies (1, 2, 30), and the disease was described to occur in 100% of the VRQ/VRQ sheep born in the flock between 20 and 25 months of age.
PG127 classical scrapie oral inoculation model.
The classical scrapie isolate was derived from VRQ/VRQ sheep that were experimentally affected (PG127 isolate). Six- to 10-month-old TSE-free cheviot sheep were orally challenged with a 2-g equivalent of brain material (5% brain homogenate in glucose). This isolate has previously been endpoint titrated in tg338 mice (4). Animals then were observed until the occurrence of clinical signs (around 200 days postinoculation [dpi]) and culled when exhibiting locomotor signs of the disease that impaired their feeding capacities. After culling, each animal was necropsied and a variety of lymphoid tissues (spleen, third eye lid, mesenteric lymph node, prescapular lymph node, and tonsil) and central nervous system (CNS) samples were collected.
TSE-free recipient sheep.
TSE-free VRQ/VRQ cheviot sheep were produced in the DEFRA TSE-free flock, which is a unique source able to provide VRQ/VRQ animals that can be considered free from classical scrapie (37). The animals included in our experiments were imported to France and housed before their use in experiments in a dedicated scrapie-free farm situated 30 km from the experimental facilities, and to avoid the possibility of cross-contamination, dedicated staff (who had no contact with the infected animals) were used. Five different imports of sheep were necessary to complete the experiments presented here. During this 6-year period, no contamination was observed in the 10 sentinel sheep that were kept on this site. In all cases, the PrP genotype was obtained by sequencing exon 3 of the prnp gene as previously described (5). In each case, the polymorphisms at codons 136 (A/V), 154 (H/R), and 171 (R/Q), which have been demonstrated to strongly influence the susceptibility to TSE in sheep, were indicated (27).
Blood collection and plasma/WBC preparation.
Whole blood was collected from the jugular vein using citrate dextrose (35 ml) in a 250-ml blood collection pouch (MSE3500Q; Macopharma). For plasma and WBC preparation, whole blood was transferred into 15-ml conic tubes before centrifugation at 3,600 rpm for 10 min at room temperature. Plasma was removed and the buffy coat was collected using a disposable hard bulb pipette. Buffy coat then was mixed volume/volume with ACK solution (NH4CL, 0.15 M; KHCO3, 1 mM; Na2EDTA, 0.1 mM; pH 7.4) for 5 min at room temperature (RT). The obtained white blood cells then were washed 3 times with PBS. Cell concentration was established by counting using either Mallasez's cell or an automatic cell counter. When not in use, fresh cells and plasma were aliquoted before freezing (−80°C).
Platelet preparation.
Whole blood (80 ml) was collected as previously described before being transferred into 5-ml borosilicate tubes, which were centrifuged for 15 min at 300 × g at room temperature. The top third of the plasma was collected by pipetting and transferred into a new tube for centrifugation for 30 min at 3,000 × g. The supernatant was eliminated and the pellet resuspended in PBS (pH 7.4) before two additional pelletings or washings. Pellets finally were resuspended in PBS, and the absence of both leukocytes and red cells was checked by direct microscopic examination in a Thomas' cell and Sysmex XT-2000i automat. According to these numerations, the leukocyte and red blood cell concentrations in the final preparation was below 103 cells per ml, meaning that fewer than 20 cells were inoculated into each mouse (20 μl per mouse).
PMCA.
PMCA was carried out as previously described (10, 11). The substrate consisted of 10% (wt/vol) ovine VRQ PrP transgenic mice (tg338) brain homogenate in PBS (pH 7.4), 0.1% Triton X-100, and 150 mM NaCl buffer. WBC (typically 104 cells in 7 μl of PBS) or brain homogenate dilutions (7 μl in PBS) were mixed with 63 μl of substrate in 0.2-ml thin-wall PCR tubes. Sealed tubes then were placed in the horn of a Misonix 4000 sonicator for a round of 96 cycles. Each cycle consisted of a 30-s sonication step (70% power) followed by a 29.5-min incubation. After the first amplification round, 7 μl of each amplicon was added to 63 μl of fresh substrate for a second PMCA round. Twenty μl of each amplicon was processed for PrPres detection by Western blotting (WB). Previous experiments carried out in this model indicated that additional PMCA rounds do not increase the sensitivity of detection (data not shown). To limit cross-contamination risks that are linked to serial PMCA, we employed procedures that are similar to those in place for nested PCR. In particular, PMCA substrates, amplification, and the handling of amplicons were performed in different rooms using dedicated material.
PBMC preparation and cell sorting.
Whole blood samples were diluted in equal amounts of Hanks balanced salt solution (HBSS), and 25 ml of diluted blood was decanted into a 50-ml conic tube before adding 12.5 ml of 1.077-g/ml Ficoll solution (Ficoll-Paque Plus 17-440-03; GE Health Care Life Sciences). Tubes were centrifuged for 20 min at 1,000 × g at 20°C. Annealed peripheral blood mononucleated cells (PBMC) were collected by pipetting, and cells were washed twice in HBSS and stored on ice.
Cells were labeled using fluorescein isothiocyanate (FITC)-coupled anti-ovine CD4 (MCA2213F; 1/100 diluted; Serotec), CD8 (MCA837F; 1/100 diluted; Serotec), CD45R (MC2221F; 1/50 diluted; Serotec), and CD14 (clone VPM65; 1 μg/ml). Labeled cells were incubated using anti-FITC monoclonal antibody (MAb) coupled to magnetic beads (130-048-701; Miltenyi Biotec) according to the manufacturer's recommendations. Positive selection was achieved using two successive passages on mass spectrometry (MS) separation columns (130-048-701; Miltenyi Biotec) according to the manufacturer's recommendations. Cell depletion was achieved on an Automacs system using a double-deplete program (Deplete S). The purity of sorted cell subpopulations and the viability of cells (propidium iodide [PI] labeling) were controlled by flow cytometry (with a control of 2 × 104 events per fraction). Fractions that were below 95% purity (in fluorescent label 1 [FL1]) were rejected.
Brain material and WBC PK digestion.
A 10% PG127 brain homogenate (from inoculated tg338 mice at the terminal stage; 10 ID50 intracerebral route [IC]/g) was 10-fold serially diluted into negative 10% homogenate. Half of each sample was treated with PK (4 μg of PK per mg of protein) for 2 h at 37°C, and the reaction was stopped by adding Pefabloc (4 mM final concentration). Under these conditions, normal PrP is totally degraded and 27 to 30 kDa of PrPres is generated. Buffy coat samples (108 cells) were homogenized in 300 μl of 5% glucose solution, and the protein concentration was determined by bicinchoninic acid assay. Half of each sample was digested with PK (4 μg of PK per mg of protein) for 2 h at 37°C, and the reaction was stopped by adding Pefabloc. All undigested and PK-digested samples then were frozen at −80°C until inoculation into tg338 mice.
Bioassay.
Mouse bioassays were carried out in ovine VRQ PrP transgenic mice (tg338), which are considered to be highly efficient for the detection of sheep scrapie infectivity (31). The high sensitivity of tg338 mice for the detection of both PG127 scrapie and Langlade scrapie was previously reported (3). At least six mice were intracerebrally inoculated with each sample (20 μl). Mice were clinically monitored until the occurrence of TSE clinical signs, at which time they were culled. Central nervous system (CNS) and spleen samples were individually collected and analyzed by WB. Half of the brain was formalin fixed for vacuolar brain lesion profiling (19).
PrPSc WB detection.
A Western blotting kit (TeSeE Western blot; Bio-Rad) was used by following the manufacturer's recommendations. For each sample, 250 μl of 10% brain homogenate was submitted to PrPSc extraction. In PMCA, 20 μl of amplicon was mixed with 230 μl of 10% negative brain homogenate before the extraction step. The obtained pellet was denatured in Laemmli's buffer (15 μl) before being loaded undiluted or diluted on a 12% acrylamide gel before electrophoresis and blotting. Immunodetection was performed using SHA31, which recognizes the PrP sequence of residues 145 to 152 (YEDRYYRE) (18), conjugated to horseradish peroxidase (at 0.06 μg per ml). Peroxidase activity was revealed using ECL substrate (Pierce).
RESULTS
Prionemia is a very early event.
Blood from three sheep with a genotype encoding susceptibility (VRQ/VRQ) that had been born and raised in a classical scrapie-infected flock (INRA Langlade) was collected at different time points during the incubation period, between 3 months of age and clinical onset at 22 months of age (Table 1). In all cases, the transfusion of whole blood (180 ml) into TSE-free VRQ/VRQ recipient sheep caused the occurrence of classical scrapie with incubation periods comparable to those observed in the donors.
Table 1.
Incubation period of VRQ/VRQ TSE-free recipient sheep transfused with whole blood collected from three VRQ/VRQ sheep naturally contaminated with classical scrapiea
| Donor | Incubation period (days) at blood collection day: |
|||||||
|---|---|---|---|---|---|---|---|---|
| 90 | 150 | 210 | 270 | 360 | 460 | 580 | 720 | |
| D1 | 736 | 792 | 867 | 834 | 719 | 703 | 724 | 557 |
| D2 | 835 | 765 | 735 | 787 | 653 | 621 | 643 | 690 |
| D3 | 912 | 812 | 767 | 750 | 693 | 689 | 709 | 653 |
Blood samples (180 ml) from three VRQ/VRQ donors (identified as D1, D2, and D3) born and raised in the INRA Langlade flock were drawn at different time points of the incubation phase. The three donors died at 781, 728, and 789 days of age, respectively. After death, natural scrapie occurrence was confirmed by histopathology (vacuolar changes in the central nervous system) and the detection of abnormal PrP deposits in the central nervous system and lymphoid tissues. Blood was transfused to TSE-free VRQ/VRQ recipients within 12 h following collection. Recipients were observed until the occurrence of clinical signs. The incubation periods in recipients are presented. In all recipients, abnormal PrP was detected in brain and lymphoid tissues after death.
A similar experiment was carried out in three VRQ/VRQ sheep orally inoculated with the PG127 rapid scrapie isolate (Table 2). Orally challenged sheep developed the disease after 200 to 250 days of incubation. The transfusion of 180 ml of whole blood collected at 30 or 60 days postinoculation failed to transmit disease to recipients, but the blood collected at 90 dpi and later transmitted disease to recipients with similar or shorter incubation periods than those of donors. In the same experiment, the transfusion of blood collected from an orally inoculated sheep with the ARR/ARR genotype (i.e., resistant to classical scrapie) failed to transmit the disease, indicating that blood-borne transfusion in that experimental model is not due to the residual circulation of the inoculum load in the bloodstream. Posterior brainstem homogenate, prepared from both donors and recipient sheep, were inoculated into mice that overexpress the VRQ ovine PRP gene (tg338). The vacuolar lesion profile in tg338 mice confirmed that TSE agents which developed in blood recipients were identical to the one which caused disease in the donors (Fig. 1A and B). In both experiments, none of the TSE-free VRQ/VRQ control sheep that were housed in contact with transfusion recipients developed TSE or displayed abnormal PrP accumulation in their tissues. Collectively, these data indicate an early and persistent prionemia in classical scrapie-infected sheep.
Table 2.
Incubation period of VRQ/VRQ TSE-free recipient sheep transfused with whole blood collected from three VRQ/VRQ sheep and one ARR/ARR sheep, all orally inoculated with PG127 classical scrapie isolatea
| Donors | Incubation period (days) and PrPSc status at dpi: |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 30 |
60 |
90 |
130 |
190 |
||||||
| Incubation (days) | PrPSc status | Incubation (days) | PrPSc status | Incubation (days) | PrPSc status | Incubation (days) | PrPSc status | Incubation (days) | PrPSc status | |
| D4 | >450 | − | >450 | − | >450 | − | 184 | + | 161 | + |
| D5 | >450 | − | >450 | − | 184 | + | 168 | + | 184 | + |
| D6 | >450 | − | >450 | − | 234 | + | 184 | + | 161 | + |
| C1 | >450 | − | >450 | − | >450 | − | >450 | − | >450 | − |
Three susceptible VRQ/VRQ sheep (identified as D4, D5, and D6) and one resistant ARR/ARR sheep (C1) were orally challenged with 2 g of brain homogenate (106.6 ID50/g IC in tg338 mice) between 6 and 10 months of age. The three VRQ/VRQ sheep died 217, 206, and 217 days postinoculation (dpi), whereas the ARR/ARR sheep is still alive (>1,500 days). In VRQ/VRQ donors, the occurrence of scrapie was confirmed by histopathology (vacuolar changes in the central nervous system) and the detection of abnormal PrP deposits in the central nervous system and lymphoid tissues. At different time points, whole blood (180 to 200 ml) was collected from each donor and transfused to TSE-free VRQ/VRQ recipients within the 12 h following collection. Recipients were observed until the occurrence of clinical signs. At 450 days after transfusion, recipients that were apparently still healthy were killed. The incubation periods in recipients are presented. All recipients were tested for the presence of abnormal PrP deposition in brain and various lymphoid tissues by immunohistochemistry.
Fig 1.

Lesion profiles in tg338 mice inoculated with brain material and blood fractions from sheep. Shown are lesion profiles (showing vacuolar changes) in tg338 mice inoculated by the intracerebral route with (A) 10% obex homogenate prepared from clinically affected VRQ/VRQ sheep that were naturally exposed to Langlade classical scrapie agent (▴; blood donor 2 in Table 1) or that were transfused with blood collected from donor 2 at 90 days (○) and 360 days (▿) of age; (B) 10% obex homogenate from clinically affected VRQ/VRQ sheep that were orally challenged with PG127 classical scrapie agent (△; blood donor 5 in Table 2) or that were transfused with blood collected from donor 4 at 90 days (○) and 190 days (▿) postinoculation; (C) white blood cell homogenates (white symbols) and PBMC (black symbols) prepared from VRQ/VRQ sheep that were naturally exposed to Langlade classical scrapie agent (blood donor 2 in Table 3) collected at 210 days (▿) and 360 days (○) of age; and (D) white blood cell homogenates (white symbols) and PBMC (black symbols) prepared from VRQ/VRQ sheep that were orally exposed to PG127 classical scrapie agent (blood donor 5 in Table 4) and collected 130 days (▿) and 180 days (○) postinoculation.
Infectivity in WBC and PBMC correlates with prionemia.
In naturally infected sheep (Langlade flock animals D1 to D3) and experimental orally challenged sheep (D4 to D6 and C1), WBC and PBMC were prepared using aliquots from the same blood pouch as that used in transfusion experiments.
Because of the length of the incubation periods, only parts of the tg338 bioassay results are available for the three blood donor sheep naturally exposed to scrapie (animals under 360 days old) (Table 3). WBC and PBMC prepared from two out of the three donors collected at 90 days caused TSE transmission in few inoculated tg338 mice. At 120 days and later, TSE infectivity was detected in WBC/PBMC from all three donors. A progressive increase of the attack rate was observed in tg338 mice inoculated with WBC/PBMC prepared from the blood of animals collected between 90 and 270 days of age. This suggests an increase in the infectivity titer in blood during the incubation. However, no increase in the attack rate or reduction of incubation period was observed in mice inoculated with WBC/PBMC between the samples collected at 270 and 360 days of age (Table 3).
Table 3.
Attack rate and incubation period in tg338 mice intracerebrally inoculated with WBC and PBMC prepared at different stages of the incubation period in natural classical scrapie VRQ/VRQ sheepa
| Donor and sample type | Result for samples from incubation day: |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 90 |
120 |
180 |
270 |
360 |
|||||||||||
| No. of cells inoculated/mouse | tg338 transmission |
No. of cells inoculated/mouse | tg338 transmission |
No. of cells inoculated/mouse | tg338 transmission |
No. of cells inoculated/mouse | tg338 transmission |
No. of cells inoculated/mouse | tg338 transmission |
||||||
| No. of mice positive for abnormal PrP | Incubation time (days) | No. of mice positive for abnormal PrP | Incubation time (days) | No. of mice positive for abnormal PrP | Incubation time (days) | No. of mice positive for abnormal PrP | Incubation time (days) | No. of mice positive for abnormal PrP | Incubation time (days) | ||||||
| D1 | |||||||||||||||
| WBC | 3 × 106 | 0/6 | >750 | 1.9 × 106 | 2/6 | 460, 658 | 1.8 × 106 | 5/6 | 529 ± 125 | 3.5 × 106 | 5/5 | 533 ± 54 | 5.8 × 106 | 5/5 | 543 ± 66 |
| PBMC | 8.5 × 105 | 0/6 | >750 | 1 × 106 | 1/6 | 623 | 1 × 106 | 3/6 | 563, 653, 721 | 2.2 × 106 | 6/6 | 732 ± 64 | 1.6 × 106 | 5/5 | 594 ± 84 |
| D2 | |||||||||||||||
| WBC | 3.4 × 106 | 0/6 | >750 | 1.9 × 106 | 1/6 | 425 | 1.2 × 106 | 5/5 | 612 ± 133 | 4.8 × 106 | 6/6 | 605 ± 139 | 3.7 × 106 | 5/5 | 516 ± 101 |
| PBMC | 7.7 × 105 | 1/6 | 627 | 1.1 × 106 | 4/6 | 616 ± 70 | 8.1 × 105 | 5/6 | 570 ± 114 | 2.4 × 106 | 6/6 | 570 ± 38 | 1.5 × 106 | 6/6 | 641 ± 27 |
| D3 | |||||||||||||||
| WBC | 2.6 × 106 | 0/6 | >750 | 2.3 × 106 | 5/5 | 661 ± 67 | 5.1 × 106 | 5/6 | 646 ± 114 | 4.2 × 106 | 6/6 | 629 ± 98 | 6.7 × 106 | 6/6 | 585 ± 94 |
| PBMC | 4.7 × 105 | 1/6 | 783 | 1.2 × 106 | 4/5 | 691 ± 117 | 8.5 × 105 | 5/6 | 689 ± 77 | 2.7 × 106 | 6/6 | 678 ± 55 | 1.9 × 106 | 6/6 | 644 ± 89 |
Blood samples (100 ml) from three VRQ/VRQ donors (identified as D1, D2, and D3) born and raised in the INRA Langlade flock were taken at different time points of the incubation phase. The three donors died at 781, 728, and 789 days, respectively. After death, natural scrapie occurrence was confirmed by histopathology (vacuolar changes in the central nervous system) and the detection of abnormal PrP deposits in the central nervous system and lymphoid tissues. After preparation, cells were counted and pelleted. Each pellet was resuspended in 200 μl of a 5% glucose solution before grinding. Each homogenate was inoculated intracerebrally into tg338 mice (n = 6; 20 μl per mouse). Equivalent numbers of cells inoculated into each mouse are indicated. Mice then were observed until the occurrence of clinical signs or killed after 750 days postinoculation. Mice were considered positive when abnormal PrP deposition was detected in the brain. Incubation periods are presented as means ± standard deviations (SD), except when less than 50% of mice were positive. In those cases, the incubation times of the positive mice are individually presented.
In the orally challenged scrapie model, no transmission was observed in mice inoculated with WBC and PBMC prepared from sheep at 30 dpi. For all three donors, a progressive increase of the attack rate was observed in tg338 mice inoculated with the samples collected between 60 and 190 dpi (Table 4). WBC prepared from the orally challenged ARR/ARR genotype control did not transmit disease into any inoculated mice, ruling out the possibility that infectivity detected by the tg338 bioassay corresponded to residual circulating inocula. Lesion profiles in tg338 mice inoculated with either WBC or PBMC that succumbed to disease were undistinguishable from those recorded in mice inoculated with blood donors' brainstem homogenate (Fig. 1C and D).
Table 4.
Attack rate and incubation period in tg338 mice intracerebrally inoculated with WBC and PBMC prepared at different stages of the incubation period of sheep orally inoculated with PG127 scrapiea
| Donor and sample type | Result on dpi: |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 30 |
60 |
90 |
130 |
190 |
|||||||||||
| No. of cells inoculated/mouse | tg338 transmission |
No. of cells inoculated/mouse | tg338 transmission |
No. of cells inoculated/mouse | tg338 transmission |
No. of cells inoculated/mouse | tg338 transmission |
No. of cells inoculated/mouse | tg338 transmission |
||||||
| No. of mice positive for abnormal PrP | Incubation time (days) | No. of mice positive for abnormal PrP | Incubation time (days) | No. of mice positive for abnormal PrP | Incubation time (days) | No. of mice positive for abnormal PrP | Incubation time (days) | No. of mice positive for abnormal PrP | Incubation time (days) | ||||||
| D4 | |||||||||||||||
| WBC | 5.3 × 106 | 0/6 | >250 | 1.2 × 107 | 0/6 | >250 | 107 | 1/6 | 137 | 6.7 × 106 | 1/6 | 115 | 1.7 × 107 | 6/6 | 85 ± 3 |
| PBMC | 2 × 107 | 0/6 | >250 | 2.8 × 107 | 1/6 | 115 | 2.3 × 107 | 0/6 | >250 | 3.8 × 107 | 3/6 | 113 ± 14 | 8.1 × 106 | 6/6 | 81 ± 3 |
| D5 | |||||||||||||||
| WBC | 7.4 × 106 | 0/6 | >250 | 1.7 × 107 | 1/6 | 85 | 7.2 × 106 | 2/6 | 126, 134 | 7.2 × 106 | 6/6 | 112 ± 3 | 8.2 × 106 | 6/6 | 91 ± 7 |
| PBMC | 1.3 × 107 | 0/6 | >250 | 2.8 × 107 | 0/6 | 116 | 9.2 × 106 | 0/5 | >250 | 9.2 × 106 | 6/6 | 102 ± 8 | 1.2 × 107 | 6/6 | 86 ± 7 |
| D6 | |||||||||||||||
| WBC | 2.7 × 106 | 0/6 | >250 | 3.6 × 106 | 0/6 | >250 | 4.4 × 106 | 0/5 | >250 | 6.4 × 106 | 1/5 | 124 | 7.5 × 106 | 6/6 | 97 ± 5 |
| PBMC | 6 × 106 | 0/6 | >250 | 5.3 × 106 | 0/6 | >250 | 4.2 × 106 | 1/5 | 116 | 7.8 × 106 | 4/5 | 122 ± 15 | 6.3 × 106 | 6/6 | 85 ± 2 |
| C1 | |||||||||||||||
| WBC | 8.3 × 106 | 0/6 | >250 | 1.2 × 107 | 0/6 | >250 | 9.6 × 106 | 0/6 | >250 | 2.1 × 107 | 0/6 | >250 | 9.3 × 106 | 0/6 | >250 |
Three susceptible VRQ/VRQ sheep (identified as D4, D5, and D6) and one resistant ARR/ARR sheep (C1) were orally challenged with 2 g of brain homogenate (106.6 ID50/g IC in tg338 mice). The three VRQ/VRQ sheep died 217, 206, and 217 dpi, respectively, whereas the ARR/ARR sheep is still alive (>1,500 days). At different time points of the incubation period, whole blood (150 to 180 ml) was collected from each donor and WBC and PBMC were prepared, counted, and pelleted. Each pellet was resuspended in 200 μl of a 5% glucose solution and inoculated intracerebrally into tg338 mice (n = 6; 20 μl per mouse). Mice then were observed until the occurrence of clinical signs or killed after 250 days postinoculation. Mice were considered positive when abnormal PrP deposition was detected in the brain. Incubation periods are shown as means ± SD, except when less than 50% of mice were positive. In those cases, incubation times of the positive mice are individually presented.
WBC infectivity titers.
WBC homogenates prepared from donors D4, D5, and D6 at the clinical stage were used for evaluating the infectious titer in tg338 mice (Table 5). These samples displayed an infectious titer varying between 5 and 21 ID50 per ml IC tg338, which was equivalent to between 1 and 6 μg of a reference PG127 posterior brain stem homogenate prepared from symptomatic VRQ/VRQ sheep.
Table 5.
Endpoint titration in tg338 mice of one 10% brain homogenate sample and WBC samples collected from VRQ/VRQ sheep orally inoculated with PG127 scrapiea
| Inoculum dilution | Results from 10% brain homogenate |
Results for WBC from sheep: |
||||||
|---|---|---|---|---|---|---|---|---|
| D4 |
D5 |
D6 |
||||||
| No. of mice positive for abnormal PrP | Length of incubation (days) | No. of mice positive for abnormal PrP | Length of incubation (days) | No. of mice positive for abnormal PrP | Length of incubation (days) | No. of mice positive for abnormal PrP | Length of incubation (days) | |
| Undiluted | 6/6 | 64 ± 4 | 6/6 | 104 ± 6 | 6/6 | 102 ± 6 | 6/6 | 107 ± 12 |
| 10−1 | 6/6 | 76 ± 3 | 4/6 | 116 ± 10 | 1/6 | 131 | 1/6 | 118 |
| 10−2 | 6/6 | 87 ± 2 | 1/6 | 132 | 0/6 | >250 | 0/6 | >250 |
| 10−3 | 6/6 | 97 ± 5 | 0/6 | >250 | 0/6 | >250 | 0/6 | >250 |
| 10−4 | 3/6 | 110 ± 4 | ND | ND | ND | |||
| 10−5 | 2/6 | 117-121 | ND | ND | ND | |||
| 10−6 | 0/6 | >200 | ND | ND | ND | |||
| 10−7 | 0/6 | >200 | ND | ND | ND | |||
Homogenate (10%, wt/vol) was prepared using posterior brainstem from VRQ/VRQ sheep inoculated with PG127 scrapie at the terminal stage of disease. Groups of 6 mice that overexpress the VRQ ovine PrP (tg338) were intracerebrally (20 μl) inoculated with successive 1/10 dilutions of this homogenate. These data were already used in a previous publication (3). Three susceptible VRQ/VRQ sheep (identified as D4, D5, and D6) were orally challenged with 2 g of brain homogenate (106.6 ID50/g IC in tg338 mice) between 6 and 10 months of age. These animals died at 217, 206, and 217 dpi, respectively, and classical scrapie occurrence was confirmed by histopathology (vacuolar changes in the central nervous system) and the detection of abnormal PrP deposits in the central nervous system and lymphoid tissues. Whole-blood (30 ml) samples were collected at 206 dpi, and WBC were prepared. WBC samples equivalent to 10 ml of starting blood were resuspended in 200 μl of 5% glucose solution before homogenization. Successive 1/10 dilutions of homogenate were inoculated intracerebrally into tg338 mice (n = 6; 20 μl per mouse). Mice were observed until the occurrence of clinical signs or were sacrificed after 250 dpi. Mice were considered positive when abnormal PrP deposition was detected in the brain. Incubation periods are presented as means ± SD except for dilutions with which less than 50% of mice were positive. In those cases, incubation times of the positive mice are individually presented. ND, not determined.
Brain homogenates displaying different infectivity titers and WBC lysates, both prepared from PG127-affected sheep, were submitted to PK digestion using a standard procedure for PrPres Western blot (WB) detection. PK concentrations were adjusted to the total amount of protein in each homogenate, and both PK-treated and untreated samples were intracerebrally inoculated into tg338 mice (Table 6).
Table 6.
Intracerebral inoculation of tg338 mice with PK-treated or untreated PG127 scrapie brain homogenate and WBC from PG127-infected VRQ/VRQ sheepa
| PK treatment status | Results for PG127 brain homogenate at dose (ID50/IC tg338) of: |
Results for WBC from sheep: |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 103 |
102 |
10 |
D7 |
D8 |
D9 |
D10 |
D11 |
|||||||||
| No. of mice positive for abnormal PrP | Length of incubation (days) | No. of mice positive for abnormal PrP | Length of incubation (days) | No. of mice positive for abnormal PrP | Length of incubation (days) | No. of mice positive for abnormal PrP | Length of incubation (days) | No. of mice positive for abnormal PrP | Length of incubation (days) | No. of mice positive for abnormal PrP | Length of incubation (days) | No. of mice positive for abnormal PrP | Length of incubation (days) | No. of mice positive for abnormal PrP | Length of incubation (days) | |
| Untreated | 6/6 | 83 ± 6 | 6/6 | 102 ± 5 | 6/6 | 122 ± 14 | 5/5 | 111 ± 13 | 6/6 | 109 ± 5 | 6/6 | 98 ± 7 | 6/6 | 101 ± 3 | 6/6 | 99 ± 4 |
| Treated | 6/6 | 87 ± 3 | 6/6 | 107 ± 10 | 5/6 | 127 ± 18 | 6/6 | 123 ± 17 | 5/6 | 121 ± 16 | 6/6 | 107 ± 8 | 6/6 | 117 ± 15 | 6/6 | 111 ± 12 |
Aliquots of PG127 scrapie brain homogenate (previously titrated in tg338 mice) and WBC homogenate prepared from five susceptible VRQ/VRQ sheep (identified as D7, D8, D9, D10, and D11) orally challenged with the same scrapie agent (collected 224 dpi) were prepared. The blood donors died at 228, 248, 256, 242, and 244 dpi, respectively, and classical scrapie occurrence was confirmed by histopathology (vacuolar changes in the central nervous system) and the detection of abnormal PrP deposits in the central nervous system and lymphoid tissues. Aliquots of the brain material and WBC homogenate were submitted to PK digestion under standard conditions (4 μg of PK per mg of protein for 2 h at 37°C) that allow the complete digestion of PrPC. Aliquots of both PK-treated and untreated material then were inoculated in tg338 mice (n = 6; 20 μl per mice). Mice then were observed until the occurrence of clinical signs or killed after 200 dpi. Mice were considered positive when abnormal PrP deposition was detected in the brain. Incubation periods are presented as means ± SD except when less than 50% of mice were positive. For brain material dilutions, the infectivity amount (number of ID50/IC in tg338) inoculated in each mouse is indicated.
For both brain and WBC samples, PK treatment resulted into an apparent, but not statistically different (P > 0.05 by Wilcoxon-Mann-Whitney nonparametric test; data not shown), increase of the incubation period in tg338 mice. In both types of samples the effect of PK digestion on the incubation period was less than a 1/10 dilution, as observed by the endpoint titration of brain homogenate or WBC (Table 6). These results support the view that TSE agents in WBC and CNS do not display fundamental differences of sensitivity to PK digestion.
PrPres detection in WBC by protein misfolding cyclic amplification.
Aliquots from the same WBC samples (D4 to D6 and C1; sheep were challenged orally with PG127) used for inoculating tg338 mice were submitted to serial PMCA amplification (two rounds) using tg338-negative brain homogenate as the substrate. The sensitivity and specificity of the method were evaluated using 1/10 dilution series (106.6 ID50/g IC in tg338 mice) of PG127 brain and TSE-free brain homogenate from VRQ/VRQ sheep (n = 2). While no PrPres was detected in the reactions seeded with the TSE-free controls, PMCA reactions of the samples seeded with PG127-infected sheep brain homogenate allowed PrPres detection at up to 10−6 and 10−8 dilutions after one or two amplification rounds, respectively (Fig. 2A and B).
Fig 2.
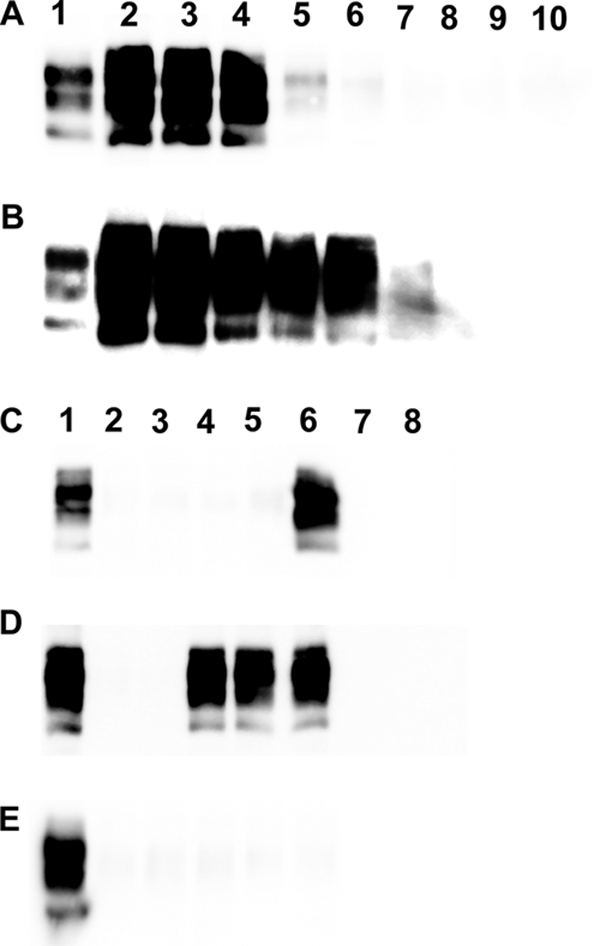
PrPres Western blot of PG127 infected sheep brain and white blood cells amplified by PMCA. (A and B) Dilution series (1/10) in PBS of 10% brain homogenates prepared from a PG127 scrapie-affected sheep (using the same material as that used for titrations in Table 5) and two scrapie-negative VRQ/VRQ sheep were submitted to two successive PMCA rounds (rounds 1 [A] and 2 [B]). Tubes seeded with different dilutions were processed for PrPres Western blotting detection (Sha31 anti-PrP antibody). Lane 1, Western blotting-positive control; lanes 2 to 8, 1/10 dilution series of PG127-positive brain homogenate (from 10−3 to 10−9); lanes 9 and 10, VRQ/VRQ TSE-free sheep (10−1 dilution). (C to E) White blood cells from one VRQ/VRQ sheep (C and D, lanes 2 to 6) (donor 4 in Tables 2, 3, and 5) or one ARR/ARR sheep (E, lanes 2 to 6) (control 1 in Tables 2 and 4) that had been orally challenged with PG127 scrapie were prepared at different time points of the incubation period. WBC from two TSE-free VRQ/VRQ (C and D, lanes 7 and 8) and two TSE-free ARR/ARR sheep (E, lanes 7 and 8) were used as controls. All WBC samples were submitted to two successive PMCA rounds (rounds 1 [C] and 2 [D and E]), and amplicons then were processed for PrPSc Western blotting detection (Sha31 anti-PrP antibody). Lane 1, positive Western blotting control. Lanes 2 to 6 correspond to WBC prepared at 30, 60, 90, 130, and 190 days postinoculation, respectively, in PG127-challenged animals. Lanes 9 and 10 correspond to WBC prepared from TSE-free controls.
After one PMCA round, PrPres was detected in all tubes seeded with WBC collected at 190 dpi from orally challenged VRQ/VRQ donors (Fig. 2C, lane 6). After a second round of PMCA, tubes seeded with WBC collected at 90 dpi and later were PrPres positive (Fig. 2D, lanes 2 to 6). No PrPres was detected in tubes seeded with WBC samples collected from inoculated ARR/ARR control sheep (Fig. 2E, lanes 2 to 6) or from ARR/ARR (n = 5) (Fig. 2E, lanes 7 and 8) and VRQ/VRQ (n = 15) (Fig. 2C and D, lanes 7 and 8) TSE-free sheep.
Platelet and leukocyte infectivity.
Since WBC fractions contain both leukocytes and platelets, a specific preparation procedure was applied for assessing the infectivity in each of these components. Blood was collected from five VRQ/VRQ sheep orally challenged with PG127 scrapie (Table 7). Platelets were obtained directly from plasma-rich platelets, and the presence of contaminant leukocytes/red blood cells was carefully examined (see Materials and Methods). In parallel, WBC were submitted to successive washings (n = 10; PBS-5 mM EDTA) and centrifugation cycles to eliminate platelets. After 5 and 10 washes, flow cytometry and microscopic examinations of WBC showed no evidence of platelets. The inoculation of platelets prepared from plasma resulted in clinical TSE transmission in tg338 mice (Table 7). The inoculation of tg338 mice with unwashed and washed WBC (corresponding to the same starting amount of whole blood) resulted in 100% attack rate transmission with no major modification of the incubation periods. These results support the hypothesis that the infectivity is associated with both platelets and leukocytes. Considering the loss of platelets during the preparation step, providing an estimate of platelet infectious titer on the basis of our data would not be pertinent.
Table 7.
Intracerebral inoculation of tg338 mice with platelets, washed or unwashed WBC prepared from VRQ/VRQ sheep orally inoculated with PG127 scrapiea
| Sheep | Result by sample |
|||||||
|---|---|---|---|---|---|---|---|---|
| Platelets |
WBC (unwashed) |
WBC (5 washes) |
WBC (10 washes) |
|||||
| No. of mice positive for abnormal PrP | Length of incubation (days) | No. of mice positive for abnormal PrP | Length of incubation (days) | No. of mice positive for abnormal PrP | Length of incubation (days) | No. of mice positive for abnormal PrP | Length of incubation (days) | |
| D7 | 6/6 | 95 ± 10 | 6/6 | 84 ± 3 | 6/6 | 87 ± 4 | 6/6 | 85 ± 3 |
| D8 | 6/6 | 97 ± 4 | 6/6 | 87 ± 4 | 6/6 | 92 ± 7 | 6/6 | 96 ± 3 |
| D9 | 3/5 | 108 ± 6 | 5/5 | 84 ± 3 | 5/5 | 86 ± 4 | 5/5 | 85 ± 3 |
| D10 | 5/5 | 98 ± 8 | 5/5 | 93 ± 9 | 5/5 | 95 ± 3 | 5/5 | 106 ± 6 |
| D11 | 4/5 | 113 ± 12 | 5/5 | 88 ± 2 | 5/5 | 97 ± 3 | 5/5 | 93 ± 3 |
Five susceptible VRQ/VRQ sheep (identified as D7, D8, D9, D10, and D11) were orally challenged with 2 g of brain homogenate (106.6 ID50/g IC in tg338 mice) between 6 and 10 months of age. These animals died at 228, 248, 256, 242, and 244 dpi, respectively. Classical scrapie occurrence was confirmed by histopathology (vacuolar changes in the central nervous system) and the detection of abnormal PrP deposits in the central nervous system and lymphoid tissues. Whole-blood samples (15 ml) were collected at 220 dpi, and platelets (derived from platelet-rich plasma) and WBC were prepared. WBC was washed 5 or 10 times in PBS-5 mM EDTA. WBC (washed and unwashed) corresponding to 5 ml of starting blood and platelets corresponding to a starting volume of 15 ml were resuspended into 120 μl of 5% glucose before homogenization. Each homogenate was inoculated (20 μl) intracerebrally into tg338 mouse groups (n = 6). Mice then were observed until the occurrence of clinical signs or killed after 200 days postinoculation. Mice were considered positive when abnormal PrP deposition was detected in the brain. Incubation periods are presented as means ± SD.
Infectivity in mononucleated cell subpopulations.
PBMC prepared from blood collected from five VRQ/VRQ sheep challenged orally with PG127 scrapie at the late stage of incubation (210 dpi) were inoculated into tg338 mice. Using the same PBMC preparations, a variety of mononucleated cell subpopulations were sorted by magnetic bead technology using antibodies raised against different leukocyte phenotypic markers (CD14, CD4/CD8, and CD45R). The double-positive selection and depletion method used ensured that the sorted cell fractions had purity greater than 95% as assessed by flow cytometry. The inoculation of the different cell subpopulations (CD14+, CD14−, CD4/CD8+, CD4/CD8−, CD45R+, and CD45R−) in tg338 mice resulted in positive TSE transmission (Table 8). The transmission rate and incubation periods observed after the inoculation of 105 to 106 CD4/8+, CD45R+, or CD14+ cells in tg388 mice suggested the presence of low and not fundamentally dissimilar infectious titers in these different cell subpopulations.
Table 8.
Intracerebral inoculation of tg338 mice with PBMC and cell-sorted mononuclear cell subpopulations prepared from VRQ/VRQ sheep orally inoculated with PG127 scrapiea
| Sample type | Results for samples from sheep: |
|||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D7 |
D9 |
D10 |
D11 |
D12 |
||||||||||||||||
| Purity (%) | No. of cells inoculated/mouse | tg338 transmission |
Purity (%) | No. of cells inoculated/mouse | tg338 transmission |
Purity (%) | No. of cells inoculated/mouse | tg338 transmission |
Purity (%) | No. of cells inoculated/mouse | tg338 transmission |
Purity (%) | No. of cells inoculated/mouse | tg338 transmission |
||||||
| No. of mice positive for abnormal PrP | Incubation time (days) | No. of mice positive for abnormal PrP | Incubation time (days) | No. of mice positive for abnormal PrP | Incubation time (days) | No. of mice positive for abnormal PrP | Incubation time (days) | No. of mice positive for abnormal PrP | Incubation time (days) | |||||||||||
| PBMC | 4 × 106 | 6/6 | 96 ± 5 | 4 × 106 | 6/6 | 100 ± 3 | 4 × 106 | 6/6 | 86 ± 2 | 4 × 106 | 6/6 | 105 ± 14 | 4 × 106 | 6/6 | 104 ± 14 | |||||
| CD14+ | 98.2 | 1.5 × 105 | 3/6 | 119 ± 17 | 96.4 | 2 × 105 | 3/7 | 107, 133, 138 | 98.2 | 1.8 × 105 | 6/6 | 111 ± 17 | 95.2 | 1.7 × 105 | 4/6 | 123 ± 9 | 96.3 | 1.7 × 105 | 3/6 | 124 ± 17 |
| CD14− | 99 | 1.7 × 106 | 5/6 | 112 ± 14 | 99.3 | 2.8 × 106 | 6/6 | 113 ± 7 | 99.2 | 2.2 × 106 | 6/6 | 107 ± 6 | 99.6 | 3.1 × 106 | 6/6 | 117 ± 9 | 99.7 | 1.5 × 106 | 6/6 | 107 ± 13 |
| CD4/CD8+ | 88.7 | NA | 96.1 | 6 × 105 | 5/6 | 130 ± 15 | 95.3 | 5.8 × 105 | 1/6 | 119 | 95.5 | 7.8 × 105 | 2/6 | 128, 150 | 96.4 | 5.3 × 105 | 2/6 | 117, 119 | ||
| CD4/CD8− | 98.7 | NA | 97.8 | 3.1 × 105 | 4/6 | 124 ± 11 | 98.2 | 5 × 105 | 5/6 | 110 ± 6 | 99.4 | 6.1 × 105 | 4/6 | 123 ± 20 | 98.6 | 3.8 × 105 | 5/6 | 104 ± 8 | ||
| CD45R+ | 91.1 | NA | 95.7 | 3 × 105 | 0/6 | > 200 | 98.4 | 3.5 × 105 | 3/6 | 127 ± 15 | 97.8 | 4 × 105 | 0/6 | >200 | 97.2 | 4.6 × 105 | 1/6 | 112 | ||
| CD45R− | 87.3 | NA | 98.2 | 4 × 105 | 6/6 | 125 ± × 10 | 99.2 | 7.2 × 105 | 4/6 | 112 ± 21 | 97.3 | 8 × 105 | 3/6 | 116 ± 11 | 98.4 | 3.2 × 105 | 7/7 | 101 ± 7 | ||
Five susceptible VRQ/VRQ sheep (identified as D7, D9, D10, D11, and D12) aged between 6 and 10 months were orally challenged with 2 g of brain homogenate (106.6 ID50/g IC in tg338 mice). These animals were sampled at 210 dpi and died at 228, 256, 242, 244, and 248 dpi, respectively. Classical scrapie occurrence was confirmed by histopathology (vacuolar changes in the central nervous system) and the detection of abnormal PrP deposits in the central nervous system and lymphoid tissues. Whole-blood samples (150 ml) were collected for PBMC preparations. CD14+, CD14− (FITC-labeled VPM65 MAb), CD4/CD8+, CD4/CD8− (FITC-labeled SBU/T4 and SBU/T8 MAb), CD45R+, and CD45R− (FITC-labeled 20.29 MAb) subpopulations were sorted using anti-FITC-coated magnetic beads and two passages on magnetically activated cell sorting minicolumns. The purity of sorted cells was assessed by flow cytometry (percentage of FL1-labeled cells for 2 × 104 events). After preparation, cells were counted and pelleted. Each fraction was resuspended in 200 μl of a 5% glucose solution before grinding. Each homogenate was inoculated intracerebrally into tg338 mice (n = 6; 20 μl per mouse). Equivalent numbers of cells inoculated into each mouse are indicated. Mice then were observed until the occurrence of clinical signs or were killed after 200 days postinoculation. Mice were considered positive when abnormal PrP deposition was detected in the brain. Incubation periods are presented as means ± SD except for dilutions with which less than 50% of mice were found positive. In those cases, incubation times of the positive mice are individually presented.
DISCUSSION
The range of the v-CJD incubation period in patients that were dietarily exposed to BSE remains open to debate. On the basis of v-CJD cases that had occurred in prnp homozygous methionine 129 patients, it was estimated to exceed 10 years (14). In that context, the prionemia in the preclinical phase represents a major concern in transfusion medicine due to the capacity of blood collected from v-CJD-incubating patients to transmit disease.
Our results indicate that in naturally and experimentally infected sheep models, blood collected as early as 3 months after birth or after inoculation can efficiently transmit the disease by transfusion in the homologous host. They also indicate that once established in an incubating individual, prionemia persists throughout the incubation period. In agreement with the data obtained from whole-blood transfusion in sheep, both WBC and PBMC showed infectivity when they were obtained from blood collected as early as 60 to 90 days after birth or TSE exposure.
Our observations contrast with those previously reported by Houston et al., who used another natural scrapie infection model (Neuropathogenesis Unit's [NPU's] scrapie-infected flock) (25). In those experiments, a single blood collection was performed from sheep that were at different stages of the incubation period before transfusion into homologous-genotype recipient sheep. Whole blood (n = 3) collected from infected sheep during the first third of the incubation period apparently failed to transmit the disease, whereas the majority of the recipients who received whole blood collected after the first half of the incubation period developed scrapie. These apparent discrepancies between the study of Houston et al. and our study could be explained by the nature of the scrapie agents involved, which affect the kinetics of TSE agent dissemination and accumulation in host tissues and on the final incubation period (around 740 days in this model and 1,300 days in Houston's experiment) (6). This hypothesis is further reinforced by the results obtained by the same group in similarly designed experiments using sheep orally challenged with BSE, in which whole blood collected at 27% of the incubation period transmitted the disease by transfusion (34). Under the hypothesis that the blood transfusion models in sheep represent a pertinent model for v-CJD blood-borne TSE transmission, these data collectively support the view that blood collected from v-CJD-infected patients represents a risk from a very early stage of the incubation period.
The presence of infectivity in the buffy coat of sheep affected with either natural scrapie or experimental BSE was clearly established by previous studies (25, 26). However, the nature of the elements that contain infectivity was not assessed. Our results demonstrate that in ovine cells, cellular elements belonging to different hematopoietic lineages, such as platelets, monocytes (CD14+) (7), and lymphocytes (CD4/CD8+ and CD45R+), can bear TSE infectivity. The presence of infectivity in B cells supports the observation reported for CWD-affected white-tailed deer. This finding was interpreted as the consequence of lymphocyte recirculation from prion-accumulating lymphoid germinal centers (33). However, it does not explain the presence of infectivity in platelets and CD14+ cells. Indeed, these two cellular subsets do not reach and/or recirculate from the lymphoid germinal centers (42). Considering the short life of platelets (3 days) (36) and short residence time of monocytes in blood (3 days) (41), an alternative hypothesis is that a certain cell lineage in bone marrow is the origin of infectivity in these cells. The expression of PrPC by long-term hematopoietic stem cells (43) and the previously reported infectivity in scrapie-infected sheep bone marrow (22) could be considered supportive elements for this hypothesis. However, these results do not constitute definitive proof that bone marrow is a source of the prionemia, and specific experiments will have to be designed to answer this question.
Despite 15 years of effort, the development of a TSE diagnostic blood assay relying on the direct detection of PK-resistant PrP remains an unachieved goal. As an explanation, it was proposed that misfolded PrP levels in blood are insufficient to allow PrPres detection. Alternatively, it was proposed that TSE agents in blood are associated with abnormal PrP species that would differ from those accumulating in CNS and lymphoid tissues. The discovery of PK-sensitive PrP aggregates, which specifically accumulate in the tissues of infected individuals (40), was considered supportive for this statement, even if there is no definitive evidence that the described PK-sensitive PrP aggregates are associated with infectivity (15). This hypothesis is in direct contradiction to our findings which indicate that WBC and brain-associated infectivity do not display fundamentally dissimilar PK resistance. The presence of the TSE agent in WBC fractions following PK digestion under conditions that ensure PrPC depletion also suggest that the direct detection of the PrPres fraction in blood remains a valid approach to develop a blood diagnostic assay. However, the infectious titer that we measured in WBC (corresponding to 1 ml of blood) was very low, i.e., equivalent to the infectious load contained in a few micrograms of posterior brain stem from an affected individual. This value gives a clear indication of the sensitivity requirement and difficulty for direct PrPSc detection in the blood.
In contrast to direct PrPSc detection, PMCA followed by PrPres detection typically allows us to reach the necessary level of sensitivity. Within the limits of our model (PG127 scrapie and sample numbers), this approach displayed a high specificity and sensitivity equivalent to those of a bioassay in transgenic mice that overexpress PrP for TSE agent detection in blood. Even if further experiments including other TSE agents and host species are required before drawing definitive conclusions, these results support the contention that PMCA on WBC represents a promising approach for the early and reliable diagnosis of TSE infection.
In contrast to the results reported for hamsters (32), our study demonstrated that the extensive washing of sheep WBC had no apparent effect on their infectivity. In addition, our results strongly support the contention that ovine platelets are associated with limited but consistent levels of infectivity. These data concur and reinforce those recently obtained with the BSE-infected sheep model, where leucodepleted or nonleucodepleted platelet concentrates prepared from infected donors transmitted the disease following intravenous administration in ovine recipients (34). These results are clearly discrepant with observations reported for both hamster (23) and mouse (12) TSE models, in which platelets do not contain detectable infectivity. As in the ovine model, infectivity in platelets was reported for CWD-affected white-tailed deer (33). There are significant differences between the PrPC expression patterns and levels in blood cells, in particular in platelets, between rodents and other species (35). These variations in PrPC expression could at least partly explain the differences in infectivity distribution between rodents and ovine/cervids. This raises some concerns about the rationale of measures that were implemented to prevent v-CJD blood-borne transmission risk, which essentially rely on risk assessment models that were developed using TSE rodent models. It underlines the need to generate data on the distribution and the level of infectivity in the human blood using samples that were collected from v-CJD-affected patients for a more accurate assessment of risk.
ACKNOWLEDGMENTS
This work was funded in part by DEFRA and the EU 7th Framework project priority.
Footnotes
Published ahead of print 7 December 2011
REFERENCES
- 1. Andréoletti O, et al. 2000. Early accumulation of PrP(Sc) in gut-associated lymphoid and nervous tissues of susceptible sheep from a Romanov flock with natural scrapie. J. Gen. Virol. 81:3115–3126 [DOI] [PubMed] [Google Scholar]
- 2. Andréoletti O, et al. 2002. PrP(Sc) accumulation in placentas of ewes exposed to natural scrapie: influence of foetal PrP genotype and effect on ewe-to-lamb transmission. J. Gen. Virol. 83:2607–2616 [DOI] [PubMed] [Google Scholar]
- 3. Andréoletti O, et al. 2011. Atypical/Nor98 scrapie infectivity in sheep peripheral tissues. PLoS Pathog. 7:e1001285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andréoletti O. Atypical/Nor98 scrapie infectivity in sheep peripheral tissues. PLoS Pathog. 7:e1001285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arsac JN, et al. 2007. Similar biochemical signatures and prion protein genotypes in atypical scrapie and Nor98 cases, France and Norway. Emerg. Infect. Dis. 13:58–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bellworthy SJ, et al. 2008. Oral transmission of BSE to VRQ/VRQ sheep in an experimental flock. Vet. Rec. 162:130–131 [DOI] [PubMed] [Google Scholar]
- 7. Berthon P, Hopkins J. 1996. Ruminant cluster CD14. Vet. Immunol. Immunopathol. 52:245–248 [DOI] [PubMed] [Google Scholar]
- 8. Brown P, et al. 1998. The distribution of infectivity in blood components and plasma derivatives in experimental models of transmissible spongiform encephalopathy. Transfusion 38:810–816 [DOI] [PubMed] [Google Scholar]
- 9. Bruce ME, et al. 1997. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 389:498–501 [DOI] [PubMed] [Google Scholar]
- 10. Castilla J, et al. 2006. Protein misfolding cyclic amplification for diagnosis and prion propagation studies. Methods Enzymol. 412:3–21 [DOI] [PubMed] [Google Scholar]
- 11. Castilla J, Saa P, Soto C. 2005. Detection of prions in blood. Nat. Med. 11:982–985 [DOI] [PubMed] [Google Scholar]
- 12. Cervenakova L, et al. 2003. Similar levels of infectivity in the blood of mice infected with human-derived vCJD and GSS strains of transmissible spongiform encephalopathy. Transfusion 43:1687–1694 [DOI] [PubMed] [Google Scholar]
- 13. Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. 1996. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 383:685–690 [DOI] [PubMed] [Google Scholar]
- 14. Collinge J, et al. 2006. Kuru in the 21st century–an acquired human prion disease with very long incubation periods. Lancet 367:2068–2074 [DOI] [PubMed] [Google Scholar]
- 15. Cronier S, et al. 2008. Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem. J. 416:297–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Edgeworth JA, et al. 2011. Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet 377:487–493 [DOI] [PubMed] [Google Scholar]
- 17. Elsen JM, et al. 1999. Genetic susceptibility and transmission factors in scrapie: detailed analysis of an epidemic in a closed flock of Romanov. Arch. Virol. 144:431–445 [DOI] [PubMed] [Google Scholar]
- 18. Féraudet C, et al. 2005. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J. Biol. Chem. 280:11247–11258 [DOI] [PubMed] [Google Scholar]
- 19. Fraser H, Dickinson AG. 1968. The sequential development of the brain lesion of scrapie in three strains of mice. J. Comp. Pathol. 78:301–311 [DOI] [PubMed] [Google Scholar]
- 20. Grassi J, Maillet S, Simon S, Morel N. 2008. Progress and limits of TSE diagnostic tools. Vet. Res. 39:33. [DOI] [PubMed] [Google Scholar]
- 21. Gregori L, et al. 2006. Reduction in infectivity of endogenous transmissible spongiform encephalopathies present in blood by adsorption to selective affinity resins. Lancet 368:2226–2230 [DOI] [PubMed] [Google Scholar]
- 22. Hadlow WJ, Kennedy RC, Race RE. 1982. Natural infection of Suffolk sheep with scrapie virus. J. Infect. Dis. 146:657–664 [DOI] [PubMed] [Google Scholar]
- 23. Holada K, et al. 2002. Scrapie infectivity in hamster blood is not associated with platelets. J. Virol. 76:4649–4650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Houston F, Foster JD, Chong A, Hunter N, Bostock CJ. 2000. Transmission of BSE by blood transfusion in sheep. Lancet 356:999–1000 [DOI] [PubMed] [Google Scholar]
- 25. Houston F, et al. 2008. Prion diseases are efficiently transmitted by blood transfusion in sheep. Blood 112:4739–4745 [DOI] [PubMed] [Google Scholar]
- 26. Hunter N, et al. 2002. Transmission of prion diseases by blood transfusion. J. Gen. Virol. 83:2897–2905 [DOI] [PubMed] [Google Scholar]
- 27. Hunter N, et al. 1996. Natural scrapie in a closed flock of Cheviot sheep occurs only in specific PrP genotypes. Arch. Virol. 141:809–824 [DOI] [PubMed] [Google Scholar]
- 28. Hunter N, Houston F. 2002. Can prion diseases be transmitted between individuals via blood transfusion: evidence from sheep experiments. Dev. Biol. 108:93–98 [PubMed] [Google Scholar]
- 29. Ironside JW, Head MW. 2004. Variant Creutzfeldt-Jakob disease: risk of transmission by blood and blood products. Haemophilia 10(Suppl. 4):64–69 [DOI] [PubMed] [Google Scholar]
- 30. Lacroux C, et al. 2007. Dynamics and genetics of PrPSc placental accumulation in sheep. J. Gen. Virol. 88:1056–1061 [DOI] [PubMed] [Google Scholar]
- 31. Le Dur A, et al. 2005. A newly identified type of scrapie agent can naturally infect sheep with resistant PrP genotypes. Proc. Natl. Acad. Sci. U. S. A. 102:16031–16036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lefrère JJ, Hewitt P. 2009. From mad cows to sensible blood transfusion: the risk of prion transmission by labile blood components in the United Kingdom and in France. Transfusion 49:797–812 [DOI] [PubMed] [Google Scholar]
- 33. Mathiason CK, et al. 2010. B cells and platelets harbor prion infectivity in the blood of deer infected with chronic wasting disease. J. Virol. 84:5097–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McCutcheon S, et al. 2011. All clinically-relevant blood components transmit prion disease following a single blood transfusion: a sheep model of vCJD. PLoS One 6:e23169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rocchi MS, et al. 2007. Three-colour flow cytometric detection of PrP in ovine leukocytes. Vet. Immunol. Immunopathol. 116:172–181 [DOI] [PubMed] [Google Scholar]
- 36. Schneider MD, Miller JK, White PK, Ramsey N. 1983. Life span and tissue distribution of 111 indium-labeled blood platelets in hypomagnesemic lambs. Am. J. Vet. Res. 44:806–810 [PubMed] [Google Scholar]
- 37. Simmons HA, et al. 2009. Atypical scrapie in sheep from a UK research flock which is free from classical scrapie. BMC Vet. Res. 5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thorne L, Terry LA. 2008. In vitro amplification of PrPSc derived from the brain and blood of sheep infected with scrapie. J. Gen. Virol. 89:3177–3184 [DOI] [PubMed] [Google Scholar]
- 39. Turner ML, Ludlam CA. 2009. An update on the assessment and management of the risk of transmission of variant Creutzfeldt-Jakob disease by blood and plasma products. Br. J. Haematol. 144:14–23 [DOI] [PubMed] [Google Scholar]
- 40. Tzaban S, et al. 2002. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 41:12868–12875 [DOI] [PubMed] [Google Scholar]
- 41. Whitelaw DM. 1966. The intravascular lifespan of monocytes. Blood 28:455–464 [PubMed] [Google Scholar]
- 42. Young AJ, Hay JB. 1995. Rapid turnover of the recirculating lymphocyte pool in vivo. Int. Immunol. 7:1607–1615 [DOI] [PubMed] [Google Scholar]
- 43. Zhang CC, Steele AD, Lindquist S, Lodish HF. 2006. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc. Natl. Acad. Sci. U. S. A. 103:2184–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]