

Abstract
It is known that cytotoxic T lymphocytes (CTLs) recognizing HIV-1 escape mutants are elicited in HIV-1-infected individuals, but their role in the control of HIV-1 replication remains unclear. We investigated the antiviral ability of CTLs recognizing the HLA-A*24:02-restricted Gag28 -36 (KYKLKHIVW) epitope and/or its escape mutant (KYRLKHIVW) elicited in the early and chronic phases of the infection. Wild-type (WT)-epitope-specific CTLs, as well as cross-reactive CTLs recognizing both WT and K30R (3R) epitopes, which were predominantly elicited at early and/or chronic phases in HLA-A*24:02+ individuals infected with the WT virus, suppressed the replication of the WT virus but failed to suppress that of the 3R virus, indicating that the 3R virus was selected by these 2 types of CTLs. On the other hand, cross-reactive and 3R-specific CTLs, which were elicited in those infected with the 3R virus, did not suppress the replication of either WT or 3R virus, indicating that these CTLs did not contribute to the control of 3R virus replication. High accumulation of the 3R mutation was found in a Japanese population recently recruited. The selection and accumulation of this 3R mutation resulted from the antiviral ability of these Gag28-specific CTLs and high prevalence of HLA-A*24:02 in a Japanese population. The present study highlighted the mechanisms for the roles of cross-reactive and mutant-epitope-specific CTLs, as well as high accumulation of escape mutants, in an HIV-1-infected population.
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1)-specific cytotoxic T lymphocytes (CTLs) play an important role in the control of HIV-1 during the acute and chronic phases of an HIV-1 infection (22, 40). However, HIV-1-specific CTLs cannot completely eliminate HIV-1-infected cells, because HIV-1 escapes from CTL-mediated immune pressure by various mechanisms, such as selection of escape mutations, Nef-mediated HLA class I downregulation, and skewed maturation of memory HIV-specific CD8+ T lymphocytes (5, 8, 9). The most documented escape mechanism is acquisition of amino acid mutations within the CTL epitope and/or its flanking regions. These mutations lead to reduced ability of peptide to bind to HLA class I molecules, impaired T cell receptor (TCR) recognition, and defective epitope generation (21, 31). These escape mechanisms are involved in impaired activities of HIV-1-specific CTLs to kill target cells infected with escape mutant virus and to suppress HIV-1 replication, contributing to the selection of escape mutant viruses (5, 10, 13, 20, 29, 35, 41).
There is growing evidence that escape mutations selected by HLA class I-restricted CTLs accumulate at the population level (7, 28, 36). The accumulation of escape mutants may affect the clinical outcomes for HIV-1-infected individuals (11, 37, 38). On the other hand, it is known that CTLs recognizing escape mutants are elicited after the emergence of the escape mutant selected by wild-type (WT) epitope-specific CTLs (2, 4, 12, 15, 33, 39). The escape mutant-specific CTLs were also elicited in new hosts carrying the same restricted HLA allele when they were infected with the mutant (15). Several studies showed that CTLs cross-recognizing the WT and its escape mutant epitopes are elicited before or after the emergence of the escape mutant in the same hosts (18, 25, 26, 33, 34). However, the antiviral abilities of these cross-reactive CTLs remain unknown, since the recognition of cross-reactive CTLs for synthesized epitope peptides was characterized by using the enzyme-linked immunosorbent spot assay (ELISPOT) or 51Cr cytotoxic assay in those studies. We previously showed that HLA-A*24:02-restricted Nef 138-specific CTLs recognizing an escape mutant had weaker ability to suppress the replication of the mutant virus than that of the WT virus (15). However, it still remains unclear whether cross-reactive or escape mutant-specific CTLs contribute to the control of HIV-1, since the CTLs have not been analyzed in detail.
To clarify the abilities of cross-reactive and escape mutant-specific CTLs to recognize HIV-1-infected cells, we analyzed CTLs specific for HLA-A*24:02-restricted HIV-1 Gag28-36 (KYKLKHIVW; Gag28), which is the only immunodominant Gag epitope presented by this HLA class I allele (24). Since HLA-A*24:02 is found in approximately 70% of the Japanese population (42), the mutants of HLA-A*24:02-restricted epitopes may accumulate in HIV-1-infected Japanese individuals. We previously suggested that K30R (3R) in the Gag28 epitope is an escape mutation from HLA-A*24:02-restricted Gag28-specific CTLs (30) and that CTLs recognizing 3R are elicited in HIV-1-infected HLA-A*24:02+ individuals (46). From these studies, we hypothesized that cross-reactive CTLs recognizing WT and 3R mutant epitopes and/or 3R-specific CTLs are elicited in HLA-A*24:02+ HIV-1-infected individuals after the 3R mutant is selected and in new 3R virus-infected hosts carrying HLA-A*24:02. Here, we investigated the elicitation of Gag28-specific CTLs in 12 HLA-A*24:02+ HIV-1-infected Japanese individuals who could be monitored from the early phase to the chronic phase of an HIV-1 infection, as well as the abilities of cross-reactive, 3R mutant-specific, and WT-specific CTLs to kill WT or 3R virus-infected cells and to suppress the replication of the WT or 3R virus. In addition, we investigated the accumulation of the 3R mutation in HIV-1-infected nonhemophiliac Japanese individuals, as well as in Japanese hemophiliacs who had been infected around 1983. The results clarified the role of CTLs recognizing the WT and/or 3R epitope in high accumulation of the 3R mutant in HIV-1-infected Japanese individuals.
MATERIALS AND METHODS
Samples from HIV-1-infected individuals.
This study was approved by the ethics committee of Kumamoto University and the National Center for Global Health and Medicine. Informed consent was obtained from all individuals according to the Declaration of Helsinki. For sequence analysis, blood specimens were collected in EDTA. Plasma and peripheral blood mononuclear cells (PBMCs) were separated from whole blood. HLA types were determined by standard sequence-based genotyping. Twelve HLA-A*24:02+ individuals who could be monitored from the early to the chronic phase of an HIV-1 infection were recruited for CTL analysis. Early HIV-1 infection was confirmed by seroconversion within 6 months or by an increasing number and density of bands on Western blots. Four-hundred fifty-one chronically HIV-1-infected individuals were also recruited for sequence analysis.
Cells.
C1R cells expressing HLA-A*24:02 (C1R-A2402) and 721.221 cells expressing CD4 and HLA-A*24:02 (721.221-CD4-A2402) were previously generated (27, 30). These cells were cultured in RPMI 1640 medium containing 5 to 10% fetal bovine serum (FBS) and 0.15 mg/ml hygromycin B. MAGIC-5 cells (CCR5-transfected HeLa-CD4/long terminal repeat–β-galactosidase [LTR–β-Gal] cells) were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS as described previously (17).
Induction of Gag28-specific T cells.
PBMCs from HIV-1-infected HLA-A*24:02+ individuals were stimulated with WT or 3R peptide (1 μM) in culture medium (RPMI 1640 containing 10% FBS and 200 U/ml human recombinant interleukin-2 [rIL-2]). After 14 days, the cultured PBMCs were tested for gamma interferon (IFN-γ) production by performing an intracellular cytokine staining (ICC) assay.
ICC assay.
C1R-A2402 cells were prepulsed or not with the WT or 3R peptide at concentrations from 0.1 to 1,000 nM at 37°C for 1 h and then were washed twice with RPMI 1640 containing 10% FBS. PBMCs cultured for 2 weeks after peptide stimulation were incubated with the C1R-A2402 cells in a 96-U plate (Nunc) at 37°C. Brefeldin A (10 μg/ml) was added after a 2-h incubation, and then the cells were incubated for an additional 4 h. Subsequently, the cells were stained with Pacific-blue-conjugated anti-CD8 monoclonal antibody (MAb) (BD Biosciences) and 7-amino-actinomycin D (7-AAD) (BD Biosciences) at 4°C for 30 min, after which the cells were fixed with 4% paraformaldehyde solution and rendered permeable with permeabilization buffer (0.1% saponin and 10% FBS in phosphate-buffered saline) at 4°C for 10 min. Thereafter the cells were stained with fluorescein isothiocyanate (FITC)-conjugated anti-IFN-γ MAb (BD Biosciences) at 4°C for 30 min and then washed twice with the permeabilization buffer. The percentage of CD8+ cells producing IFN-γ was analyzed by flow cytometry (FACSCanto II).
Generation of Gag28-specific CTL clones.
Gag28-specific CTL clones were generated from Gag28-specific bulk-cultured T cells by limiting dilution in 96-U plates, together with 200 μl of cloning mixture (1 × 106 irradiated allogeneic PBMCs from healthy donors and 1 × 105 irradiated C1R-A2402 cells prepulsed with the WT or 3R peptide at a concentration of 1 μM in RPMI 1640 containing 10% FBS, 200 U/ml rIL-2, and 2.5% phytohemagglutinin [PHA] soup). After 14 to 21 days in culture, the growing cells were tested for cytotoxic activity by performing the standard chromium release assay. Since TCRs on these CTL clones were not sequenced, it is still possible that they were oligonucleotide clones.
HIV-1 clones.
An infectious provirus, HIV-1 pNL-432, was reported previously (1). NL-432gagSF2 and NL-432gagSF2-3R were previously generated (30).
Assay of cytotoxicity of CTL clones toward target cells prepulsed with the epitope peptide.
The cytotoxic activities of Gag28-specific CTL clones were determined by use of the standard chromium release assay, as described previously (15). Briefly, 721.221-CD4-A2402 cells were incubated with 100 μCi of Na251CrO4 in saline for 1 h and then washed 3 times with RPMI 1640 containing 10% newborn calf serum. The labeled target cells (2 × 103/well) were prepulsed with the WT or 3R peptide at concentrations of 1 to 1,000 nM for 1 h and then cocultured at 37°C for 4 h with effector cells at an effector-to-target (E:T) ratio of 1:1 in 96-U plates (Nunc). The supernatants were collected and analyzed with a gamma counter. Spontaneous 51Cr release was determined by measuring the counts per minute in supernatants from wells containing only target cells (cpm spn). Maximum 51Cr release was determined by measuring the cpm in supernatants from wells containing target cells in the presence of 2.5% Triton X-100 (cpm max). Specific lysis was defined as (cpm exp − cpm spn)/(cpm max − cpm spn) × 100, where “cpm exp” is the counts per minute in the supernatant in the wells containing both target and effector cells.
Assay of cytotoxicity of CTL clones toward target cells infected with HIV-1.
721.221-CD4-A2402 cells were infected with WT or 3R virus, and then the infection rates were determined by detecting intracellular p24 antigen (Ag)-positive cells stained with FITC-conjugated anti-p24 Ag MAb (KC57-FITC; BD Biosciences). When approximately 50% of the total cells were p24 Ag-positive cells, they were used as target cells. The 51Cr-labeled target cells (2 × 103/well) were cocultured with effector cells at E:T ratios of 0:1 to 2:1 in 96-U plates at 37°C for 6 h. The supernatants were collected and analyzed with a gamma counter.
Generation of HLA-peptide tetrameric complexes.
HLA class I-peptide tetrameric complexes (tetramers) were synthesized as previously described (3). The WT or 3R peptide was added to the refolding solution containing the biotinylation sequence-tagged extracellular domain of the HLA-A*24:02 molecule and β2 microglobulin. The purified monomer complexes were mixed with phycoerythrin (PE)-labeled streptavidin (Molecular Probes) at a molar ratio of 4:1.
Tetramer binding assay.
CTL clones were stained with PE-conjugated tetramer at concentrations of 1 to 100 nM at 37°C for 30 min. After 2 washes with RPMI 1640 containing 10% FBS (R10), the cells were stained with FITC-conjugated anti-CD8 MAb and 7-AAD at 4°C for 30 min. Thereafter, the cells were washed twice with R10 and then analyzed by flow cytometry (FACSCanto II). The mean fluorescence intensity (MFI) of tetramer-positive cells among CD8-positive cells was calculated.
Replication suppression assay.
The ability of Gag28-specific CTLs to suppress HIV-1 replication was examined as previously described (43). CD4+ T cells were isolated from PBMCs of healthy HLA-A*24:02+ donors and incubated with a given HIV-1 clone at 37°C for 6 h. After 3 washes with R10, the cells (3 × 104/well) were cocultured with Gag28-specific CTL clones at E:T ratios of 0.1:1 to 1:1 in R10 containing 1% nonessential amino acid solution and, 1% 100 mM sodium pyruvate (complete medium) plus 200 U/ml rIL-2. From day 3 to day 7 postinfection, a 30-μl volume of culture supernatant was collected, and the volume removed was replaced with fresh medium. The concentration of p24 Ag was measured by using an enzyme-linked immunosorbent assay (ELISA) (HIV-1-p24-Ag ELISA kit; ZeptoMetrix).
Replication kinetics assay.
The replication kinetics of the WT and 3R viruses were examined as previously described (17). After CD4+ T cells (2 × 106) had been exposed to each infectious virus preparation (500 blue cell-forming units in MAGIC-5 cells) for 2 h and washed twice with R10, they were cultured in 1 ml of R10 containing 1% nonessential amino acid solution and 1% 100 mM sodium pyruvate (complete medium) plus 200 U/ml rIL-2. Then, 0.1 ml of the culture supernatant was collected from day 2 to day 10 postinfection, and the volume removed was replaced with fresh medium. The concentration of p24 Ag in the supernatant was measured by using ELISA. Replication kinetics assays were performed in triplicate.
Sequence of autologous virus.
Viral RNA was extracted from plasma samples from HIV-1-infected individuals by using a QIAamp MinElute virus spin kit (Qiagen). For clone sequencing, cDNA was synthesized from the RNA with SuperScript III and Random Primers (Invitrogen), and the Gag region was amplified by nested PCR with Taq DNA polymerase (Promega). Then, the PCR products were gel purified and cloned with a TOPO TA cloning kit (Invitrogen). For bulk sequencing, the Gag region was amplified from the RNA by using the SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase (Invitrogen) and Gag-specific primers, and then the second PCR was done. We prepared the Gag-specific primer sets shown below. For clone sequencing, 5′-TTTTTGACTAGCGGAGGCTAGAA-3′ and 5′-CACAATAGAGGGTTGCTACTGT-3′ were used for the first PCR and 5′-GGGTGCGAGAGCGTCGGTATTAAGC-3′ and 5′-TAAGTTCTTCTGATCCTGTCTG-3′ for the second PCR. For bulk sequence, 5′-TCTCTCGACGCAGGACTC-3′ and 5′-AGGGTTCCTTTGGTCCTTGT-3′ were employed for the reverse transcription (RT)-PCR and 5′-TCTCTCGACGCAGGACTC-3′ and 5′-TCTCCTACTGGGATAGGTG-3′ for the second PCR. All DNA sequencing was performed by using a BigDye Terminator cycle-sequencing kit (Applied Biosystems) and an ABI Prism 310 or 3100 genetic analyzer.
RESULTS
Selection of the 3R mutation by WT epitope-specific CD8+ T cells in individuals infected with WT virus.
We investigated 12 HIV-1-infected HLA-A*24:02+ individuals who could be monitored from the early to the chronic phases of their infections. We first analyzed the sequence of the Gag28 epitope at an early phase in the 12 HIV-1-infected HLA-A*24:02+ individuals. The WT sequence of the Gag28 epitope was detected in 4 of these individuals, whereas 3R was found in the other 8, suggesting that the former and the latter individuals had been infected with WT and 3R viruses, respectively (Table 1). This is consistent with a previous finding that the 3R mutant is found in approximately 70% of HIV-1-infected HLA-A*24:02+ individuals (30). We investigated the elicitation of Gag28-specific CD8+ T cells in the individuals infected with WT virus. PBMCs from these individuals at early and chronic phases were stimulated with WT or 3R peptide and then cultured for 2 weeks. The frequency of Gag28-specific CD8+ T cells among the cultured cells was measured by performing the ICC assay using WT and 3R peptides. Gag28-specific CD8+ T cells were detected at the early phase in 3 of the 4 individuals when the PBMCs were stimulated with WT peptide (Table 2). In 2 individuals, i.e., KI-092 and KI-161, Gag28-specific CD8+ T cells were much more WT specific than 3R mutant specific, whereas in KI-158 they recognized both peptides, but especially the WT peptide (Fig. 1). On the other hand, cross-reactive CD8+ T cells were induced in KI-092 and KI-161 when their PBMCs had been stimulated with 3R peptide, although the frequency of cross-reactive CD8+ T cells induced by stimulation with 3R peptide was lower than that of WT-specific cells induced by stimulation with WT peptide. The 3R peptide failed to induce Gag28-specific CD8+ T cells in PBMCs from KI-158. Thus, WT-specific CD8+ T cells were predominantly elicited at an early phase in the individuals infected with WT virus, although a small but significant number of cross-reactive T cells were also elicited in them.
Table 1.
Sequence at Gag30 in 12 HLA-A*24:02+ individuals with an early-phase HIV-1 infection
| Patient IDa | Sampling date (mo/day/yr) | Gag30 sequence | Method |
|---|---|---|---|
| KI-091 | 12/13/2000 | 3R | Cloning |
| 12/27/2000 | 3R | Direct | |
| 1/7/2002 | 3R | Direct | |
| 7/9/2003 | 3R | Cloning | |
| 9/29/2004 | 3R | Cloning | |
| 8/4/2005 | 3R | Cloning | |
| KI-092 | 1/22/2001 | WT | Cloning |
| 11/21/2001 | WT | Cloning | |
| 12/10/2002 | WT/3R | Cloning | |
| 8/14/2003 | 3R | Cloning | |
| KI-102 | 5/11/2001 | WT | Direct |
| 7/5/2004 | WT | Direct | |
| 3/28/2005 | WT | Direct | |
| KI-126 | 7/19/2001 | 3R | Direct |
| 1/18/2002 | 3R | Direct | |
| 11/15/2004 | 3R | Direct | |
| 9/12/2005 | 3R | Direct | |
| KI-134 | 10/25/2001 | 3R | Direct |
| 6/30/2004 | 3R | Direct | |
| KI-136 | 10/29/2001 | 3R | Direct |
| 7/10/2003 | 3R | Direct | |
| KI-140 | 11/08/2001 | 3R | Direct |
| KI-151 | 5/2/2001 | 3R | Direct |
| 8/28/2003 | 3R | Direct | |
| KI-154 | 4/12/2002 | 3R | Direct |
| KI-158 | 6/14/2002 | WT | Direct |
| 10/11/2002 | WT | Direct | |
| 8/25/2003 | WT | Direct | |
| 11/14/2003 | WT/3R | Direct | |
| 2/23/2004 | 3R/WT | Direct | |
| 11/1/2004 | 3R | Direct | |
| 4/4/2005 | 3R | Direct | |
| KI-161 | 2/15/2002 | WT | Direct |
| 9/12/2002 | WT | Direct | |
| 3/4/2003 | WT | Direct | |
| 9/30/2003 | WT/3R | Direct | |
| 5/6/2004 | 3R | Direct | |
| 1/27/2005 | 3R | Direct | |
| 6/16/2005 | 3R | Cloning | |
| KI-163 | 8/30/2002 | 3R | Direct |
| 9/27/2004 | 3R | Direct |
ID, identifier.
Table 2.
Responses of CD8+ T cells from individuals infected with WT virus to WT or 3R peptide
| Patient ID | Virus sequence [mo/day/yr (type)] |
PBMC sampling date (mo/day/yr) | PBMCs cultured with: | % IFN-γ-producing cells specific for each peptide among CD8+ T cellsa |
|||
|---|---|---|---|---|---|---|---|
| Early phase | Chronic phase | Without | WT | 3R | |||
| KI-092 | 1/22/2001 (WT) | 8/14/2003 (3R) | 5/24/2001 | WT | 0.2 | 34.4 | 13.7 |
| 3R | 0.1 | 12.1 | 16.8 | ||||
| 2/3/2003 | WT | 0.2 | 5.8 | 4.2 | |||
| 3R | 0.6 | 0.3 | 0.3 | ||||
| KI-102 | 5/11/2001 (WT) | 3/28/2005 (WT) | 7/11/2001 | WT | 1.0 | 0.6 | 1.1 |
| 3R | 1.1 | 1.5 | 2.0 | ||||
| 7/5/2004 | WT | 0.2 | 28.7 | 9.3 | |||
| 3R | 0.6 | 0.7 | 0.6 | ||||
| KI-158 | 6/14/2002 (WT) | 4/4/2005 (3R) | 10/11/2002 | WT | 1.4 | 19.3 | 24.6 |
| 3R | 0.1 | 0.5 | 0.4 | ||||
| 4/4/2005 | WT | 0.3 | 23.3 | 23.8 | |||
| 3R | 0.4 | 18.8 | 20.9 | ||||
| KI-161 | 2/15/2002 (WT) | 6/16/2005 (3R) | 7/26/2002 | WT | 0.0 | 74.5 | 8.0 |
| 3R | 0.2 | 55.1 | 41.8 | ||||
| 5/6/2004 | WT | 0.1 | 21.4 | 4.9 | |||
| 3R | 0.2 | 42.5 | 43.9 | ||||
Without, without peptide. Boldface, positive IFN-γ-producing response.
Fig 1.

Gag28-specific CD8+ T cells from individuals infected with WT virus at early and chronic phases. Gag28-specific CD8+ T cells were induced by stimulating PBMCs from early and chronic phases in 4 WT-virus-infected HLA-A*24:02+ individuals with WT or 3R peptide. The responses of these bulk-cultured cells to C1R-A2402 cells prepulsed with WT or 3R peptide at concentrations of 0.1 to 1,000 nM were analyzed by using the ICC assay.
To clarify the specificity of Gag28-specific CD8+ T cells at the early phase in KI-092 and KI-161, we generated Gag28-specific CTL clones by stimulating early-phase PBMCs from KI-092 and KI-161 with the WT peptide. The CTL clones from KI-092 showed a much greater ability to kill cells prepulsed with WT peptide than to kill those prepulsed with the 3R peptide (Fig. 2A), suggesting that they were WT-specific CTLs. To further clarify the specificity of these T cell clones, we investigated the binding affinity of the clones for WT peptide-binding HLA-A*24:02 tetramer (WT tetramer) and 3R peptide-binding HLA-A*24:02 tetramer (3R tetramer). These clones exhibited much greater binding ability to the WT tetramer than to the 3R tetramer (Fig. 2B). These results together indicate that these were WT-specific CTL clones. We further analyzed the abilities of these clones to recognize HIV-1-infected cells. These CTL clones effectively killed WT-virus-infected cells, but not the 3R virus-infected cells (Fig. 2C), and showed the ability to suppress the replication of WT virus, but not to suppress that of the 3R virus (Fig. 2D). WT-specific CD8+ T cell clones established from early-phase PBMCs of KI-161 also showed a similar ability to kill WT virus-infected and 3R virus-infected cells (Fig. 3). In these individuals, the 3R mutant virus became dominant 1 to 2 years after the early phase (Table 1). Taken together, these findings suggest that the 3R mutation was selected by WT-specific CTLs.
Fig 2.

Antiviral activity of Gag28-specific CTL clones generated from early-phase PBMCs from patient KI-092, infected with WT virus. Gag28-specific CTL clones were generated from early-phase PBMCs from KI-092 by stimulating them with WT peptide. The activities of 3 CTL clones (n = 3) were analyzed. (A) Cytotoxic activity toward 721.221-CD4-A2402 cells prepulsed with the WT or 3R peptide at concentrations of 1 to 1,000 nM. The cytotoxic activity was measured at an E:T ratio of 1:1. (B) Binding affinity to WT and 3R tetramers at concentrations of 1 to 100 nM. The MFI values of the T cell clones are shown. (C) Cytotoxic activity against 721.221-CD4-A2402 cells infected with NL-432gagSF2 (WT virus) or NL-432gagSF2-3R (3R virus). WT-virus-infected (49.1% of total cells were p24 Ag+) and 3R virus-infected (48.6% of total cells were p24 Ag+) cells were used as target cells. The cytotoxic activity was measured at E:T ratios of 0.5:1, 1:1, and 2:1. (D) Abilities of the clones to suppress the replication of WT or 3R viruses. The ability was tested at different E:T ratios. The error bars indicate standard deviations.
Fig 3.

Antiviral activities of Gag28-specific CTL clones generated from PBMCs of patient KI-161, infected with WT virus. Gag28-specific CTL clones were generated from early-phase and chronic-phase PBMCs isolated from KI-161 after stimulating them with the WT and 3R peptides, respectively. Three types of Gag28-36-specific CTL clones, i.e., WT specific (left), cross-reactive (middle), and WT dominant (right), were generated from the early-phase PBMCs. (A) Cytotoxic activity against 721.221-CD4-A2402 cells prepulsed with the WT or 3R peptide at concentrations of 1 to 1,000 nM. The cytotoxic activity was measured at an E:T ratio of 1:1. (B) Binding affinity toward WT and 3R tetramers at concentrations of 1 to 100 nM. The MFIs of the T cell clones are shown. (C) Cytotoxic activity against 721.221-CD4-A2402 cells infected with WT virus or 3R virus. WT-virus-infected (49.0% of total cells were p24 Ag+) and 3R-virus-infected (50.0% of total cells were p24 Ag+) cells were used as target cells. The cytotoxic activity was measured at E:T ratios of 0.5:1, 1:1, and 2:1. (D) Abilities of the clones to suppress the replication of WT or 3R virus. The ability was tested at different E:T ratios. n, number of clones tested. The error bars indicate standard deviations.
The 3R virus was not detected by approximately 4 years postinfection in KI-102, who had been infected with the WT virus (Table 1). This individual did not have Gag28-specific CD8+ T cells at an early phase of the HIV-1 infection (Fig. 1). Interestingly, only WT-specific CD8+ T cells were induced from PBMCs of this patient 2.5 year later. Thus, WT-specific CD8+ T cells did not select 3R within about 2 years after the WT-specific CD8+ T cells had been elicited in the patient.
Cross-reactive CD8+ T cells in individuals who had been infected with WT virus and had selected 3R virus.
We investigated whether the 3R-specific or cross-reactive CD8+ T cells were elicited after the 3R mutant had been selected in individuals who had been infected with the WT virus. In KI-158, no Gag28-specific CD8+ T cells were induced from early-phase PBMCs stimulated with the 3R peptide, whereas cross-reactive CD8+ T cells were induced from chronic-phase PBMCs stimulated with WT peptide or 3R peptide (Fig. 1). In KI-161, Gag28-specific CD8+ T cells recognizing WT peptide more than the 3R peptide were induced from early-phase PBMCs stimulated with WT peptide or the 3R peptide, whereas cross-reactive CD8+ T cells were predominantly induced from chronic-phase PBMCs stimulated with the 3R peptide (Fig. 1). These results indicate that cross-reactive CD8+ T cells became dominant in the Gag28-specific CD8+ T cell population after the emergence of the 3R virus in these 2 individuals.
To investigate the function of these cross-reactive CD8+ T cells, we generated Gag28-specific CTL clones from PBMCs at a chronic phase in KI-161 by stimulating them with the 3R peptide. The CTL clones evenly recognized both WT and the 3R peptides (Fig. 3A) and showed the same binding affinity to the 2 tetramers (Fig. 3B). These results suggest that the two peptides had the same binding affinity for HLA-A*24:02. They effectively killed WT-virus-infected cells and weakly killed the 3R virus-infected cells (Fig. 3C), whereas they suppressed the replication of the WT virus but not that of the 3R virus (Fig. 3D). These results indicate that these cross-reactive CTLs contributed to the selection of the 3R virus. In addition, the results strongly suggest weak presentation of the 3R peptide in the cells infected with 3R virus, because the cross-reactive CTL clones had TCR with the same binding affinity for both HLA-A*24:02-WT peptide and HLA-A*24:02-3R peptide complexes and because WT and 3R peptides had the same binding affinity for HLA-A*24:02. This reduced presentation may have affected the control of 3R virus by the cross-reactive CTLs.
Gag28-specific T cell repertoire in an individual infected with WT virus.
The results in Fig. 1 suggest that both WT-specific and cross-reactive CD8+ T cells were elicited at an early phase of HIV-1 infection in 3 individuals infected with WT virus (KI-092, KI-158, and KI-161). To characterize Gag28-specific CTLs elicited at that time, we established Gag28-specific CTL clones from PBMCs at an early phase in KI-161 by stimulating them with the WT peptide. We found 3 types of CTL clones among the 8 clones analyzed. As shown in Fig. 3A, 3 clones effectively recognized the WT peptide but not the 3R peptide (WT specific), 3 clones recognized the WT peptide more than the 3R peptide (WT dominant), and 2 clones evenly recognized both peptides (cross-reactive). We next investigated the binding affinity of TCRs on these clones to WT tetramer and 3R tetramer. The results confirmed the specificity of these 3 types of CTL clones (Fig. 3B). These results together indicate that KI-161 had a multiple T cell repertoire for the Gag28 epitope before the 3R virus had been selected.
Next, we analyzed the abilities of these T cell clones to kill HIV-1-infected cells. The WT-specific and WT-dominant CTL clones effectively killed the target cells infected with WT virus but failed to kill those infected with the 3R virus (Fig. 3C, left and right graphs under early phase). On the other hand, cross-reactive CTL clones weakly killed the target cells infected with the 3R virus and effectively killed those infected with the WT virus (Fig. 3C, middle graphs under early phase). Then, we analyzed the abilities of these CTL clones to suppress HIV-1 replication. Both WT-specific and cross-reactive CTL clones effectively suppressed the replication of the WT virus, whereas WT-specific and cross-reactive CTL clones exhibited no and weak ability, respectively, to suppress that of the 3R virus (Fig. 3D). These results indicate that WT-specific and cross-reactive CTLs could suppress the replication of the WT virus but that the former CTLs could not suppress the 3R virus in vivo. The latter CTLs may weakly suppress 3R virus in vivo. Interestingly, the WT-dominant CTL clones exhibited much weaker ability to suppress the replication of WT virus than did the WT-specific and cross-reactive CTLs (Fig. 3D), although no difference in killing activity against WT-virus-infected cells was found among these 3 CTL clones. Overall, KI-161 had a multiple Gag28-specific CTL repertoire at an early phase of HIV-1 infection, but only 2 types of Gag28-specific CTLs, which were the majority among the Gag28-specific CTLs, contributed to the suppression of WT virus replication.
Cross-reactive CD8+ T cells and 3R-specific CD8+ T cells in individuals who were infected with 3R virus.
Next, we analyzed the elicitation of Gag28-specific CD8+ T cells in 5 individuals infected with the 3R virus. Gag28-specific CD8+ T cells were detected at both early and chronic phases in 3 individuals, whereas they were found at only the chronic phase in the other 2 (Table 3). Cross-reactive CD8+ T cells were induced by stimulating KI-091 PBMCs from both early and chronic phases, not only with 3R peptide, but also with WT peptide. To characterize Gag28-specific CD8+ T cells in KI-091, we generated Gag28-specific CTL clones from PBMCs at a chronic phase in KI-091 by stimulating them with 3R peptide. We investigated the recognition of 3 CTL clones for WT and 3R peptides. These CTL clones evenly recognized both peptides (Fig. 4A) and revealed the same binding affinity for the 2 tetramers (Fig. 4B), indicating that they were cross-reactive CTLs. They moderately killed target cells infected with either WT or 3R virus (Fig. 4C) but did not suppress the replication of the WT and 3R viruses (Fig. 4D). Thus, Gag28-specific CD8+ T cells elicited in KI-091 had no ability to suppress the replication of WT and 3R viruses. Further analysis of 13 other clones revealed similar characteristics (data not shown), supporting the data indicating that cross-reactive CTLs were predominantly elicited in KI-091.
Table 3.
Responses of CD8+ T cells from individuals infected with 3R virus to WT or 3R peptide
| Patient ID | Virus sequence [mo/day/yr (type)] |
PBMC sampling date (mo/day/yr) | PBMCs cultured with: | % IFN-γ-producing cells specific for each peptide among CD8+ T cellsa |
|||
|---|---|---|---|---|---|---|---|
| Early phase | Chronic phase | Without | WT | 3R | |||
| KI-091 | 12/13/2000 (3R) | 8/4/2005 (3R) | 12/13/2000 | WT | 0.2 | 74.6 | 71.2 |
| 3R | 0.3 | 55.4 | 71.9 | ||||
| 9/29/2004 | WT | 0.2 | 77.7 | 65.5 | |||
| 3R | 0.2 | 61.1 | 69.3 | ||||
| KI-134 | 10/25/2001 (3R) | 6/30/2004 (3R) | 10/25/2001 | WT | 0.4 | 0.6 | 0.8 |
| 3R | 1.0 | 1.1 | 5.7 | ||||
| 1/21/2004 | WT | 0.8 | 1.0 | 0.7 | |||
| 3R | 0.7 | 0.6 | 2.0 | ||||
| KI-136 | 10/29/2001 (3R) | 7/10/2003 (3R) | 10/29/2001 | WT | 0.1 | 0.4 | 0.2 |
| 3R | 0.1 | 0.2 | 0.2 | ||||
| 5/15/2003 | WT | 0.4 | 0.8 | 0.4 | |||
| 3R | 0.1 | 0.2 | 24.8 | ||||
| KI-151 | 2/15/2002 (3R) | 6/16/2005 (3R) | 11/21/2001 | WT | 0.3 | 0.7 | 0.8 |
| 3R | 0.7 | 0.6 | 10.8 | ||||
| 7/28/2004 | WT | 0.4 | 0.7 | 1.3 | |||
| 3R | 0.1 | 0.1 | 44.5 | ||||
| KI-163 | 8/30/2002 (3R) | 9/27/2004 (3R) | 8/30/2002 | WT | 0.2 | 0.3 | 0.2 |
| 3R | 0.2 | 0.4 | 0.2 | ||||
| 8/29/2005 | WT | 0.3 | 0.5 | 0.2 | |||
| 3R | 0.4 | 0.6 | 6.9 | ||||
Without, without peptide. Boldface, positive IFN-γ-producing response.
Fig 4.

Antiviral activities of cross-reactive and 3R-specific CTL clones generated from patients KI-091 and KI-163 infected with 3R virus. Gag28-specific CTL clones were generated from chronic-phase PBMCs isolated from patients KI-091 and KI-163 after their stimulation with 3R peptide. The following activities of these CTL clones were analyzed. (A) Cytotoxic activity against 721.221-CD4-A2402 cells prepulsed with the WT or 3R peptide at concentrations of 1 to 1,000 nM. The cytotoxic activity was measured at an E:T ratio of 1:1. (B) Binding affinity toward WT and 3R tetramers at concentrations of 1 to 100 nM. The MFIs of the T cell clones are shown. (C) Cytotoxic activity against 721.221-CD4-A2402 cells infected with WT virus or 3R virus. WT-virus-infected and 3R virus-infected cells were used as target cells. The frequency of p24 Ag+ cells among the HIV-1-infected cells was as follows: WT-virus-infected cells, 49.1% and 43.1% for CTL clones from KI-091 and KI-163, respectively, and 3R-virus-infected cells, 48.6% and 45.6% for CTL clones from KI-091 and KI-163, respectively. The cytotoxic activity was measured at E:T ratios of 0.5:1, 1:1, and 2:1. (D) Abilities of the clones to suppress the replication of WT or 3R virus. The abilities were tested at different E:T ratios. n, number of clones tested. The error bars indicate standard deviations.
In the chronic phase, KI-091 had cross-reactive CD8+ T cells, whereas 3R-specific CD8+ T cells were found in 4 other individuals (Table 3). To characterize these 3R-specific CD8+ T cells, we generated 3R-specific CTL clones from KI-163 PBMCs at the chronic phase by stimulating them with 3R peptide. All 3 clones recognized the 3R peptide much more effectively than the WT peptide (Fig. 4A). These CTL clones bound to 3R tetramer, but not to WT tetramer (Fig. 4B), indicating that these CTL clones carried a 3R-specific TCR. In addition, we analyzed the abilities of these CTL clones to recognize virus-infected cells and found that they effectively killed target cells infected with 3R virus, but not those infected with WT virus (Fig. 4C). However, they failed to suppress the replication of either 3R or WT virus (Fig. 4D). These results indicate that Gag28-specific CD8+ T cells elicited in all individuals infected with 3R virus had no ability to suppress the replication of WT or 3R virus. Thus, Gag28-specific CD8+ T cells seem to have failed to control the 3R virus, although they were elicited in individuals infected with the 3R virus.
High accumulation of the 3R variant in the Japanese population.
The results described above strongly suggest that WT-specific and cross-reactive CD8+ T cells selected the 3R mutation in the individuals infected with the WT virus and that 3R-specific and cross-reactive CD8+ T cells failed to control the 3R virus in the individuals infected with it. Therefore, we assume that this 3R mutation has accumulated in the HLA-A*24:02+ individuals. In addition, since HLA-A*24:02 is found in approximately 70% of Japanese, we speculate that the mutation has accumulated to high levels in the Japanese population.
A previous study analyzed the frequency of 3R in only 32 HLA-A*24:02+ and 26 HLA-A*24:02− individuals chronically infected with HIV-1 and showed that the frequency of 3R was significantly higher in HLA-A*24:02+ individuals than in the HLA-A*24:02− individuals (30). To confirm the association of this mutation with HLA-A*24:02, we analyzed a large number of chronically HIV-1-infected nonhemophiliac individuals (220 HLA-A*24:02+ and 154 HLA-A*24:02− individuals) recruited from April 2008 to March 2011 (2008 to 2011 cohort). The results confirmed that the frequency of 3R was significantly higher in HLA-A*24:02+ individuals than in the HLA-A*24:02− individuals (P < 0.0005) (Fig. 5). Since 3R was found in 74.7% of the HLA-A*24:02− individuals in this cohort, we speculate that the mutation has been accumulating in the Japanese population. Therefore, we analyzed HIV-1-infected nonhemophiliac Japanese individuals who had been recruited from 1996 to 2002 (1996 to 2002 cohort), as well as Japanese hemophiliacs who had been infected around 1983 (hemophiliac cohort), and then compared them to the 2008 to 2011 cohort (Fig. 5). The association of this mutation with HLA-A*24:02 was also found in both the 1996 to 2002 cohort and the hemophiliac cohort (P < 0.01 and P = 7.4 × 10−7, respectively). The frequency of this mutation in HLA-A*24:02− individuals significantly increased from 0% in the hemophiliac cohort to 50.0% in the 1996 to 2002 cohort (P = 0.0084) and to 74.7% in the 2008 to 2011 cohort (P = 2.6 × 10−7). These results indicate that the 3R mutation was strongly selected by Gag28-specific CTLs and has been accumulating during the past 30 years in the Japanese population.
Fig 5.

Frequencies of the 3R mutation in a Japanese hemophiliac cohort and nonhemophiliac cohorts recruited from 1996 to 2002 and from 2008 to 2011. The frequencies of mutations at position 3 of the Gag28 epitope in chronically HIV-1-infected HLA-A*24:02+ or HLA-A*24:02− hemophiliac individuals and nonhemophiliac individuals recruited from 1996 to 2002 or from 2008 to 2011 are shown. The consensus sequence of this epitope in HIV-1 subtype B is KYKLKHIVW. The frequency of the 3R mutation between HLA-A*24:02+ and HLA-A*24:02− subjects in each cohort or that in HLA-A*24:02+ or HLA-A*24:02− subjects among the 3 cohorts was statistically analyzed by using Fisher's exact test.
It is well known that some escape mutations affect replication capacity and that HIV-1 containing such mutations reverts to WT in individuals not carrying HLA class I restriction alleles (23, 32). We previously showed that the 3R mutation does not affect replication capacity when 2 T cell lines are used in an assay measuring it (46). Since a different effect of mutations on replication capacity between cell lines and CD4+ T cells from a healthy individual is known (23), we measured the replication capacity of the 3R virus by using CD4+ T cells from a healthy individual. The results confirm that this mutation did not affect the replication capacity (Fig. 6), suggesting that the 3R mutant could not revert in HLA-A*24:02− individuals.
Fig 6.
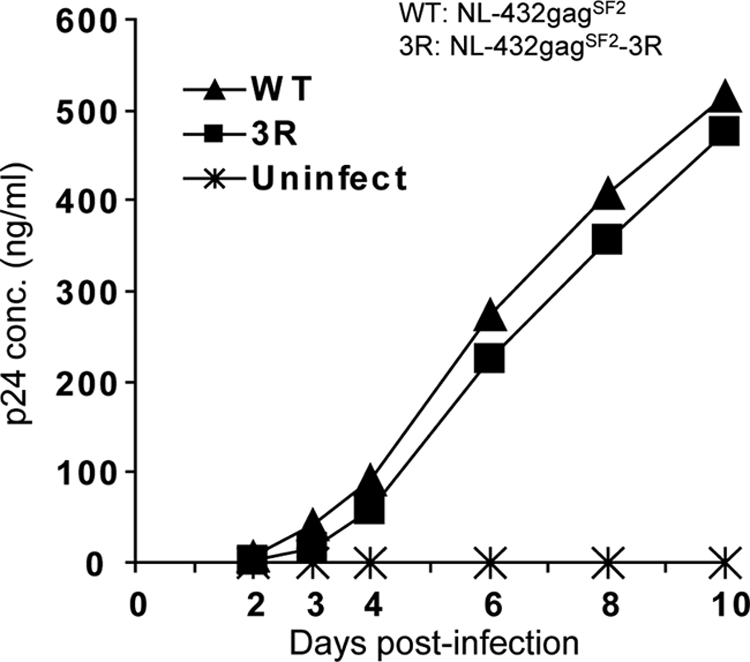
Replication kinetics of WT and 3R viruses in CD4+ T cells. CD4+ T cells (2 × 105) isolated from PBMCs from a healthy donor were infected with WT or 3R virus in triplicate at a blue-cell-forming unit of 500 (in MAGIC-5 cells) in a total volume of 0.2 ml and then incubated at 37°C for 2 h. The infected cells were washed twice with R10 and then cultured in 1 ml of complete medium plus rIL-2 at 37°C. A 0.1-ml volume of the culture supernatants was collected at days 2 to 10 postinfection. The concentration of p24 Ag was measured by using ELISA.
DISCUSSION
It is known that CTLs recognizing escape mutants are elicited after the escape mutant had been selected by WT epitope-specific CTLs (2, 4, 12, 15, 33, 39) or in new escape mutant virus-infected hosts having the same restricted HLA allele (15). However, since the CTLs recognizing escape mutants have been not well analyzed, the role of these CTLs in the control of HIV-1 infections remains unclear. In the present study, we investigated 2 groups, HLA-A*24:02+ individuals infected with WT virus and those infected with 3R escape mutant virus. We found that both WT-specific and cross-reactive CD8+ T cells were elicited in individuals infected with WT virus. Interestingly, cross-reactive T cells had been elicited before the emergence of the 3R escape mutant virus, though a similar finding was made in previous studies that analyzed other epitope-specific CTLs (18, 25, 26, 34). The present study shows that WT-specific CD8+ T cells were predominantly elicited in an early phase of the infection and that the number of cross-reactive CD8+ T cells increased in the chronic phase. The CTL clones from early and chronic phases in KI-161 showed similar abilities to kill WT virus-infected or 3R virus-infected cells and activities to suppress both viruses, suggesting that cross-reactive CD8+ T cells elicited at the early phase were expanded via antigen presentation by 3R virus-infected cells at the chronic phase.
WT-specific and cross-reactive CTL clones from KI-092 and KI-161 at an early phase of the infection effectively killed WT-virus-infected cells and suppressed the replication of the WT virus, whereas they exhibited no and weak ability, respectively, to suppress that of the 3R virus. Cross-reactive CTL clones had the same ability to suppress the replication of WT virus as did the WT-specific CTL clones. These results strongly suggest that both CTLs selected the 3R virus in these individuals infected with the WT virus. The 3R virus was not selected within at least 1 year after Gag28-specific CTLs had been detected in the individuals infected with the WT virus. This finding indicates that the 3R mutation was more slowly selected by these CTLs than escape mutants selected at an acute phase of the infection (16, 19, 34, 44, 45). On the other hand, a previous study suggested that acute accumulation of mutations in this epitope occurs after an HIV-1 infection (6). However, the data shown in that study concerned mutations contained at position 1 of the epitope. In addition, those data may have included cases in which the individuals had been infected with the 3R mutant virus, because it may be assumed that 3R virus had accumulated in the cohorts analyzed. Cross-reactive CTL clones established from PBMCs at both early and chronic phases of KI-161 killed 3R virus-infected cells, though the killing activity against the 3R virus-infected cells was weaker than that against the WT virus-infected cells. These CTL clones weakly suppressed the replication of the 3R virus (Fig. 3C). This weak ability to suppress it might have delayed the emergence of the 3R mutation in these patients.
WT-specific CTLs were not induced by stimulation of early- or chronic-phase PBMCs from the 5 individuals in which the 3R mutation had been detected at the early phase with WT peptides. This finding supports the possibility that these individuals had been infected with the 3R virus. Only KI-091 had cross-reactive T cells at early and chronic phases of the infection. All CTL clones established from this patient had cross-reactivity, implying that the patient had been infected with WT virus and that 3R had been selected at an early phase. However, WT-specific CTL clones were not established from this patient. In addition, the cross-reactive CTL clones established from KI-091 did not have the ability to suppress the replication of the WT virus, although the CTL clones from individuals who had been infected with the WT virus had strong ability to suppress it. These findings suggest that this patient had been infected with the 3R virus rather than with the WT virus. However, it remains unknown why 3R-specific CTLs were elicited in the other 4 individuals but not in this patient. Thus, the abilities of CTLs to respond to WT peptide and to suppress the replication of WT virus together supported the idea that the individuals who had 3R virus in the early phase had been infected with 3R virus, although the possibility that they had been infected with WT virus cannot be completely excluded.
The 3R mutant epitope peptide would have been processed and presented to 3R-specific CTLs in 3R virus-infected cells, since 3R-specific and cross-reactive CTL clones effectively killed 3R virus-infected cells. However, these CTL clones failed to suppress the replication of the 3R virus. 721.221-CD4-A2402 cell lines were used as target cells for the killing assay, whereas CD4+ T cells from healthy individuals were used for the replication suppression assay. The former cells express HLA-A*24:02 to a much higher degree than the latter cells. This difference between the 2 cell lines may account for the discrepancy of the results between the 2 assays. 3R-specific CTL clones failed to suppress the replication of the 3R virus, whereas cross-reactive CTLs from the individuals infected with WT virus effectively suppressed the replication of the WT virus but failed to suppress that of the 3R virus. These findings suggest that 3R virus-infected CD4+ T cells could not effectively present the 3R mutant epitope. This finding also suggests that 3R virus-infected CD4+ T cells were not the main source of antigen-presenting cells in 3R virus-infected individuals. A previous study showed that HIV-1-infected macrophages effectively present HIV-1 epitopes more than HIV-1-infected CD4+ T cells (14), implying that 3R virus-infected macrophages are the main antigen-presenting cells and contribute to the elicitation of 3R-specific and cross-reactive CTLs in 3R virus-infected individuals. A further study should clarify the role of macrophages in the elicitation of 3R-specific and cross-reactive CTLs in 3R virus-infected individuals.
Cross-reactive CTLs were found in individuals infected with the WT virus or with the 3R virus. The CTL clones established from individuals infected with the WT virus had a strong ability to kill WT-virus-infected cells and to suppress the replication of the WT virus, whereas those established from an individual infected with the 3R virus showed moderate ability to kill WT-virus-infected cells and no ability to suppress the replication of WT virus. These findings indicate that cross-reactive CTLs from an individual infected with the 3R virus may have had less ability to recognize the WT epitope than those from an individual infected with the WT virus. Indeed, the former CTL clones exhibited lower sensitivity to reaction with WT peptide-pulsed cells than the latter CTLs, indicating that cross-reactive CTLs elicited in individuals infected with the WT virus had higher-affinity TCRs for WT peptide than those in an individual infected with the 3R virus. In addition, the latter CTL clones weakly killed 3R virus-infected cells, whereas the former clones showed the same killing activity against 3R virus-infected cells as against WT-virus-infected cells. Thus, cross-reactive CTLs in individuals infected with 3R virus have different characteristics than those in individuals infected with the WT virus. This finding suggests that cross-reactive CTLs elicited in individuals infected with the WT virus had TCRs with higher affinity for WT and 3R peptides than those in individuals infected with the 3R virus.
Japanese hemophiliacs were infected with HIV-1 via blood products from the United States around 1983, and HLA-A*24:02 is a rare allele in North America. Therefore, it may be speculated that HIV-1 in the blood product had not yet accumulated escape mutations. Indeed, the 3R mutation was not found in the 12 HLA-A*24:02− hemophiliacs tested, though other amino acid variants at position 3 were detected in 2 of these hemophiliacs. This mutation was found in 50.0% of HLA-A*24:02− individuals in the 1996 to 2002 cohort and in 74.7% of those in the 2008 to 2011 cohort, indicating that the mutation had accumulated in the Japanese population. The frequency of this mutation in HLA-A*24:02− individuals thus increased about 1.5-fold during the approximately 10-year period between these 2 nonhemophiliac cohorts. Thus, the mutation greatly accumulated over the last 10 years. Since HLA-A*24:02 is found in approximately 70% of Japanese, the high prevalence of the allele is the cause of the high accumulation of the 3R mutation in the Japanese population. In addition, this high accumulation resulted not only from a strong selection of the 3R mutation by WT-specific and cross-reactive CTLs elicited in the donors infected with WT virus, but also from a lack of reversion of the mutation in the HLA-A*24:02− individuals.
Our previous study concerning HLA-A*24:02-restricted Nef138-specific CTLs demonstrated that only WT epitope-dominant CTLs, which suppress the replication of WT virus but fail to suppress that of mutant virus, are elicited at an early phase in HLA-A*24:02+ individuals infected with the WT virus and that mutant-epitope-dominant CTLs but not cross-reactive CTLs are elicited after the emergence of the mutant virus in them (15). In addition, only mutant-epitope-dominant CTLs are elicited in those individuals infected with the mutant virus. The mutant-epitope-dominant CTLs suppress the replication of WT virus but weakly suppress that of mutant virus (15). Thus, Nef138-specific CTLs elicited in individuals infected with WT or mutant viruses had different characteristics in terms of the recognition of WT and mutant epitopes than the Gag28-specific CTLs analyzed in the present study. The difference between Nef138-specific and Gag28-specific CTLs might be explained by a different CTL repertoire elicited at an early phase. These 2 studies suggest the elicitation of various HIV-1-specific CTLs in regard to recognition of escape mutations.
In the present study, we demonstrated that WT-specific and cross-reactive CTLs were elicited at an early phase in individuals infected with the WT virus and that cross-reactive CTLs were dominant in Gag28-specific CTLs after the emergence of the 3R virus. On the other hand, 3R-specific and cross-reactive CTLs were elicited in individuals infected with the 3R virus, though the former CTLs were predominantly elicited in these individuals. The CTLs elicited in the individuals infected with the WT virus, which had a strong ability to suppress the replication of WT virus, played a central role in the accumulation of the 3R mutation. In contrast, the CTLs elicited in those infected with 3R virus, which failed to suppress the replication of WT and 3R viruses, did not contribute to the control of the 3R virus infection. In addition, the high prevalence of HLA-A*24:02 and lack of effect of the 3R mutation on viral fitness may have strongly contributed to the high accumulation of the mutation in HIV-1-infeceted Japanese individuals.
ACKNOWLEDGMENTS
This research was supported by the Global COE program Global Education and Research Center Aiming at the Control of AIDS, launched as a project commissioned by the Ministry of Education, Science, Sports, and Culture, Japan; by a grant-in-aid for scientific research from the Ministry of Health (no. 18390141), Japan; and by a grant-in-aid for scientific research from the Ministry of Education, Science, Sports, and Culture (no. 20390134), Japan.
The authors have no conflicting financial interests.
We thank Sachiko Sakai for her secretarial assistance.
Footnotes
Published ahead of print 7 December 2011
REFERENCES
- 1. Adachi A, et al. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Allen TM, et al. 2005. De novo generation of escape variant-specific CD8+ T-cell responses following cytotoxic T-lymphocyte escape in chronic human immunodeficiency virus type 1 infection. J. Virol. 79:12952–12960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Altman JD, et al. 1996. Phenotypic analysis of antigen-specific T lymphocytes. Science 274:94–96 [PubMed] [Google Scholar]
- 4. Bailey JR, Williams TM, Siliciano RF, Blankson JN. 2006. Maintenance of viral suppression in HIV-1-infected HLA-B*57+ elite suppressors despite CTL escape mutations. J. Exp. Med. 203:1357–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Borrow P, et al. 1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 3:205–211 [DOI] [PubMed] [Google Scholar]
- 6. Brumme ZL, et al. 2008. Marked epitope- and allele-specific differences in rates of mutation in human immunodeficiency type 1 (HIV-1) Gag, Pol, and Nef cytotoxic T-lymphocyte epitopes in acute/early HIV-1 infection. J. Virol. 82:9216–9227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carlson JM, Brumme ZL. 2008. HIV evolution in response to HLA-restricted CTL selection pressures: a population-based perspective. Microbes Infect. 10:455–461 [DOI] [PubMed] [Google Scholar]
- 8. Champagne P, et al. 2001. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature 410:106–111 [DOI] [PubMed] [Google Scholar]
- 9. Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. 1998. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391:397–401 [DOI] [PubMed] [Google Scholar]
- 10. Couillin I, et al. 1995. HLA-dependent variations in human immunodeficiency virus Nef protein alter peptide/HLA binding. Eur. J. Immunol. 25:728–732 [DOI] [PubMed] [Google Scholar]
- 11. Crum-Cianflone N, et al. 2009. Is HIV becoming more virulent? Initial CD4 cell counts among HIV seroconverters during the course of the HIV epidemic: 1985–2007. Clin. Infect. Dis. 48:1285–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feeney ME, et al. 2005. HIV-1 viral escape in infancy followed by emergence of a variant-specific CTL response. J. Immunol. 174:7524–7530 [DOI] [PubMed] [Google Scholar]
- 13. Feeney ME, et al. 2004. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J. Virol. 78:8927–8930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fujiwara M, Takiguchi M. 2007. HIV-1-specific CTLs effectively suppress replication of HIV-1 in HIV-1-infected macrophages. Blood 109:4832–4838 [DOI] [PubMed] [Google Scholar]
- 15. Fujiwara M, et al. 2008. Different abilities of escape mutant-specific cytotoxic T cells to suppress replication of escape mutant and wild-type human immunodeficiency virus type 1 in new hosts. J. Virol. 82:138–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ganusov VV, et al. 2011. Fitness costs and diversity of CTL response determine the rate of CTL escape during the acute and chronic phases of HIV infection. J. Virol. 85:10518–10528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gatanaga H, Hachiya A, Kimura S, Oka S. 2006. Mutations other than 103N in human immunodeficiency virus type 1 reverse transcriptase (RT) emerge from K103R polymorphism under non-nucleoside RT inhibitor pressure. Virology 344:354–362 [DOI] [PubMed] [Google Scholar]
- 18. Geels MJ, et al. 2003. Identification of sequential viral escape mutants associated with altered T-cell responses in a human immunodeficiency virus type 1-infected individual. J. Virol. 77:12430–12440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goonetilleke N, et al. 2009. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J. Exp. Med. 206:1253–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goulder PJ, et al. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 3:212–217 [DOI] [PubMed] [Google Scholar]
- 21. Goulder PJ, Watkins DI. 2004. HIV and SIV CTL escape: implications for vaccine design. Nat. Rev. Immunol. 4:630–640 [DOI] [PubMed] [Google Scholar]
- 22. Goulder PJ, Watkins DI. 2008. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat. Rev. Immunol. 8:619–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Honda K, et al. 2011. Selection of escape mutant by HLA-C-restricted HIV-1 Pol-specific cytotoxic T lymphocytes carrying strong ability to suppress HIV-1 replication. Eur. J. Immunol. 41:97–106 [DOI] [PubMed] [Google Scholar]
- 24. Ikeda-Moore Y, et al. 1998. Identification of a novel HLA-A24-restricted cytotoxic T-lymphocyte epitope derived from HIV-1 Gag protein. AIDS 12:2073–2074 [DOI] [PubMed] [Google Scholar]
- 25. Jamieson BD, et al. 2003. Epitope escape mutation and decay of human immunodeficiency virus type 1-specific CTL responses. J. Immunol. 171:5372–5379 [DOI] [PubMed] [Google Scholar]
- 26. Jones NA, et al. 2004. Determinants of human immunodeficiency virus type 1 escape from the primary CD8+ cytotoxic T lymphocyte response. J. Exp. Med. 200:1243–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karaki S, et al. 1993. HLA-B51 transgenic mice as recipients for production of polymorphic HLA-A, B-specific antibodies. Immunogenetics 37:139–142 [DOI] [PubMed] [Google Scholar]
- 28. Kawashima Y, et al. 2009. Adaptation of HIV-1 to human leukocyte antigen class I. Nature 458:641–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kelleher AD, et al. 2001. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J. Exp. Med. 193:375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koizumi H, et al. 2009. Escape mutation selected by Gag28-36-specific cytotoxic T cells in HLA-A*2402-positive HIV-1-infected donors. Microbes Infect. 11:198–204 [DOI] [PubMed] [Google Scholar]
- 31. Lazaro E, et al. 2011. Variable HIV peptide stability in human cytosol is critical to epitope presentation and immune escape. J. Clin. Invest. 121:2480–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leslie AJ, et al. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 10:282–289 [DOI] [PubMed] [Google Scholar]
- 33. Lichterfeld M, et al. 2007. A viral CTL escape mutation leading to immunoglobulin-like transcript 4-mediated functional inhibition of myelomonocytic cells. J. Exp. Med. 204:2813–2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Y, McNevin JP, Holte S, McElrath MJ, Mullins JI. 2011. Dynamics of viral evolution and CTL responses in HIV-1 infection. PLoS One 6:e15639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McMichael AJ, Rowland-Jones SL. 2001. Cellular immune responses to HIV. Nature 410:980–987 [DOI] [PubMed] [Google Scholar]
- 36. Moore CB, et al. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296:1439–1443 [DOI] [PubMed] [Google Scholar]
- 37. Muller V, et al. 2009. Increasing clinical virulence in two decades of the Italian HIV epidemic. PLoS Pathog. 5:e1000454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nakamura H, et al. 2011. Clinical symptoms and courses of primary HIV-1 infection in recent years in Japan. Intern. Med. 50:95–101 [DOI] [PubMed] [Google Scholar]
- 39. O'Connell KA, Hegarty RW, Siliciano RF, Blankson JN. 2011. Viral suppression of multiple escape mutants by de novo CD8+ T cell responses in a human immunodeficiency virus-1 infected elite suppressor. Retrovirology 8:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ogg GS, et al. 1998. Quantitation of HIV-1-specific cytotoxic T lymphocytes and plasma load of viral RNA. Science 279:2103–2106 [DOI] [PubMed] [Google Scholar]
- 41. Price DA, et al. 1997. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. U. S. A. 94:1890–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saito S, Ota S, Yamada E, Inoko H, Ota M. 2000. Allele frequencies and haplotypic associations defined by allelic DNA typing at HLA class I and class II loci in the Japanese population. Tissue Antigens 56:522–529 [DOI] [PubMed] [Google Scholar]
- 43. Tomiyama H, Akari H, Adachi A, Takiguchi M. 2002. Different effects of Nef-mediated HLA class I down-regulation on human immunodeficiency virus type 1-specific CD8(+) T-cell cytolytic activity and cytokine production. J. Virol. 76:7535–7543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Turnbull EL, et al. 2009. Kinetics of expansion of epitope-specific T cell responses during primary HIV-1 infection. J. Immunol. 182:7131–7145 [DOI] [PubMed] [Google Scholar]
- 45. Wang YE, et al. 2009. Protective HLA class I alleles that restrict acute-phase CD8+ T-cell responses are associated with viral escape mutations located in highly conserved regions of human immunodeficiency virus type 1. J. Virol. 83:1845–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yokomaku Y, et al. 2004. Impaired processing and presentation of cytotoxic-T-lymphocyte (CTL) epitopes are major escape mechanisms from CTL immune pressure in human immunodeficiency virus type 1 infection. J. Virol. 78:1324–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]