

Abstract
To establish a cell culture system for chimeric hepatitis C virus (HCV) genotype 2b, we prepared a chimeric construct harboring the 5′ untranslated region (UTR) to the E2 region of the MA strain (genotype 2b) and the region of p7 to the 3′ UTR of the JFH-1 strain (genotype 2a). This chimeric RNA (MA/JFH-1.1) replicated and produced infectious virus in Huh7.5.1 cells. Replacement of the 5′ UTR of this chimera with that from JFH-1 (MA/JFH-1.2) enhanced virus production, but infectivity remained low. In a long-term follow-up study, we identified a cell culture-adaptive mutation in the core region (R167G) and found that it enhanced virus assembly. We previously reported that the NS3 helicase (N3H) and the region of NS5B to 3′ X (N5BX) of JFH-1 enabled replication of the J6CF strain (genotype 2a), which could not replicate in cells. To reduce JFH-1 content in MA/JFH-1.2, we produced a chimeric viral genome for MA harboring the N3H and N5BX regions of JFH-1, combined with a JFH-1 5′ UTR replacement and the R167G mutation (MA/N3H+N5BX-JFH1/R167G). This chimeric RNA replicated efficiently, but virus production was low. After the introduction of four additional cell culture-adaptive mutations, MA/N3H+N5BX-JFH1/5am produced infectious virus efficiently. Using this chimeric virus harboring minimal regions of JFH-1, we analyzed interferon sensitivity and found that this chimeric virus was more sensitive to interferon than JFH-1 and another chimeric virus containing more regions from JFH-1 (MA/JFH-1.2/R167G). In conclusion, we established an HCV genotype 2b cell culture system using a chimeric genome harboring minimal regions of JFH-1. This cell culture system may be useful for characterizing genotype 2b viruses and developing antiviral strategies.
INTRODUCTION
Hepatitis C virus (HCV) is a major cause of chronic liver disease (5, 13), but the lack of a robust cell culture system to produce virus particles has hampered the progress of HCV research (2). Although the development of a subgenomic replicon system has enabled research into HCV RNA replication (15), infectious virus particle production has not been possible. Recently, an HCV cell culture system was developed using a genotype 2a strain, JFH-1, cloned from a fulminant hepatitis patient (14, 29, 32), thereby allowing investigation of the entire life cycle of this virus. However, several groups of investigators have reported genotype- and/or strain-dependent effects of some antiviral reagents (6, 17) and neutralizing antibodies (7, 25). Therefore, efficient virus production systems using various genotypes and strains are indispensable for HCV research and the development of antiviral strategies.
The JFH-1 strain is the first HCV strain that can efficiently produce HCV particles in HuH-7 cells (29). Other strains can replicate and produce infectious virus by HCV RNA transfection, but the efficiency is far lower than that of JFH-1 (24, 31). In the case of replication-incompetent strains, chimeric virus containing the JFH-1 nonstructural protein coding region is useful for analyses of viral characteristics (6, 9, 14, 23, 30, 31).
In this study, we developed a genotype 2b chimeric infectious virus production system using the MA strain (accession number AB030907) (19) harboring minimal regions of JFH-1 and cell culture-adaptive mutations that enhance infectious virus production.
MATERIALS AND METHODS
Cell culture.
Huh7.5.1 cells (a kind gift from Francis V. Chisari) (32) and Huh7-25 cells (1) were cultured at 37°C in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum under 5% CO2 conditions. For follow-up study, RNA-transfected cells were passaged every 2 to 5 days depending on cell status.
Full-length genomic HCV constructs.
Plasmids used in the analysis of genomic RNA replication were constructed based on pJFH1 (29) and pMA (19). For convenience, an EcoRI recognition site was introduced upstream of the T7 promoter region of pMA by PCR, and an XbaI recognition site was introduced at the end of the 3′ untranslated region (UTR). To construct MA/JFH-1, the EcoRI-BsaBI (nucleotides [nt] 1 to 2570; 5′ UTR to E2) fragment of pMA was substituted into pJFH1 (Fig. 1A). Replacement of the 5′ UTR was performed by exchanging the EcoRI-AgeI (nt 1 to 159) fragment. A point mutation in the core region (R167G) was introduced into MA chimeric constructs by PCR using the following primers: sense, 5′-TTA TGC AAC GGG GAA TTT ACC CGG TTG CTC T-3′; antisense, 5′-GGT AAA TTC CCC GTT GCA TAA TTT ATC CCG TC-3′. G167R substitution in the JFH-1 construct was performed by PCR using the following primers: sense, 5′-ATT ATG CAA CAA GGA ACC TAC CCG GTT TCC C-3′; antisense, 5′-GGT AGG TTC CTT GTT GCA TAA TTA ACC CCG TC-3′. Point mutations (L814S, R1012G, T1106A, and V1951A) were introduced into MA chimeric constructs by PCR using the following primers: L814S, 5′-GCT TAC GCC TCG GAC GCC GCT GAA CAA GGG G-3′ (sense) and 5′-AGC GGC GTC CGA GGC GTA AGC CTG CTG CGG C-3′ (antisense); R1012G, 5′-GAG GCT AGG TGG GGA AGT TCT GCT CGG CCC T-3′ (sense) and 5′-AGA ACT TCC CCA CCT AGC CTC GCG GAA ACC G-3′ (antisense); T1106A, 5′-CAG ATG TAC GCC AGC GCA GAG GGG GAC CTC-3′ (sense) and 5′-CTG CGC TGG CGT ACA TCT GGG TGA CTG GTC-3′ (antisense); and V1951A, 5′-GTG ACG CAG GCG TTA AGC TCA CTC ACA ATT ACC-3′ (sense) and 5′-TGA GCT TAA CGC CTG CGT CAC GCG CAG CGA G-3′ (antisense). To construct the MA chimeric virus harboring minimal regions of JFH-1 (MA/N3H+N5BX-JFH1), ClaI (nt 3930), EcoT22I (nt 5294), and BsrGI (nt 7782) recognition sites were introduced into pMA by site-directed mutagenesis. The 5′ UTR (EcoRI-AgeI), the region of the NS3 helicase (N3H; ClaI-EcoT22I), and the region of NS5B to 3′ X (N5BX; BsrGI-XbaI) were then replaced with the corresponding regions from JFH-1.
Fig 1.

Replication and virus production by MA/JFH-1 chimeras in Huh7.5.1 cells. (A) Schematic structures of JFH-1, MA, and two MA/JFH-1 chimeras (MA/JFH-1.1 and MA/JFH-1.2). The junction of JFH-1 and MA in the 5′ UTR is an AgeI site, and the junction of MA and JFH-1 in the NS2 region is a BsaBI site. A, AgeI; B, BsaBI; X, XbaI. (B to E) Chimeric HCV RNA replication in Huh7.5.1 cells. HCV core protein level in cells (B) and culture medium (C) and HCV RNA levels in medium (D) and infectivity of culture medium (E) from HCV RNA-transfected Huh7.5.1 cells are shown. Ten micrograms of HCV RNA was transfected into Huh7.5.1 cells, and cells and culture medium were harvested on days 1, 2, and 3. n.d., not determined. Assays were performed three times independently, and data are presented as means ± standard deviation. Dashed line indicates detection limit. wt, wild type.
RNA synthesis, transfection, and determination of infectivity.
RNA synthesis and transfection were performed as described previously (12, 22). Determination of infectivity was also performed as described previously, with infectivity expressed as the number of focus-forming units per milliliter (FFU/ml) (12, 22). When necessary, culture medium was concentrated 20-fold in Amicon Ultra-15 spin columns (100-kDa molecular-weight-cutoff; Millipore, Bedford, MA) in order to determine infectivity.
Quantification of HCV core protein and HCV RNA.
In order to estimate the concentration of HCV core protein in culture medium, we performed a chemiluminescence enzyme immunoassay (Lumipulse II HCV core assay; Fujirebio, Tokyo, Japan) in accordance with the manufacturer's instructions. HCV RNA from harvested cells or culture medium was isolated using an RNeasy Mini RNA kit (Qiagen, Tokyo, Japan) or QiaAmp Viral RNA Minikit (Qiagen), respectively. Copy number of HCV RNA was determined by real-time quantitative reverse transcription-PCR (qRT-PCR), as described previously (28).
HCV sequencing.
Total RNA in culture supernatant was extracted with Isogen-LS (Nippon Gene Co., Ltd., Tokyo, Japan). cDNA was synthesized using Superscript III Reverse Transcriptase (Invitrogen, Carlsbad, CA). cDNA was subsequently amplified with LA Taq DNA polymerase (TaKaRa, Shiga, Japan). Four separate PCR primer sets were used to amplify the fragments of nt 130 to 2909, 2558 to 5142, 4784 to 7279, and 7081 to 9634 covering the entire open reading frame and part of the 5′ UTR and 3′ UTR of the MA strain. Sequences of amplified fragments were determined directly.
Immunostaining.
Infected cells were cultured on Multitest Slides (MP Biomedicals, Aurora, OH) and were fixed in acetone-methanol (1:1, vol/vol) for 15 min at −20°C. After a blocking step, infected cells were visualized with anti-core protein antibody (clone 2H9) (29) and Alexa Fluor 488 goat anti-mouse IgG (Invitrogen), and nuclei were visualized with 4′,6′-diamidino-2-phenylindole (DAPI).
Assessment of interferon sensitivity.
Two micrograms of in vitro transcribed RNA was transfected into 3 × 106 Huh7.5.1 cells. Four hours after transfection, cells were placed in fresh medium or medium containing 0.1, 1, 10, 100, and 1,000 IU/ml of interferon α-2b (Intron A; Schering-Plough Corporation, Osaka, Japan). Culture medium was then harvested on day 3, and HCV core levels in the cells and in the medium were measured.
Statistical analysis.
Significant differences were evaluated by Student's t test. A P value of <0.05 was considered significant.
RESULTS
Transient replication and production of 2b/2a chimeric virus.
We first tested whether the MA strain (genotype 2b) (19) was able to replicate and produce infectious virus in cultured cells. When the in vitro transcribed RNA of MA was transfected into Huh7.5.1 cells, a highly HCV-permissive cell line, replication and virus production were not observed (Fig. 1A to C). We then tested whether 2b/2a chimeric RNA harboring the structural region (5′ UTR to E2) of the MA strain and the nonstructural region (p7 to 3′ UTR) of JFH-1 (Fig. 1A, MA/JFH-1.1) was able to replicate in the cells. After MA/JFH-1.1 RNA transfection, time-dependent accumulation of core protein in the cells (Fig. 1B) and culture medium (Fig. 1C) was observed, indicating that MA/JFH-1.1 RNA was able to replicate in the cells autonomously. HCV RNA levels in the medium were determined by qRT-PCR, and time-dependent increases in HCV RNA level were also observed (Fig. 1D). Infectious virus production was observed on day 3, but infectivity was 17.6-fold lower than that of JFH-1 (Fig. 1E).
In order to improve the level of infectious virus production, we tested another chimeric construct, MA/JFH-1.2, which contained an additional MA-to-JFH-1 replacement of the 5′ UTR (Fig. 1A), as a 5′ UTR replacement from J6CF (genotype 2a) to JFH-1 enhanced virus production of chimeric J6CF virus harboring the region of NS2 to 3′ X of JFH-1 (J6/JFH-1) (A. Murayama et al., unpublished data). The core protein accumulation levels with MA/JFH-1.2 RNA-transfected cells were higher than those with MA/JFH-1.1 (P < 0.05) (Fig. 1B). Similarly, core protein and HCV RNA levels in the medium of MA/JFH-1.2 RNA-transfected cells were higher than those of MA/JFH-1.1 (P < 0.05) (Fig. 1C and D). Infectivity on day 3 was also higher than with MA/JFH-1.1 (P < 0.05) (Fig. 1E), indicating that the 5′ UTR of JFH-1 enhanced virus production. However, infectivity of medium from MA/JFH-1.2 RNA-transfected cells on day 3 remained 6.4-fold lower than that of JFH-1 although HCV RNA levels in the medium were similar to those of JFH-1 (Fig. 1D and E).
These results indicate that 2b/2a chimeric RNA is able to replicate autonomously in Huh7.5.1 cells and produce infectious virus although infectivity remains lower than that of JFH-1.
Assembly-enhancing mutation in core region introduced during long-term culture.
Because MA/JFH-1.1 and MA/JFH-1.2 replicated efficiently but produced small amounts of infectious virus, we performed long-term culture of these RNA-transfected cells in order to examine whether these chimeric RNAs would continue replicating and producing infectious virus over the long term. We prepared two RNA-transfected cell lines for each construct (MA/JFH-1.1 and MA/JFH-1.2) as both of these replicated and produced infectious virus at different levels.
Immediately after transfection, core protein levels and infectivity in culture medium were low (1.06 × 103 to 2.15 × 103 fmol/liter and 5.00 × 101 to 9.30 × 101 FFU/ml, respectively) (Fig. 2A and Table 1) although a considerable number of core protein-positive cells were observed by immunostaining (Fig. 2B, frames a to d). Subsequently, core protein levels in the culture medium decreased gradually (Fig. 2A), and core protein-positive cells were rare (Fig. 2B, frames e to h). However, at 30 to 40 days after transfection, core protein levels in the supernatants of all chimeric RNA-transfected cells increased and reached 2.27 × 105 to 4.93 × 105 fmol/liter (Fig. 2A and Table 1). Infectivity in the culture medium also increased (1.61 × 105 to 3.27 × 105 FFU/ml) (Table 1), and at this point, most of the cells were core protein positive (Fig. 2B, frame i to l).
Fig 2.

Long-term culture of MA/JFH-1.1 and MA/JFH-1.2 RNA-transfected cells. Ten micrograms of HCV RNA was transfected into Huh7.5.1 cells, and cells were passaged every 2 to 5 days, depending on cell status. Culture medium was collected after every passage, and HCV core protein levels were measured. Transfection was performed twice for each chimeric RNA (1 and 2 for each construct). (A) HCV core protein levels in culture medium from MA/JFH-1.1 and MA/JFH-1.2 RNA-transfected cells. (B) Immunostained cells at 3 days after transfection (a to d), at 21 days after transfection (e to h), and at the time of peak core levels (days 42 to 49). Infected cells were visualized with anti-core protein antibody (green), and nuclei were visualized with DAPI (blue). (C) Infection of naïve cells by culture medium at an MOI of 0.001. (D) Immunostained cells at 15 days after infection with medium at peak core protein levels (Fig. 2A) at an MOI of 0.001. Infected cells were visualized with anti-core antibody (green), and nuclei were visualized with DAPI (blue).
Table 1.
HCV core protein levels and infectivity in culture medium immediately after RNA transfection (day 3) and after long-term culture (days 35 to 49)
| Sample period and virus | Sample no. | Day no.a | HCV core (fmol/liter) | Infectivity (FFU/ml) |
|---|---|---|---|---|
| After transfection | ||||
| MA/JFH-1.1 | 1 | 3 | 1.06 × 103 | 5.00 × 101 |
| 2 | 3 | 1.14 × 103 | 5.70 × 101 | |
| MA/JFH-1.2 | 1 | 3 | 2.14 × 103 | 7.30 × 101 |
| 2 | 3 | 2.15 × 103 | 9.30 × 101 | |
| After long-term culture | ||||
| MA/JFH-1.1 | 1 | 42 | 3.38 × 105 | 1.62 × 105 |
| 2 | 42 | 4.70 × 105 | 3.23 × 105 | |
| MA/JFH-1.2 | 1 | 35 | 2.27 × 105 | 1.61 × 105 |
| 2 | 49 | 4.93 × 105 | 3.27 × 105 |
For the long-term culture, the days are those of peak core protein levels.
As the infectivity of culture supernatant of MA/JFH-1 RNA-transfected cells appeared to increase after long-term culture, we compared viral spread by infection with these supernatants on day 3 (immediately after transfection) and for each peak in core protein levels (after long-term culture). When naïve Huh7.5.1 cells were infected with supernatant on days corresponding to a peak in core protein levels at a multiplicity of infection (MOI) of 0.001, core protein levels in the medium increased rapidly and reached 0.64 × 106 to 1.13 × 106 fmol/liter by day 15 after infection (Fig. 2C). Immunostained images showed that most cells were HCV core protein positive on day 15 (Fig. 2D). When naïve Huh7.5.1 cells were infected with supernatant from day 3 at an MOI of 0.001, core protein levels in the medium did not increase under these conditions (Fig. 2C). These results indicate that both MA/JFH-1 chimeric viruses (MA/JFH-1.1 and MA/JFH-1.2) acquired the ability to spread rapidly after long-term culture.
As the characteristics of the MA/JFH-1 virus changed in long-term culture, we analyzed the possible mutations in the viral genome from the supernatant at each peak in core protein levels (Table 1, days at peak core levels). Nine- to 12-nucleotide mutations were found in the viral genome from each supernatant, and the detected mutations were distributed along the entire genome. Among these mutations, a common nonsynonymous mutation was found in the core region (Arg to Gly at amino acid [aa]167, R167G).
In order to test the effects of R167G on virus production, an R167G substitution was introduced into MA/JFH-1.2 as MA/JFH-1.2 replicated and produced infectious virus more efficiently than MA/JFH-1.1. HCV core protein levels in cells and medium of MA/JFH-1.2 with R167G (MA/JFH-1.2/R167G) were higher than with MA/JFH-1.2 (P < 0.05) (Fig. 3A and B). HCV RNA levels in the medium of MA/JFH-1.2/R167G RNA-transfected cells were also higher than with MA/JFH-1.2 (P < 0.05) (Fig. 3C). Infectious virus production was also increased by the R167G mutation (P < 0.05) (Fig. 3D) and was 8.7-fold higher than that of JFH-1 RNA-transfected cells on day 3 (P < 0.05) (Fig. 3D).
Fig 3.

Effects of R167G on replication and virus production of MA/JFH-1.2 in Huh7.5.1 cells. Ten micrograms of HCV RNA was transfected into Huh7.5.1 cells, and cells and medium were harvested on days 1, 2, and 3. HCV core protein levels in the cells (A) and culture medium (B) and HCV RNA levels in the medium (C) and the infectivity of culture medium (D) from HCV RNA-transfected Huh7.5.1 cells are shown. n.d., not determined. Dashed line indicates the detection limit. Assays were performed three times independently, and data are presented as means ± standard deviation. (E) HCV core protein levels in culture medium from cells infected with medium at 3 days posttransfection at an MOI of 0.005. (F) Immunostained cells at 19 days postinfection. Infected cells were visualized with anti-core antibody (green), and nuclei were visualized with DAPI (blue).
We then tested whether R167G was responsible for the rapid spread observed in culture supernatant after long-term culture by monitoring virus spread after infection of naïve Huh7.5.1 with culture medium taken 3 days after RNA transfection of MA/JFH-1.2 and MA/JFH-1.2/R167G at an MOI of 0.005. Core protein levels in medium from MA/JFH-1.2/R167G-infected cells increased with the same kinetics as levels of JFH-1 (Fig. 3E), and the population of core protein-positive cells was almost the same as with JFH-1-infected cells (Fig. 3F), indicating that MA/JFH-1.2/R167G virus spread as rapidly as JFH-1 virus. In contrast, we observed no infectious foci in the MA/JFH-1.2 virus-inoculated cells (Fig. 3F). These data suggest that the R167G mutation in the core region was a cell culture-adaptive mutation and that it enhanced infectious MA/JFH-1.2 virus production.
In order to determine whether R167G enhances RNA replication or other steps in the viral life cycle, we performed a single-cycle virus production assay (11) using Huh7-25 cells, a HuH-7-derived cell line lacking CD81 expression on the cell surface (1). This cell line can support replication and infectious virus production upon transfection of HCV genomic RNA but cannot be reinfected by progeny virus, thereby allowing observation of a single cycle of infectious virus production without the confounding effects of reinfection. R167G did not affect HCV core protein levels in the chimeric RNA-transfected Huh7-25 cells (Fig. 4A), demonstrating that R167G did not enhance RNA replication. Nevertheless, R167G increased HCV core protein levels in the medium (P < 0.05 on days 2 and 3) and infectivity (Fig. 4B and C). These results suggest that R167G did not affect RNA replication but affected other steps such as virus assembly and/or virus secretion.
Fig 4.

Effects of R167G on replication and virus production of MA/JFH-1.2 and JFH-1 in Huh7-25 cells. Ten micrograms of HCV RNA was transfected into Huh7-25 cells, and cells and medium were harvested on days 1, 2, and 3. HCV core protein levels in cells (A and D) and in medium (B and E) were measured, and infectivity of medium (C and F) was determined. n.d., not determined. Dashed line indicates the detection limit. (G) Intracellular specific infectivity and virus secretion efficiency of chimeric HCV RNA-transfected cells. Intracellular and extracellular infectivity of day 3 samples was determined, and specific infectivity and virus secretion rate were calculated. Assays were performed three times independently, and data are presented as means ± standard deviation. NA, not available.
Virus particle assembly efficiency was then assessed by determining intracellular-specific infectivity from infectivity and RNA titer in the cells, as reported previously (11). As shown in Fig. 4G, R167G enhanced intracellular-specific infectivity of MA/JFH-1.2 virus 10.2-fold. Virus secretion efficiency was also calculated from the amount of intracellular and extracellular infectious virus, but R167G had no effect (Fig. 4G).
To confirm the effects of Arg167 in other HCV strains, we tested its effects on JFH-1. As aa 167 of JFH-1 is Gly, we replaced it with Arg (G167R). HCV core protein levels in the cells were not affected by G167R (Fig. 4D), and no effects on RNA replication were confirmed. HCV core protein levels in the medium and infectivity decreased after G167R mutation (Fig. 4E and F). As the G167R mutation decreased intracellular infectious virus production of JFH-1 to undetectable levels, we were unable to determine the intracellular-specific infectivity and virus secretion efficiency of JFH-1 G167R (Fig. 4G). These results indicate that Gly is favored over Arg at core position 167 for infectious virus assembly in multiple HCV strains.
MA harboring the R167G mutation, 5′ UTR, and N3H (NS3 helicase) and N5BX (NS5B to 3′ X) regions of JFH-1 replicated and produced infectious chimeric virus.
In order to establish a genotype 2b cell culture system with the MA strain with minimal regions of JFH-1, we attempted to reduce JFH-1 content in MA/JFH-1.2. We previously reported that replacement of the N3H and N5BX regions of JFH-1 allowed efficient replication of the J6CF strain, which normally cannot replicate in cells (21). Thus, we tested whether the N3H and N5BX regions of JFH-1 could also support MA RNA replication.
We prepared two chimeric MA constructs harboring the 5′ UTR and N3H and N5BX regions of JFH-1, MA/N3H+N5BX-JFH1 (Fig. 5A) and MA/N3H+N5BX-JFH1/R167G. After in vitro transcribed RNA was transfected into Huh7.5.1 cells, intra-cellular core protein levels of MA/N3H+N5BX-JFH1 and MA/N3H+N5BX-JFH1/R167G RNA-transfected cells increased in a time-dependent manner and reached almost the same levels as with MA/JFH-1.2 RNA-transfected cells on day 5 (Fig. 5B). Extracellular core protein and HCV RNA levels of MA/N3H+N5BX-JFH1 and MA/N3H+N5BX-JFH1/R167G RNA-transfected cells also increased in a time-dependent manner (Fig. 5C and D). However, they were more than 10 times lower than with MA/JFH-1.2 RNA-transfected cells although intracellular core levels were comparable on day 5 (Fig. 5B to D).
Fig 5.

Replication and virus production of MA/N3H+N5BX-JFH1/R167G in Huh7.5.1 cells. (A) Schematic structures of JFH-1, MA, and MA/N3H+N5BX-JFH1. The junction of JFH-1 and MA in the 5′ UTR is an AgeI site; the junctions of MA and JFH-1 in the NS3 regions are ClaI and EcoT22I sites, and the junction in the NS5B region is a BsrGI site. A, AgeI; X, XbaI. (B to G) Chimeric HCV RNA replication in Huh7.5.1 cells. Ten micrograms of HCV RNA was transfected into Huh7.5.1 cells, and cells and medium were harvested on days 1, 3, and 5. HCV core protein levels in cells (B) and in medium (C) and HCV RNA levels in medium (D) were measured, and infectivity of medium (E) was determined. Assays were performed three times independently, and data are presented as means ± standard deviation. n.d., not determined. Dashed line indicates the detection limit. (F) Immunostained cells. Huh7.5.1 cells were infected with concentrated medium from RNA-transfected cells on day 5. Infected cells were visualized with anti-core antibody (green), and nuclei were visualized with DAPI (blue). (G) Infectivity of concentrated culture medium from HCV RNA-transfected cells. Culture medium was concentrated by 20 times. Infectivities of original and concentrated culture media were determined. Dashed line indicates detection thelimit.
We then tested whether the medium from MA/N3H+N5BX-JFH1 and MA/N3H+N5BX-JFH1/R167G RNA-transfected cells was infectious. Infectivity of the medium from MA/N3H+N5BX-JFH1 RNA-transfected cells was below the detection limit, and that of MA/N3H+N5BX-JFH1/R167G RNA-transfected cells on day 5 was very low (3.3 × 101 ± 2.1 × 101 FFU/ml) (Fig. 5E). To confirm infectivity, the culture media were concentrated, and their infectivity was determined. Infected foci were observed after infection with concentrated medium in MA/N3H+N5BX-JFH1/R167G RNA-transfected cells (Fig. 5F), and infectivity was found to be 7.27 × 102 ± 7.57 × 101 FFU/ml (Fig. 5G). No infected foci were observed after infection of MA/N3H+N5BX-JFH1 RNA-transfected cells, even when medium was concentrated (Fig. 5F), although intracellular and extracellular core protein levels were comparable to those with MA/N3H+N5BX-JFH1/R167G RNA-transfected cells (Fig. 5B and C). These results indicate that replacement of the 5′ UTR and N3H and N5BX regions in JFH-1 were necessary to rescue autonomous replication in the replication-incompetent MA strain and for secretion of infectious chimeric virus. However, the secretion and infection efficiencies of the virus were low.
Cell culture-adaptive mutations enhanced infectious virus production of MA/N3H+N5BX-JFH1/R167G.
Because MA/N3H+N5BX-JFH1/R167G replicated efficiently but produced very small amounts of infectious virus, we performed a long-term culture of the RNA-transfected cells in order to induce cell culture-adaptive mutations that could enhance infectious virus production. We prepared RNA-transfected cells using two constructs, MA/N3H+N5BX-JFH1 and MA/N3H+N5BX-JFH1/R167G; both of these replicated efficiently, and MA/N3H+N5BX-JFH1/R167G produced infectious virus at low levels while MA/N3H+N5BX-JFH1 did not. Immediately after transfection, the HCV core protein levels in the medium of each RNA-transfected cell culture peaked at 3.0 × 103 fmol/liter and declined thereafter. However, the core protein level in the medium with MA/N3H+N5BX-JFH1/R167G RNA-transfected cells continued to increase and reached a peak of 2.7 × 105 fmol/liter 54 days after transfection, at which point most cells were core protein positive (Fig. 6B). The core protein level in the medium with MA/N3H+N5BX-JFH1 RNA-transfected cells did not increase and core-positive cells were scarce on day 54 (Fig. 6B). We analyzed the viral genome in the culture supernatants from day 54 for possible mutations and identified four nonsynonymous mutations in the MA/N3H+N5BX-JFH1/R167G genome: L814S (NS2), R1012G, (NS2), T1106A (NS3), and V1951A (NS4B). In order to test whether these amino acid substitutions enhance infectious virus production, L814S, R1012G, T1106A, and V1951A were introduced into MA/N3H+N5BX-JFH1/R167G, and the product was designated MA/N3H+N5BX-JFH1/5am (where am indicates adaptive mutation). On day 1, although HCV core protein levels in the MA/N3H+N5BX-JFH1/5am RNA-transfected cells were higher than those of MA/N3H+N5BX-JFH1/R167G RNA-transfected cells, they were still lower than those of MA/JFH-1.2/R167G RNA-transfected cells; however, on days 3 and 5, they reached a level comparable to that of MA/JFH-1.2/R167G RNA-transfected cells (Fig. 6C). HCV core protein and HCV RNA levels in the medium of MA/N3H+N5BX-JFH1/5am RNA-transfected cells were higher than those of MA/JFH-1.2/R167G RNA-transfected cells (P < 0.05, Fig. 6D and 6E, respectively). MA/N3H+N5BX-JFH1/5am, containing the four additional adaptive mutations, produced infectious virus at the same level as MA/JFH-1.2/R167G on day 5 (Fig. 6F). These results indicate that the four additional adaptive mutations enhance infectious virus production and that MA/N3H+N5BX-JFH1/5am RNA-transfected cells replicate and produce infectious virus as efficiently as MA/JFH-1.2/R167G RNA-transfected cells.
Fig 6.
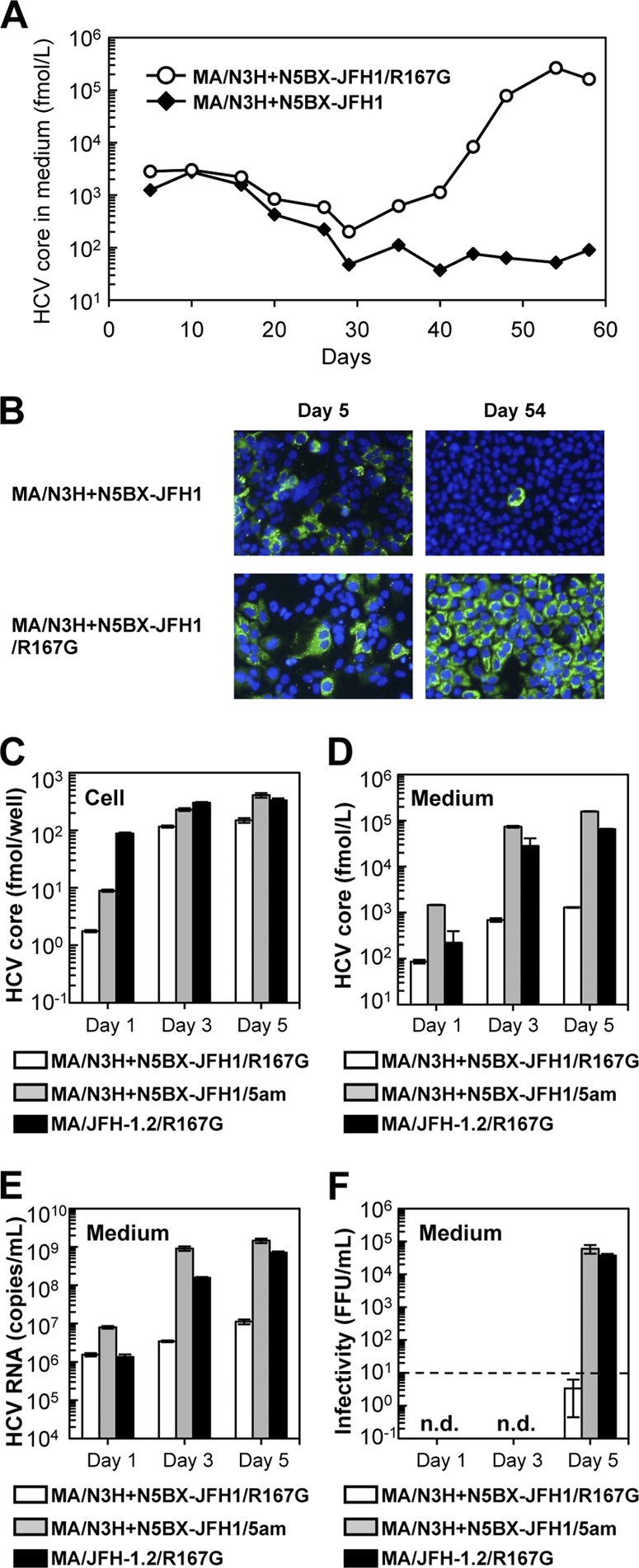
Cell culture-adaptive mutations enhanced infectious virus production of MA/N3H+N5BX-JFH1/R167G. (A) Long-term culture of MA/N3H+N5BX-JFH1 and MA/N3H+N5BX-JFH1/R167G RNA-transfected cells. Ten micrograms of HCV RNA was transfected into Huh7.5.1 cells, and cells were passaged every 2 to 5 days, depending on cell status. Culture medium was collected after every passage, and HCV core protein levels were measured. HCV core protein levels in culture medium from MA/N3H+N5BX-JFH1 and MA/N3H+N5BX-JFH1/R167G RNA-transfected cells are presented. (B) Immunostained cells on days 5 and 54 after transfection. Infected cells were visualized with anti-core antibody (green), and nuclei were visualized with DAPI (blue). (C to F) Effect of four additional cell culture-adaptive mutations on virus production. Ten micrograms of HCV RNA was transfected into Huh7.5.1 cells, and cells and medium were harvested on days 1, 3, and 5. HCV core levels in cells (C) and in medium (D) and HCV RNA levels in medium (E) were measured, and infectivity of medium (F) was determined. Assays were performed three times independently, and data are presented as means ± standard deviation. n.d., not determined. Dashed line indicates the detection limit.
Comparison of interferon sensitivity between JFH-1, MA/JFH-1.2/R167G, and MA/N3H+N5BX-JFH1/R167G.
Using the newly established genotype 2b infectious chimeric virus, we compared interferon sensitivity between the JFH-1, MA/JFH-1.2/R167G, and MA/N3H+N5BX-JFH1/5am viruses. JFH-1 or MA chimeric viral RNA-transfected Huh7.5.1 cells were treated with 0.1, 1, 10, 100, or 1,000 IU/ml interferon α-2b, and HCV core protein levels in the cells and in culture media were compared. Interferon decreased HCV core protein levels in the JFH-1 RNA-transfected cells and in the medium in a dose-dependent manner, and production was inhibited to 26.8% ± 3.0% and 45.6% ± 4.7%, respectively, of control levels (Fig. 7A and B, respectively). In contrast, HCV core protein levels in cells and medium of MA/JFH-1.2/R167G and MA/N3H+N5BX-JFH1/5am RNA-transfected cells decreased more pronouncedly in a dose-dependent manner (Fig. 7A and B, respectively). HCV core protein levels in cells and medium from MA/N3H+N5BX-JFH1/5am RNA-transfected cells were lower than those from MA/JFH-1.2/R167G RNA-transfected cells (Fig. 7A and B, respectively) (P < 0.05 at 1, 10, 100, and 1,000 IU/ml), indicating that the MA/N3H+N5BX-JFH1/5am virus was more sensitive to interferon than the MA/JFH1.2/R167G virus, which contained more regions from JFH-1.
Fig 7.

Comparisons of interferon sensitivity between JFH-1, MA/JFH-1.2/R167G and MA/N3H+N5BX-JFH1/5am. Two micrograms of HCV RNA was transfected into Huh7.5.1 cells, and interferon was added at the indicated concentrations at 4 h after transfection. HCV core protein levels in cells (A) and in medium (B) on day 3 were measured, and data are expressed as percent versus untreated cells (0 IU/ml). Assays were performed three times independently, and data are presented as means ± standard deviation.
DISCUSSION
In this study, we developed a novel infectious HCV production system using a genotype 2b chimeric virus. To improve infectious virus production, we introduced two modifications into the chimeric genome.
First, we replaced the 5′ UTR from MA with that of JFH-1. Similarly to J6/JFH-1, replacement of the 5′ UTR increased core protein accumulation in both the cells and medium when these RNAs were transfected into Huh7.5.1 cells (Fig. 1). The same trend was observed when these RNAs were transfected into Huh7-25 cells (data not shown), indicating that the 5′ UTR of JFH-1 enhanced RNA replication. There are two genetic variations in J6CF and seven in MA in the region we replaced (nt 1 to 154 for J6CF and nt 1 to 155 for MA), and some of these mutations may affect RNA replication by changing the RNA secondary structure, RNA-RNA interactions, or binding of host or viral proteins.
Second, we introduced a cell culture-adaptive mutation (R167G) in the core region. This mutation was induced by long-term culture of MA/JFH-1 RNA-transfected cells (Fig. 2). MA/JFH-1 chimeric RNA (MA/JFH-1.1 and MA/JFH-1.2) replicated when synthesized RNA was transfected into the cells. However, infectious virus production was low, and virus infection did not spread over the short term. In early stages of long-term culture, the number of core protein-positive cells gradually decreased, and core protein-positive cells were scarcely detectable. Subsequently, the population of core protein-positive cells increased, reaching almost 100%. At this time point, we identified a common mutation in the core region (R167G) of the viral genome as a cell culture-adaptive mutation and found that it enhanced infectious virus production (Fig. 3). Several nonsynonymous mutations other than R167G were identified in the viral genome from each supernatant, and these mutations may enhance infectious virus production. However, there was a discrepancy between RNA levels and the infectivity of the culture media of MA/JFH-1.2 and MA/JFH-1.2/R167G RNA-transfected cells (Fig. 3C and D). The MA/JFH-1.2/R167G mutant had a 2-log increase in viral infectivity compared to that of MA/JFH-1.2 but only a 1-log increase in secreted RNA. The replication efficiency of MA/JFH-1.2 RNA-transfected cells was comparable to that of MA/JFH-1.2/R167G RNA-transfected cells, but the efficiency of infectious virus assembly within the cells was low, indicating that mainly noninfectious virus may be produced.
Infection of MA/JFH-1.2/R167G virus spreads rapidly, similarly to that of the JFH-1 virus, when it is inoculated into naïve Huh7.5.1 cells. On a single-cycle virus production assay, we found that the R167G mutation did not affect RNA replication or virus secretion but enhanced infectious virus assembly within the cells (Fig. 4). Efficient infectious virus assembly within the cells was mainly responsible for the rapid spread and high virus production of MA/JFH-1.2/R167G.
The amino acid at 167 (aa 167) is located in domain 2 of the core region, which is important for localization of the core protein (3, 8). Lipid droplet localization of the core protein and/or NS5A is important for infectious virus production (4, 18, 26). The interaction between the core protein and NS5A is also important for infectious virus production (16). Thus, aa 167 affects infectious virus production possibly by altering subcellular localization of the core protein or interaction between the core protein and NS5A. We examined the amino acid sequence of the core protein in 2,078 strains in the Hepatitis Virus Database (http://s2as02.genes.nig.ac.jp/) and found that aa 167 is Gly in all other strains. These data strongly suggest that Gly at aa 167 is important for the HCV life cycle. As the MA strain was cloned from the serum of a patient with chronic hepatitis C, the low virus production by this Gly at aa 167 may be important for persistent infection.
We then attempted to reduce the contents of JFH-1 from MA/JFH-1.2/R167G. We previously reported that the N3H and N5BX regions of JFH-1 were sufficient for replication of the J6CF strain (21). We also reported that this effect was observed only in genotype 2a strains (J6CF, JCH-1, and JCH-4). In this study, we tested whether the N3H and N5BX regions of JFH-1 could also support replication of a genotype 2b strain, MA. We constructed an MA chimeric virus harboring the N3H and N5BX regions of JFH-1 and combined this with the 5′ UTR of JFH-1 and the R167G mutation (MA/N3H+N5BX-JFH1/R167G). This chimeric RNA was able to replicate in the cells and produce infectious chimeric virus in culture medium although infectious virus production levels were low (Fig. 5).
We showed in this paper that the N3H and N5BX regions of JFH-1 were able to support RNA replication by both genotype 2a clones and genotype 2b clones, but the nucleotide sequence similarity between JFH-1 and MA was lower than that between JFH-1 and J6CF (77% versus 89%, respectively). Compared to MA/JFH-1.2/R167G, MA/N3H+N5BX-JFH1/R167G RNA showed the same levels of RNA replication and low levels of infectious virus production. To clarify whether there were any differences in the characteristics of the secreted virus, we performed density gradient ultracentrifugation with the MA/JFH-1.2/R167G and MA/N3H+N5BX-JFH1/R167G viruses. The distributions of the HCV core protein and infectivity showed similar profiles (data not shown).
The differences between MA/JFH-1.2/R167G and MA/N3H+N5BX-JFH1/R167G are the NS2, NS3 protease domain (N3P), and NS4A to NS5A regions. Nucleotide variation(s) other than aa 167 in these regions of the MA strain may be associated with reduced virus assembly. We identified four additional cell culture-adaptive mutations, L814S (NS2), R1012G (NS2), T1106A (NS3), and V1951A (NS4B), which resulted from long-term culture of MA/N3H+N5BX-JFH1/R167G RNA-transfected cells. Consequently, cells transfected with MA/N3H+N5BX-JFH1/5am constructed by insertion of these four adaptive mutations into MA/N3H+N5BX-JFH1/R167G replicated and produced infectious virus as efficiently as MA/JFH-1.2/R167G RNA-transfected cells (Fig. 6).
This system is able to contribute to studies into the development of antiviral strategies. It has been reported that HCV genotype 2a was more sensitive to interferon therapy than HCV genotype 2b in a clinical study (20). To assess the interferon resistance of genotype 2b, a cell culture system with multiple genotype 2b strains is necessary. The previously reported replicable genotype 2b chimeric virus harbored only structural regions of 2b strains (6, 27). The 2b/JFH-1 chimeric virus containing the region of the core protein to NS2 from the J8 strain (genotype 2b) and the region of NS3 to 3′ X of JFH-1 was able to replicate and showed that there were no differences in interferon sensitivity among the JFH-1 chimeric viruses of other genotypes (6, 27). Another 2b/JFH-1 chimeric virus containing the regions of the core protein to NS2 (nt 342 to 2867) of a genotype 2b strain and of NS2 to 3′ UTR (nt 2868) of JFH-1 has been reported (6, 27). The authors reported that their 2b/JFH-1 chimeric virus was more sensitive to interferon than JFH-1 (6, 27). We developed the genotype 2b HCV cell culture system with another HCV genotype 2b strain (MA). We identified a virus assembly-enhancing mutation in the core region, the minimal JFH-1 regions necessary for replication, and four additional adaptive mutations that enhance infectious virus production and demonstrated that MA harboring the five adaptive mutations and the 5′ UTR and N3H and N5BX regions of JFH-1 (MA/N3H+N5BX-JFH1/5am) could replicate and produce infectious virus efficiently.
Using these novel genotype 2b chimeric viruses, we assessed interferon sensitivity. We found that MA/JFH-1.2/R167G chimeric virus and MA/N3H+N5BX-JFH1/5am virus were more sensitive to interferon than the JFH-1 virus (Fig. 7). Furthermore, we found that MA/N3H+N5BX-JFH1/5am was more sensitive to interferon than MA/JFH-1.2/R167G, indicating that the genetic variation(s) in the NS2, N3P, and NS4A to NS5A regions affect interferon sensitivity. Although genotype 2a viruses are more sensitive to interferon than genotype 2b viruses in clinical studies, JFH-1 displayed interferon resistance in our study.
These results suggest that the JFH-1 regions in the 2b/JFH-1 virus affect the interferon sensitivity of the chimeric virus. Moreover, it was reported that amino acid variations in E2, p7, NS2, and NS5A were associated with the response to peginterferon and ribavirin therapy in genotype 2b HCV infection (10). Therefore, our MA/JFH-1 chimeric virus harboring minimal regions from JFH-1 (MA/N3H+N5BX-JFH1/5am) is more suitable for assessing the characteristics of the MA strain than the MA/JFH-1 chimeric virus, which includes a nonstructural region from JFH-1 (MA/JFH-1.2/R167G). We showed here that replacement of the 5′ UTR and N3H and N5BX regions in MA with those from JFH-1 is able to convert MA into a replicable virus. Using the same strategy, numerous HCV cell culture systems with various genotype 2b strains, as well as genotype 2a strains, may be available.
In conclusion, we established a novel HCV genotype 2b cell culture system using a chimeric genome in MA harboring minimal regions from JFH-1. This cell culture system using the chimeric genotype 2b virus will be useful for characterization of genotype 2b viruses and the development of antiviral strategies.
ACKNOWLEDGMENTS
We are grateful to Tetsuro Suzuki of Hamamatsu University School of Medicine for helpful comments and suggestions. Huh7.5.1 cells were kindly provided by Francis V. Chisari.
A.M. is partially supported by the Japan Health Sciences Foundation and Viral Hepatitis Research Foundation of Japan. This work was partially supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science, from the Ministry of Health, Labor and Welfare of Japan, from the Ministry of Education, Culture, Sports, Science and Technology, from the National Institute of Biomedical Innovation, and by Research on Health Sciences Focusing on Drug Innovation from the Japan Health Sciences Foundation.
Footnotes
Published ahead of print 7 December 2011
REFERENCES
- 1. Akazawa D, et al. 2007. CD81 expression is important for the permissiveness of Huh7 cell clones for heterogeneous hepatitis C virus infection. J. Virol. 81:5036–5045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bartenschlager R, Lohmann V. 2000. Replication of hepatitis C virus. J. Gen. Virol. 81:1631–1648 [DOI] [PubMed] [Google Scholar]
- 3. Boulant S, et al. 2006. Structural determinants that target the hepatitis C virus core protein to lipid droplets. J. Biol. Chem. 281:22236–22247 [DOI] [PubMed] [Google Scholar]
- 4. Boulant S, Targett-Adams P, McLauchlan J. 2007. Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J. Gen. Virol. 88:2204–2213 [DOI] [PubMed] [Google Scholar]
- 5. Choo QL, et al. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359–362 [DOI] [PubMed] [Google Scholar]
- 6. Gottwein JM, et al. 2009. Development and characterization of hepatitis C virus genotype 1–7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49:364–377 [DOI] [PubMed] [Google Scholar]
- 7. Griffin S, et al. 2008. Genotype-dependent sensitivity of hepatitis C virus to inhibitors of the p7 ion channel. Hepatology 48:1779–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hope RG, McLauchlan J. 2000. Sequence motifs required for lipid droplet association and protein stability are unique to the hepatitis C virus core protein. J. Gen. Virol. 81:1913–1925 [DOI] [PubMed] [Google Scholar]
- 9. Jensen TB, et al. 2008. Highly efficient JFH1-based cell-culture system for hepatitis C virus genotype 5a: failure of homologous neutralizing-antibody treatment to control infection. J. Infect. Dis. 198:1756–1765 [DOI] [PubMed] [Google Scholar]
- 10. Kadokura M, et al. 2011. Analysis of the complete open reading frame of genotype 2b hepatitis C virus in association with the response to peginterferon and ribavirin therapy. PLoS One 6:e24514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kato T, et al. 2008. Hepatitis C virus JFH-1 strain infection in chimpanzees is associated with low pathogenicity and emergence of an adaptive mutation. Hepatology 48:732–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kato T, et al. 2006. Cell culture and infection system for hepatitis C virus. Nat. Protoc. 1:2334–2339 [DOI] [PubMed] [Google Scholar]
- 13. Kiyosawa K, et al. 1990. Interrelationship of blood transfusion, non-A, non-B hepatitis and hepatocellular carcinoma: analysis by detection of antibody to hepatitis C virus. Hepatology 12:671–675 [DOI] [PubMed] [Google Scholar]
- 14. Lindenbach BD, et al. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626 [DOI] [PubMed] [Google Scholar]
- 15. Lohmann V, et al. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113 [DOI] [PubMed] [Google Scholar]
- 16. Masaki T, et al. 2008. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 82:7964–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miyamoto M, Kato T, Date T, Mizokami M, Wakita T. 2006. Comparison between subgenomic replicons of hepatitis C virus genotypes 2a (JFH-1) and 1b (Con1 NK5.1). Intervirology 49:37–43 [DOI] [PubMed] [Google Scholar]
- 18. Miyanari Y, et al. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089–1097 [DOI] [PubMed] [Google Scholar]
- 19. Murakami K, Abe M, Kageyama T, Kamoshita N, Nomoto A. 2001. Down-regulation of translation driven by hepatitis C virus internal ribosomal entry site by the 3′ untranslated region of RNA. Arch. Virol. 146:729–741 [DOI] [PubMed] [Google Scholar]
- 20. Murakami T, et al. 1999. Mutations in nonstructural protein 5A gene and response to interferon in hepatitis C virus genotype 2 infection. Hepatology 30:1045–1053 [DOI] [PubMed] [Google Scholar]
- 21. Murayama A, et al. 2007. The NS3 helicase and NS5B-to-3′X regions are important for efficient hepatitis C virus strain JFH-1 replication in Huh7 cells. J. Virol. 81:8030–8040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murayama A, et al. 2010. RNA polymerase activity and specific RNA structure are required for efficient HCV replication in cultured cells. PLoS Pathog. 6:e1000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pietschmann T, et al. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A. 103:7408–7413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pietschmann T, et al. 2009. Production of infectious genotype 1b virus particles in cell culture and impairment by replication enhancing mutations. PLoS Pathog. 5:e1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scheel TK, et al. 2008. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc. Natl. Acad. Sci. U. S. A. 105:997–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shavinskaya A, Boulant S, Penin F, McLauchlan J, Bartenschlager R. 2007. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J. Biol. Chem. 282:37158–37169 [DOI] [PubMed] [Google Scholar]
- 27. Suda G, et al. 2010. IL-6-mediated intersubgenotypic variation of interferon sensitivity in hepatitis C virus genotype 2a/2b chimeric clones. Virology 407:80–90 [DOI] [PubMed] [Google Scholar]
- 28. Takeuchi T, et al. 1999. Real-time detection system for quantification of hepatitis C virus genome. Gastroenterology 116:636–642 [DOI] [PubMed] [Google Scholar]
- 29. Wakita T, et al. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yi M, Ma Y, Yates J, Lemon SM. 2007. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 81:629–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yi M, Villanueva RA, Thomas DL, Wakita T, Lemon SM. 2006. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. U. S. A. 103:2310–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhong J, et al. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]