

Abstract
Copper (Cu) is essential for development and proliferation, yet the cellular requirements for Cu in these processes are not well defined. We report that Cu plays an unanticipated role in the mitogen-activated protein (MAP) kinase pathway. Ablation of the Ctr1 high-affinity Cu transporter in flies and mouse cells, mutation of Ctr1, and Cu chelators all reduce the ability of the MAP kinase kinase Mek1 to phosphorylate the MAP kinase Erk. Moreover, mice bearing a cardiac-tissue-specific knockout of Ctr1 are deficient in Erk phosphorylation in cardiac tissue. in vitro investigations reveal that recombinant Mek1 binds two Cu atoms with high affinity and that Cu enhances Mek1 phosphorylation of Erk in a dose-dependent fashion. Coimmunoprecipitation experiments suggest that Cu is important for promoting the Mek1-Erk physical interaction that precedes the phosphorylation of Erk by Mek1. These results demonstrate a role for Ctr1 and Cu in activating a pathway well known to play a key role in normal physiology and in cancer.
INTRODUCTION
Copper (Cu) is a metal ion that functions as a redox-active cofactor for a broad range of biochemical reactions, including mitochondrial oxidative phosphorylation, protection from reactive oxygen species, connective tissue maturation, iron absorption, neuropeptide biogenesis, and other processes (28, 43). Numerous studies point to the essentiality of Cu for normal growth and development, while aberrant Cu accumulation in tissues, as manifested in Wilson's disease patients, results in significant pathologies (33, 35, 42, 47, 60, 61). However, the precise roles Cu plays and the mechanistic processes by which Cu drives cellular proliferation and growth are not well understood.
The Ras/mitogen-activated protein kinase (MAPK) signaling pathway is an evolutionarily conserved pathway involved in the control of many fundamental biological processes, including cell proliferation, apoptosis, survival, differentiation, motility, and metabolism (26, 30). Aberrant Ras/MAPK signaling has significant consequences; loss of function of several components of the Ras/MAPK signaling cascade results in lethality, whereas gain-of-function mutations in many of the Ras/MAPK signaling components underlie cancer (2, 12, 26, 55).
Here we identify the Ctr1 high-affinity Cu+ transporter, conserved from yeast to humans, as being important for stimulation of the MAPK Erk in response to extracellular growth factor-mediated activation of the Ras signaling pathway. Moreover, genetic, physiological, and biochemical experiments point to a direct role for Cu in the ability of the MAPK kinase Mek1 to phosphorylate Erk in fruit flies, cultured cells, and mice. These studies suggest that the MAPK signaling pathway is a key cellular proliferation pathway that is stimulated by Cu and may be a direct target of potent cancer chemotherapeutics that function via Cu chelation.
MATERIALS AND METHODS
Drosophila melanogaster stocks and crosses.
Phantom Gal4, UAS mCD8::GFP/TM6, Tb flies were from Michael O'Connor, University of Minnesota (44). The UAS-Ctr1ARNAi construct was made and transgenic lines were generated as described elsewhere (36, 53). All other stocks were obtained from the Bloomington Stock Center. All crosses were performed at 25°C. All fly work, including pupal measurements, was done at the Duke University Model Systems Unit.
Pupal length experiments.
Wandering 3rd-instar larvae were placed in separate vials according to genotype and sex for pupariation; genotyping was done on the basis of green fluorescent protein (GFP) expression pattern, as well as the dominant marker Tubby. At pupation, individual images were taken using a Leica MZFL III fluorescence stereomicroscope mounted with a Qimaging Retiga Exi digital camera at the same magnification setting. Length measurements were performed by aligning the micrometer ruler image along the length of the pupal case at defined start and end points.
Indirect immunofluorescence and scanning electron microscopy (SEM) images.
Brains from wandering 3rd-instar Drosophila larvae of the desired genotype were dissected and fixed in 4% paraformaldehyde for 30 min. Staining of tissue was performed as described elsewhere (60). Images were taken on a Zeiss LSM 410 confocal microscope at the Duke University Light Microscopy Core Facility. For SEM images, adult flies of the desired genotype were subjected to a graded ethanol series. Flies were given to the Duke University Shared Materials Instrumentation Facility for critical-point drying and sputter coating. SEM images were taken at the Duke University Shared Materials Instrumentation Facility.
S2 cell metal chelation and silver treatment.
S2 cells used for the no-treatment and insulin-only treatment conditions were left in basal medium (Schneider's medium with 10% fetal bovine serum) during the preincubation. S2 cells used for the other treatment conditions were preincubated for 1 h with chelator or silver as follows: 10 μM tetrathiomolybdate (TTM) and 250 μM bathocuproine disulfonate (BCS) for Cu chelation experiments, 10 μM ferrozine, and 250 μM bathophenanthroline disulfonate (BPS) for iron chelation, and 10 μM silver nitrate. Cells were stimulated with human insulin at a concentration of 25 μg/ml of medium.
Ctr1+/+ and Ctr1−/− mouse embryonic fibroblasts (MEFs) and insulin or fibroblast growth factor (FGF) stimulation experiments.
Isolation and culture of Ctr1+/+ and Ctr1−/− cells were done as described previously (34). Insulin or FGF stimulation experiments were done with plates measuring 100 by 20 mm, with one plate per time point. Cells were allowed to reach ∼95% confluence and then serum starved for 16 to 48 h. Recombinant human insulin (Invitrogen) was added at a final concentration of 200 nM, and recombinant human basic FGF (Invitrogen) was added at a final concentration of 10 ng/ml, with the exception of the time zero plate. At the appropriate time point, medium was removed, and cells were washed with ice-cold phosphate-buffered saline (PBS), harvested, and lysed using the phosphorylation lysis buffer described above or radioimmunoprecipitation assay (RIPA) buffer consisting of 1% nonylphenoxypolyethoxylethanol (NP-40), 20 mM Tris-HCl (pH 8.0), 137 mM sodium chloride (NaCl), 10% glycerol, 10 mM sodium orthovanadate (Na3VO4), 50 mM sodium fluoride (NaF), 50 mM β-glycerophosphate (β-GP), and 1× protease inhibitor cocktail (BD Biosciences).
Immunoblot assays.
Protein was quantified using the Bio-Rad DC protein assay and run on precast Criterion Tris-HCl polyacrylamide gradient gels (Bio-Rad) or 10% SDS-PAGE. The primary antibodies used are as follows: mouse anti-BRaf, mouse anti-Mek1, rabbit anti-Mek2, rabbit anti-Erk2, mouse anti-Mek1/2, rabbit anti-p44/42 MAPK (Erk1/2), rabbit anti-Akt, rabbit anti-phospho-Mek1/2 (Ser217/221), mouse anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), rabbit anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), and rabbit anti-phospho-Akt (Thr308), which were all obtained from Cell Signaling Technology and used at a 1:1,000 dilution; goat anti-phospho-BRaf (Thr598/Ser 601) (1:500) and rabbit anti-CCS (anti-copper chaperone for superoxide dismutase 1; FL-274) from Santa Cruz Biotechnology (1:200); rat anti-myelin basic protein (anti-MBP; 1:500) and mouse anti-phospho-MBP from Millipore (1:500); rabbit anti-kinase suppressor of Ras (anti-KSR) from Abcam (1:500); mouse anti-β-actin from Sigma (1:25,000); the rabbit anti-human Ctr1 antibody, described elsewhere, was used at 1:1,000 (47). Secondary antibodies were donkey anti-rabbit and anti-mouse antibodies conjugated with horseradish peroxidase from GE Healthcare Life Sciences (1:5,000) or goat anti-mouse IgG from Invitrogen (1:10,000), goat anti-mouse IgG light chain specific from Jackson ImmunoResearch Laboratories (1:5,000), goat anti-rabbit from Invitrogen (1:10,000), mouse anti-rabbit IgG light chain specific from Jackson ImmunoResearch Laboratories (1:5,000), goat anti-rat IgG from Zymed (1:10,000), and rabbit anti-goat IgG from Invitrogen (1:5,000) conjugated with horseradish peroxidase. Metal chelate affinity purification experiments were performed as described elsewhere (46).
Generation of Ctr1−/−:CMV-Ctr1 and Ctr1−/−:CMV-Ctr1M150A stable cell lines.
The Ctr1 and Ctr1M150A coding sequences were PCR amplified using plasmid templates described elsewhere (50) and cloned into the pcDNA3.1(+) Zeocin vector from Invitrogen. MEFs genetically null for Ctr1 (34) were electroporated with these constructs using the Amaxa Nucleofector kit in accordance with the manufacturer's recommendations. Stable cell lines were generated according to standard protocols (Animal Tissue Culture Book), and Zeocin resistance was used as the selective marker.
Metal pulldown experiments.
Experiments were performed as previously described (46). Briefly, 100 μg of protein was used in the input lane; 500 μg of protein lysate was incubated with the glutathione (GSH)-Cu beads. After a 1-h incubation, lysate was removed, beads were washed several times, Laemmli buffer was added to the beads, the samples were boiled, and the entire sample volume was loaded onto the gel.
Mouse experiments.
Mice possessing the Ctr1 gene flanked by loxP elements (Ctr1flox/flox) were described elsewhere (47). Generation of cardiac-tissue-specific Ctr1 conditional-knockout mice has been described previously (29). Cardiac tissues from age-matched mice (10 days old) were dissected after perfusion with PBS (pH 7.4) and homogenized in cell lysis buffer (62.5 mM Tris [pH 6.8], 2% sodium dodecyl sulfate [SDS], 1 mM EDTA) containing protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Thermo Scientific). Anti-CCS antibody (Santa Cruz) was used at a 1:2,000 dilution. Antitubulin antibody (Sigma) was used at a 1:5,000 dilution.
Ethics statement.
Protocols for animal work was approved by the Institutional Animal Care and Usage Committee of Duke University Medical Center.
In vitro Cu-binding experiments.
The rat Mek1 coding sequence was PCR amplified using the pCMV-HAMek1 construct (gift of A. Catling) and cloned into the pGex6P-1 vector from GE Healthcare Life Sciences. The resulting plasmid, pGex6P-1Mek1, was transformed into BL21-CodonPlus (DE3)-RIPL cells from Stratagene. Recombinant, glutathione S-transferase (GST)-tagged Mek1 was purified by affinity chromatography using GSH agarose beads, followed by on-column Pre Scission protease cleavage of the GST tag. Further purification was achieved using MonoQ anion-exchange chromatography that served to remove the majority of the contaminating proteins, including cleaved GST tags that were not retained on the GSH column. SDS-PAGE of the resulting Mek1 revealed a predominant single band at ∼44 kDa. Protein concentrations were determined by quantitative amino acid analysis with a Beckman 6300 analyzer after hydrolysis in 5.7 N HCl at 110°C in vacuo. Equilibrium dialysis experiments were conducted as previously described (22). Recombinant Mek1 (2.5 to 10 μM in 20 mM Tris [pH 7.2]) was dialyzed, using a dialysis tube with a 10-kDa molecular mass cutoff, against CuCl2 concentrations ranging from 0.25 to 15 μM in 20 mM Tris (pH 7.2)–100 mM NaCl overnight at 4°C with slow stirring. Cu levels associated with Mek1 and the dialysate were quantitated by inductively coupled plasma mass spectroscopy after digestion with 50% HNO3 at 65°C. In specific experiments, Cu2+ was introduced as a Cu2+-histidine complex to preclude Cu2+ hydrolysis and precipitation. Cu2+ binding affinity for Mek1 was estimated using competition experiments similar to that described previously with the divalent metal ligand PAR [4-(2-pyridylazo)resorcinol] (66). The quantitative release of the 1:1 Cu2+−/PAR complex on titration of apo-Mek1 was monitored spectrophotometrically at 500 nm (Beckman DU 600 spectrophotometer) in 20 mM Tris (pH 7.2)–100 mM NaCl. The binding affinity of Cu2+ for PAR was calibrated using a spectroscopically silent ligand, EDTA, with a known affinity for Cu2+ of 1.6 × 10−19.
In vitro kinase assays and immunocomplex kinase assays.
Human Erk2 and human Mek1 were obtained from Addgene (catalog numbers 23498 and 21208) and cloned into pGEX4T3 and pGEX6P1 from GE Life Sciences, respectively. Recombinant GST-human Erk2 (hErk2) and GST-human Mek1 (hMek1) were purified from BL21(DE3) (New England BioLabs) bacteria as described previously (21, 37), while unphosphorylated MBP was purchased from Millipore. Specifically, 500 ml of LB was inoculated with BL21(DE3) bacteria transformed with pGEX4T3-hErk2 or pGEX6P1-hMek1 and allowed to grow to an optical density at 600 nm (OD600) of 0.6 was reached. GST-hErk2 was induced by IPTG (0.4 mM) at 37°C for 4 h, while GST-hMEK1 was induced by IPTG (1 mM) for 14 h at 25°C before collection by ultracentrifugation. GST-hErk2 and GST-hMek1 pellets were resuspended in 50 ml of 1× PBS–1% Triton X-100 plus a protease inhibitor tablet (Roche) and sonicated for bacterial lysis. The soluble fraction was obtained via ultracentrifugation and incubated with 1 ml of a 50% slurry of GSH-Sepharose 4B (GE) overnight at 4°C. Bound GST-hErk2 and GST-hMEK1 were washed 3× with lysis buffer and eluted from beads for 30 min at 4°C with elution buffer (100 mM Tris-HCl [pH 8.0], 120 mM NaCl) containing 15 mM GSH. Eluted GST proteins were dialyzed in tubing with a 12 to 14,000 molecular weight cutoff (Spectrum Laboratories) overnight at 4°C in 2 liters of elution buffer and subsequently concentrated using 10K Amicon Ultra centrifugal filter units (Millipore). The concentration was determined using the Bio-Rad DC protein assay. Modified versions of Mek1 and Erk2 in vitro kinase assays were performed as described previously (32, 37). Briefly, for Mek1 kinase assays, 0.6 μg of GST-hERK2 and 1.4 μg of GST-Mek1 were incubated in 180 μl of kinase buffer (25 mM Tris-HCl [pH 7.5], 20 mM MgCl2, 2 mM dithiothreitol [DTT], 25 mM β-GP, 0.5 mM Na3VO4, 120 μM ATP) in the presence or absence of increasing amounts of CuSO4, 50 μM TTM (Sigma) in the presence of CuSO4, or 1 μM Mek inhibitor 1 in the presence of CuSO4 (Calbiochem) at 22°C for 30 min. Reactions were quenched with 5× Laemmli buffer, and a third of the reaction mixture was analyzed by SDS-PAGE via subsequent Western blotting with phosphospecific antibodies. Briefly, for the Erk2 kinase assays, 2.0 μg of GST-Erk2 and 1.0 μg of MBP were incubated in 180 μl of kinase buffer (25 mM HEPES [pH 8.0], 20 mM MgCl2, 1 mM DTT, 20 mM β-GP, 0.1 mM Na3VO4, 100 mM ATP) at 30°C for 30 min. Reactions were quenched with 5× Laemmli buffer, and a third of the reaction mixture was analyzed by SDS-PAGE via Western blotting with phosphospecific antibodies.
Immunoprecipitation.
Ctr1+/+ and Ctr1−/− lysates were solubilized with the RIPA buffer described above, and the lysates (250 μg) were incubated with anti-Mek1 antibody (1:50; Cell Signaling) overnight and then with protein G-Sepharose 4B (GE) for 2 h. Beads were washed 3 times in RIPA buffer. Immunoprecipitates were resolved by SDS-PAGE and analyzed by Western blotting with anti-Mek1 (Cell Signaling) and anti-Erk1/2 (Cell Signaling) antibodies. Equal loading was analyzed with whole-cell extract by Western blotting with anti-Mek1 (Cell Signaling), anti-Erk1/2 (Cell Signaling), anti-CCS (Santa Cruz), and β-actin (Sigma) antibodies.
RESULTS
Cu and the Ctr1 Cu transporter in the Ras signaling pathway.
Given the broad roles for Cu in normal metazoan growth and development, the function of Ctr1A, the evolutionarily conserved Drosophila homologue of the mammalian Ctr1 high-affinity Cu+ transporter (60), was explored in the fruit fly prothoracic gland, a key organ controlling body size (45). Transgenic flies expressing a yeast Gal4 transcription factor-inducible double-stranded RNA hairpin molecule against Ctr1A (UAS-Ctr1ARNAi) were crossed to flies expressing phantom-Gal4 (phm-Gal4), which drives Gal4 transcription factor expression specifically in the prothoracic gland, resulting in organ-specific reduction of plasma membrane-localized Ctr1A levels (Fig. 1A). Flies carrying both the UAS-Ctr1ARNAi and phm-Gal4 transgenes are larger than siblings carrying either transgene alone (Fig. 1B) and quantitative measurements of pupae confirmed the increase in size observed in adult flies with a prothoracic-gland-specific Ctr1A knockdown (Fig. 1C). Similar results were obtained with fly stocks in which the UAS-Ctr1ARNAi transgene was integrated on a different chromosome (data not shown), indicating that this phenotype is not due to a locus-specific integration of the transgene.
Fig 1.

Knockdown of Ctr1A in the prothoracic gland results in a large-fly phenotype. (A) Detection of Ctr1A in dissected prothoracic glands by indirect immunofluorescence assay. The genotype is shown above each image. The prothoracic gland driver used for these experiments, phm-Gal4::UAS-mCD8GFP, also expresses membrane-localized GFP under the control of the Phantom promoter. Knockdown of Ctr1A is indicated by arrows that show reduced plasma membrane staining of Ctr1A in the prothoracic gland. (B) Images of adult Drosophila. Flies carrying only the prothoracic gland Gal4 driver, Phantom-Gal4 (phm-Gal4), are smaller than flies with knockdown of Ctr1A in the prothoracic gland (phm-Gal4:UAS-Ctr1ARNAi). The flies shown are both females. (C) Quantitative measurements of pupae. Comparisons of pupal length must be made within each sex, as female flies are larger than male flies. WT refers to the W1118 stock. Genotypes are as follows. phm-Gal4/+, pupae possessing only the prothoracic gland Gal4 driver transgene; phm-Gal4:UAS-RasDN, pupae expressing a dominant negative allele of Ras in the prothoracic gland; phm-Gal4:UAS-Ctr1ARNAi, pupae with knockdown of Ctr1A in the prothoracic gland. * indicates a P value of less than 0.01 when that genotype is compared to a WT fly of the same sex. n = 10 for all genotypes except phm-Gal4/+, where n = 8. The data shown here are for transgenic stocks with UAS-Ctr1ARNAi on the second chromosome. Similar results were obtained with flies with a UAS-Ctr1ARNAI transgene on the third chromosome (data not shown).
Appropriate Ras protein signaling in the prothoracic gland is critical for body size determination, as constitutively active Ras mutants give rise to small flies while mutations that suppress Ras signaling give rise to abnormally large flies (Fig. 1C) (7). Knockdown of Ctr1A in the prothoracic gland phenocopies the large-fly phenotype of prothoracic-gland-specific dominant negative Ras expression, suggesting a possible interaction between Ctr1A and Ras signaling in the regulation of body size in Drosophila. We genetically tested this possibility by knockdown of Ctr1A in flies expressing a constitutively active allele of Ras. While expression of constitutively active RasV12 in transgenic flies via the apterous-Gal4 driver (ap-Gal4) is lethal, coexpression of the UAS-RasV12 and UAS-Ctr1ARNAi transgenes via ap-Gal4 rescues this lethality. In some wings from viable flies, ectopic veins within the posterior compartment of the marginal cell were observed (Fig. 2A, right panel). This phenotype is also observed in Ellipse mutants possessing hyperactive alleles of the epidermal growth factor receptor that drive increased Ras signaling (3) and indicates a partial suppression of ectopic Ras signaling by Ctr1A knockdown. Moreover, expression of the UAS-RasV14 transgene in the eye using eyeless-Gal4 (ey-Gal4), which yields a rough-eye phenotype characterized by fused ommatidia and disorganized bristles, was suppressed in flies with simultaneous expression of UAS-Ctr1ARNAi and UAS-RasV14 (Fig. 2B). Taken together, these data support a potential genetic interaction between Ctr1A and Ras that occurs in multiple Drosophila tissues.
Fig 2.

Knockdown of Ctr1A suppresses constitutively active Ras phenotypes in both the fly eye and wing. (A) Bright-field images of adult Drosophila wings. Expression of UAS-RasV12 using an apterous-Gal4 (ap-Gal4) driver, which drives expression in the dorsal compartment of the wing, is lethal, presumably because apterous is also expressed in portions of the central and peripheral parts of the nervous system, while expression of both the UAS-RasV12 and UAS-Ctr1ARNAi transgenes yields viable adult flies with normal wings. Bright-field images of adult Drosophila wings. Shown on the far right are ×10 magnifications of the W1118 female wing and the ap-Gal4:UAS-RasV12, Ctr1ARNAi female wing. Note that the wings of some of the eclosed flies had an ectopic vein in the posterior portion of the marginal cell (arrow) in the ap-Gal4:UAS-RasV12, Ctr1ARNAi wing. This phenotype is reminiscent of Ellipse mutants, a hyperactive allele of the epidermal growth factor receptor upstream of Ras, that leads to ectopic vein differentiation (3). Thus, some ectopic Ras activity is visible and not suppressed in flies expressing both the UAS-RasV12 and UAS-Ctr1ARNAi transgenes in the wing. (B) SEM images of adult female Drosophila eyes, with the genotype shown above each image. The ey-Gal4 driver line expresses UAS promoter-driven transgenes in the eye. Expression of a constitutively active isoform of Rasv14 results in a rough-eye phenotype (top far right image). Knockdown of Ctr1A in the eye can suppress the Rasv14 phenotype (bottom images; compare bottom images to top far right and far left images).
Ctr1A knockdown suppresses developmental phenotypes associated with activated Ras, as well as phenocopies the size defect associated with loss of Ras activity in the prothoracic gland. These genetic interactions suggest a functional interaction with Ras or with components of the multiple signaling pathways downstream of Ras. Inspection of data from a genome-wide screen for regulators of MAPK signaling in cultured fly S2 cells revealed a diminution of Erk phosphorylation when Ctr1A was targeted by RNA interference (17). Interestingly, in this study, knockdown of Ctr1A in S2 cells resulted in downregulation of Ras pathway activation to an extent comparable to that achieved by knockdown of canonical pathway members such as the insulin receptor or Ras. Independently, we have confirmed that reduction of Ctr1A protein levels in S2 cells results in decreased Erk phosphorylation (data not shown). An additional study suggests a role for Xenopus Ctr1 in Ras/MAPK signaling whereby Ctr1 functions in a Cu+ transport-independent manner as a scaffold protein (20). To explore whether both Ctr1 and the associated Cu+ transport function are important for Ras signaling to Erk1/2, Cu+-specific chelation was used to impose Cu deficiency on cultured fly S2 cells. As shown in Fig. 3A, Cu+ chelation reduced the levels of insulin-stimulated Erk1/2 phosphorylation without altering steady-state Erk1/2 levels. For these and subsequent experiments, the phosphorylation of Erk1/2 at critical residues (threonine 202 and tyrosine 204 of Erk1 and threonine 185 and tyrosine 187 of Erk2) required for kinase activation was examined as a surrogate marker of Erk1/2 activity (57). This reduction of Erk1/2 phosphorylation by Cu chelation was not due to the chelation of all redox-active metals, as the Fe2+-specific chelator BPS or ferrozine did not reduce insulin-stimulated Erk1/2 phosphorylation (Fig. 3B). Moreover, as Ag is isoelectric to Cu+ and is a competitive inhibitor of Cu+ uptake transporters, preincubation of S2 cells with Ag clearly diminished the levels of insulin-stimulated Erk1/2 phosphorylation (Fig. 3C).
Fig 3.
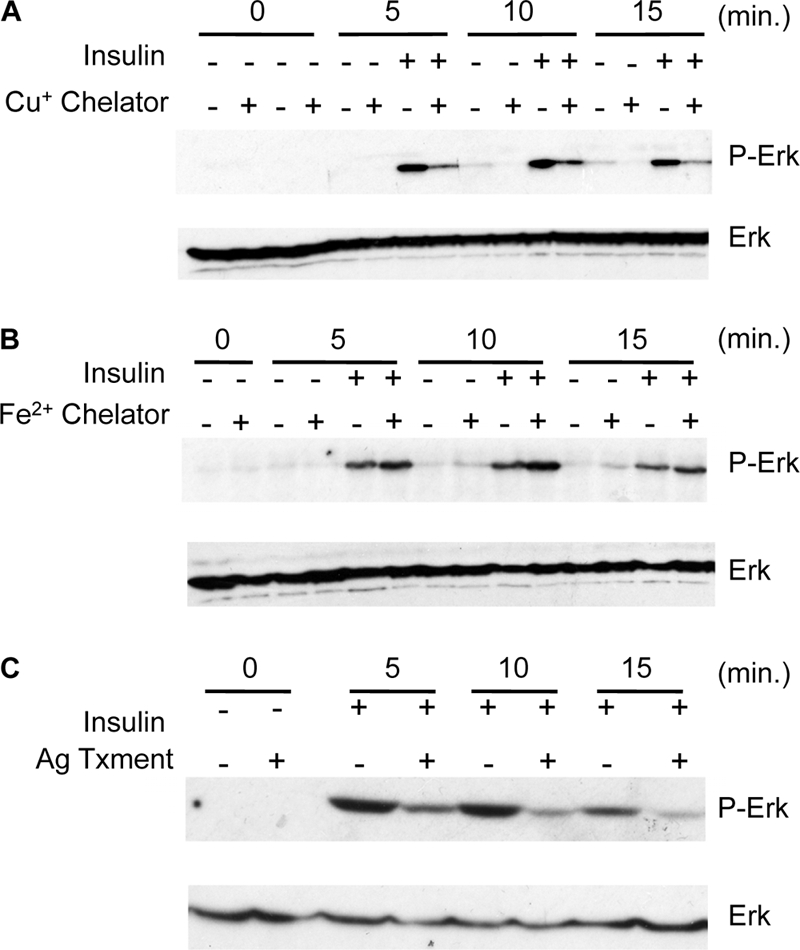
Cu chelation or competition for Ctr1A-mediated Cu+ transport compromises Ras/MAPK signaling in Drosophila S2 cells. (A) Cu chelation downregulates Ras/MAPK signaling. Cells were not pretreated (−) or pretreated (+) with the Cu+-specific membrane-impermeant chelator BCS as indicated. Cells were left untreated (−) or treated (+) with insulin from 0 to 15 min, and total protein extracts were analyzed by immunoblotting for total Erk (Erk) and phospho-Erk (P-Erk) as shown. (B) Iron chelation with the membrane-impermeant Fe2+-specific chelator BPS was used as described for panel A. (C) Competition for Ctr1A-mediated transport by silver (Ag). Cells were not pretreated (−) or pretreated (+) with Ag and assayed as described for panels A and B.
Conservation of function of Ctr1 and Cu in Erk activation in mammals.
In contrast to a report suggesting a Cu+ transport-independent function for Ctr1 in Ras/MAPK signaling (20), the experimental results presented in Fig. 3 suggest an involvement of Cu in Ras/MAPK signaling. To further ascertain a potential role for Cu and the Cu+-transporting activity of Ctr1 in Ras signaling and to determine if this functional role is conserved in mammals, Ctr1+/+ and Ctr1−/− MEFs were evaluated for insulin-stimulated Erk1/2 phosphorylation. Ctr1−/− MEFs have residual Ctr1-independent Cu transport activity that accounts for ∼30% of the total Cu accumulation (34). Ctr1+/+ MEFs demonstrated a strong insulin-stimulated Erk1/2 phosphorylation within 5 min of treatment that was maintained over a 15-min time course (Fig. 4A). In contrast, Ctr1−/− MEFs showed only marginal insulin-stimulated Erk1/2 phosphorylation. While Ctr1−/− MEFs exhibit strong reductions in the activity of Cu-dependent enzymes, such as cytochrome oxidase and lysyl oxidase, these activities can be partially rescued by exogenous Cu (34). Preincubation of Ctr1−/− MEFs with 25 μM Cu for 1 h prior to insulin stimulation resulted in increased insulin-stimulated Erk1/2 phosphorylation, though not to the same levels as Ctr1+/+ MEFs (Fig. 4A). No additional stimulation was observed in Ctr1+/+ cells when Cu was added. These results demonstrate that insulin stimulation of Erk1/2 phosphorylation in mammalian cells is heavily dependent on Ctr1 and that, in the absence of Ctr1, this defect can be partially ameliorated by exogenous Cu.
Fig 4.

Ctr1 function in Ras/MAPK signaling is dependent upon Cu+ transport activity. (A) MEFs wild type (Ctr1+/+) or null (Ctr1−/−) for Ctr1 were treated with insulin, and phospho-Erk was analyzed over time by SDS-PAGE and immunoblotting. Total Erk1/2 was evaluated as a loading control. (B) Ctr1−/− cells stably expressing either wild-type human Ctr1 (Ctr1) or a transport-defective mutant form of human Ctr1 (Ctr1M150A) were analyzed for insulin-stimulated Ras/MAPK activity in the phosphorylation of Erk. The Ctr1M150A transport-defective cell line is Cu deficient, as indicated by the increased levels of CCS, compared to those of the Ctr1 wild-type cell line. Total Erk1/2 was assessed as a loading control. Note that the same film exposures for Ctr1 wild-type and Ctr1M150A samples were used in examining protein levels; the dotted line indicates the removal of additional, superfluous controls loaded between wild-type and Ctr1M150A sample lanes.
Previous studies demonstrated that two methionine residues located in the second transmembrane domain of Ctr1 in a Met-X3-Met motif are important for Ctr1-mediated Cu+ transport but not for oligomerization or localization to the plasma membrane (50). To determine if the integrity of this motif is important for insulin-stimulated Erk1/2 phosphorylation, Ctr1−/− MEFs were stably transfected with plasmids expressing either wild-type human Ctr1 or Ctr1 in which the first methionine in this motif, M150, had been altered to alanine and evaluated for insulin-stimulated Erk1/2 phosphorylation (Fig. 4B). While the Ctr1−/− cells rescued with wild-type Ctr1 showed robust insulin-induced Erk1/2 phosphorylation, this was strongly reduced in MEFs stably expressing the Cu transport-defective Ctr1M150A protein. Although both Ctr1 wild-type and Ctr1M150A MEFs expressed approximately equivalent amounts of Ctr1, the Ctr1M150A cells remained more Cu deficient, as indicated by the increased steady-state levels of CCS, which is subject to ubiquitin-mediated proteolysis in the presence of elevated Cu levels and stabilized during Cu deficiency (8). Taken together with the findings on Cu chelation, Ag competition, and exogenous Cu rescue of Ctr1−/− MEFs, these results strongly suggest that Cu and the Cu+-transporting activity of Ctr1 are important for normal activation of Erk1/2 phosphorylation in flies and mice.
Ras is a central cellular regulator that lies upstream of multiple signaling pathways and proteins that include phosphatidylinositol 3 kinase (PI3K), RalGEF, Raf, TIAM1, and others (55). Genetic and biochemical experiments have demonstrated the involvement of Ctr1A in flies and Ctr1 in mammals in the Ras-to-Erk signaling pathway. If Ras represents the key intersection point for Ctr1 and Cu, then Ctr1 and Cu status would be predicted to alter the activity of multiple signaling pathways that are downstream of Ras. To test this hypothesis, we evaluated the Ras/PI3K/Akt kinase signaling pathway. As shown in Fig. 5, there are no significant changes in phosphorylation at Thr308 of Akt1, which is the key residue phosphorylated by PDK1 in response to PI3K pathway activation (1) in either the Ctr1+/+ or the Ctr1−/− cell line. These results suggest that the Ctr1 and Cu-responsive components of Ras signaling lie downstream of Ras and do not impact the Ras/PI3K/AKT signaling network.
Fig 5.
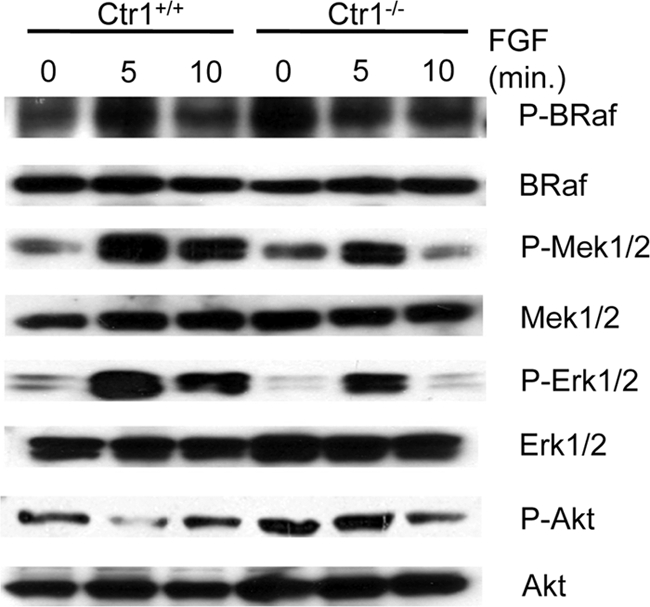
In Ctr1−/− cells, Ras/MAPK pathway signal transduction is intact through Mek1/2 activation. Ctr1+/+ and Ctr1−/− cells were serum starved for 16 h and subsequently stimulated with FGF for the times indicated, and the phosphorylated and total levels of B-Raf, Mek1/2, Erk1/2, and Akt1 were assessed by SDS-PAGE and immunoblotting of the total protein extracts.
The observation that Erk1/2 phosphorylation, but not that of Akt, is heavily dependent on Cu transport-competent Ctr1 and adequate levels of intracellular Cu suggests that Cu may influence the Ras/Raf/Mek/Erk signaling pathway. To further test this hypothesis, the steady-state levels and phosphorylation status of components of this pathway downstream of FGF-stimulated Ras activation were evaluated in Ctr1+/+ and Ctr1−/− cells by immunoblotting (Fig. 5). Activation of the main Raf kinase in MEFs, B-Raf (14), occurred to a similar extent in both Ctr1+/+ and Ctr1−/− cells as assessed by evaluating phosphorylation of Thr598 and Ser601 in B-Raf, two key residues that become phosphorylated upon Ras activation (64). Intriguingly, we observed increased phosphorylation of B-Raf on Thr598 and Ser601 in unstimulated Ctr1−/− MEFs. We speculate that the increase in phosphorylation in the knockout versus wild-type MEFs is due to the absence of active Erk1/2-mediated negative feedback on the MAPK signaling pathway that disrupts Raf-1/B-Raf dimerization (53a). Given the similar levels of Akt phosphorylation in Ctr1 wild-type versus Ctr1 knockout cells, Ras activity is likely not affected by loss of Ctr1 or reductions in intracellular Cu levels. Active Ras binds to and activates the Raf kinases that phosphorylate and activate the serine threonine MAPK kinases Mek1 and Mek2. Phosphorylation of Mek1 and Mek2 is observed in both Ctr1+/+ and Ctr1−/− cells, suggesting that Raf activity is not affected by loss of Ctr1 or reductions of intracellular Cu levels. Activated Mek1/2 phosphorylate Erk1 and Erk2, and signal transduction events downstream of Erk ultimately result in the dephosphorylation and inactivation of Mek1/2 (31, 57). As observed previously upon insulin stimulation, FGF-stimulated phosphorylation of Erk1/2 was diminished in Ctr1−/− cells compared to that in cells expressing Ctr1, consistent with a defect in Erk1/2 activation (Fig. 5). Similar effects on Ras/MAPK pathway activation were also obtained when insulin was used as the stimulus (data not show). The results of this study demonstrate that loss of the Ctr1 Cu+ transporter or reductions in Cu accumulation result in a diminution of Erk1/2 phosphorylation without altering the upstream signatures of Raf activation. These observations suggest that Cu may play a role in the Ras/MAPK signaling pathway at the juncture where Mek1/2 phosphorylate Erk1/2.
Mek1 directly binds Cu.
Loss of the Ctr1 Cu+ transporter or reductions in Cu accumulation result in a diminution of insulin or FGF-stimulated Erk1/2 phosphorylation without altering the upstream signatures of Ras and Raf activation. These observations suggest a defect in Mek activation of Erk via Mek-dependent phosphorylation when cells are Cu deficient. To test whether Mek1 itself may be a Cu-binding protein, extracts from wild-type MEFs were incubated with beads conjugated with the metal-binding tripeptide GSH that was either uncharged or charged with Cu. GSH beads alone were unable to purify Mek1, Erk1/2, or glyceraldehyde 3-phosphate dehydrogenase (GAPDH) from the lysate (Fig. 6A). However, when GSH beads were preloaded with Cu, Mek1, but not Erk1/2 or GAPDH, was enriched from total cell extracts. To further test the specificity of this metal-binding resin and the metal specificity of Mek1 binding, the broad-spectrum metal-binding ligand pentadentate was used in affinity purification with MEF protein extracts. As shown in Fig. 6B, while metal-free or Zn-loaded pentadentate beads were unable to purify Mek1 protein, Mek1 was partitioned from the extract with Cu2+-loaded pentadentate. Moreover, the Mek1/2 scaffold protein KSR1 (30) was not bound to Cu-pentadentate, suggesting that the interaction between the Cu chelate complex and Mek1 may be direct (Fig. 6B). To test for a potential direct interaction, Mek1 protein was expressed in and purified from Escherichia coli and applied to pentadentate beads for Mek1 partitioning and immunoblotting experiments. As shown in Fig. 6C, recombinant Mek1 could be adsorbed onto Cu-loaded pentadentate beads but not metal free or Zn-loaded beads. Taken together, these results strongly suggest that Mek1 interacts directly with Cu, with the ability to discriminate between Cu and Zn.
Fig 6.

Mek1 is affinity purified by Cu-chelated resins. (A) GSH resin, either alone or preincubated and loaded with Cu, was incubated with total protein extracts from wild-type MEFs. The levels of Mek1, GAPDH, and Erk1/2 were assayed from the input proteins, GSH resin affinity-purified proteins, and Cu-charged GSH resin-purified proteins by SDS-PAGE and immunoblotting. (B) Pentadentate-chelated beads complexed with no metal, zinc, or Cu were incubated with lysates from Ctr1+/+ cells, and Mek1 and the KSR1 scaffold protein were analyzed by immunoblotting as for panel A. (C) Purified recombinant rat Mek1 was added to pentadentate beads that were uncharged (metal free) or charged with zinc (Zn) or Cu, and affinity-purified Mek1 was analyzed by SDS-PAGE and immunoblotting.
To further investigate the nature of Cu binding to Mek1, purified recombinant Mek1 was used for in vitro Cu binding stoichiometry and binding affinity experiments. As shown in Fig. 7A, purified Mek1 protein was subjected to dialysis against several concentrations of CuCl2, with and without the presence of competitors, to determine the Cu2+-to-protein binding ratio under equilibrium conditions. Dialysis against 2 μM Cu2+ increased the bound Cu content to ∼2.5 molar equivalents (mol equiv) from the as-isolated ratio of ∼0.5. To prevent metal hydrolysis by water, Cu was also introduced as a Cu-His complex in the dialysate; this did not alter the bound Cu content. However, the inclusion of 0.1 mM EDTA reduced the Cu content to ∼1.5 mol equiv. Dialysis of Mek1 against a 2 μM Cu2+–4 μM His complex, followed by a subsequent dialysis step in 0.1 mM EDTA, reduced the Cu content to approximately 2 mol equiv. Inclusion of Zn(II) and Fe(II) as competitors in the dialysate gave a Cu/Mek1 ratio of 2.1, compared to 2.6 when only Cu2+ was present. Taken together, these dialysis experiments suggest the presence of specific high-affinity Cu2+-binding sites in Mek1 and a site with low-affinity interaction. In a separate series of equilibrium dialysis experiments in which the Cu2+ content was varied from 0.11 to 15 μM, the maximal Cu content associated with Mek1 plateaued near ∼2.5 mol equiv (Fig. 7B). The hyperbolic nature of the resulting curve indicated saturation at higher CuCl2 concentrations, and the maximal ratio agreed well with that of dialysis experiments. Both sets of experiments indicated more than one Cu binding site. Under these same conditions, Cu2+ binding is observed with albumin, a known Cu2+-binding protein, but not lysozyme or thyroglobulin (data not shown).
Fig 7.

Recombinant Mek1 metal-binding characteristics. (A) Dialysis experiments under the indicated equilibrium conditions showing a Cu/Mek1 ratio of 2 to 3, depending on competitive or noncompetitive conditions. Competition experiments decreased the ratio by unity, suggesting the presence of a low-affinity site. (B) Demonstration of saturation binding by equilibrium dialysis with increasing CuCl2 concentrations in the dialysate using an independent set of purified rat Mek1. Mek1 was used at concentrations of 4 to 6 μM. (C) Determination of the Cu2+ dissociation constant, KD, of Mek1 using the probe PAR showing overall spectral changes of the Cu-PAR complex on Mek1 titration. The inset shows the decrease at 500 nm relative to Mek1 additions for a [Cu-PAR]total of 3.9 μM and a [PAR]total of 9.3 μM. (D) Apparent KDs at pH 7.4 derived from competition titration using Cu2+-PAR. The asterisk indicates Mek1 dialyzed against a Cu2+-His complex overnight and then dialyzed against 0.5 M NaCl and 100 mM EDTA.
To obtain a precise binding affinity, a series of ligand competition studies using PAR were conducted (66). PAR is a chromogenic chelator forming colored complexes with metal ions. The affinity of the Cu2+-PAR complex (formation constant [β]) is 3.2 ×1017, and the equilibrium concentration of the complex is measurable at 500 nm (extinction coefficient [ε], 35,500 M−1 cm−1) with an isosbestic point at 445 nm. Bidentate PAR forms a 1:1 complex with Cu2+. Titration of apo-Mek1 with the Cu-PAR complex revealed a concentration-dependent attenuation of the Cu2+-PAR concentration, consistent with equilibration of Cu2+ from PAR to Mek1 (Fig. 7C). The initial rapid decrease at 500 nm on increasing apo-Mek1 addition is indicative of a strong affinity of Mek1 for Cu2+ (Fig. 7C, inset). This decrease at 500 nm was observed until the Mek1/Cu ratio reached ∼0.5, which showed that effective competition between the PAR ligand and Mek1 was induced.
Control titrations under the same pH and ionic strength buffer conditions were performed with EDTA and bovine serum albumin (BSA) to validate the Cu2+-PAR titration study. Both EDTA and BSA are spectrally silent, with known dissociation constants. EDTA also served to calibrate Cu2+-PAR affinity relative to the reaction condition used for our experiments. Calculations of the Cu2+-binding affinities of EDTA and BSA confirmed literature values for both ligands (Fig. 7D). Calculations of Mek1 data revealed an apparent dissociation constant of ∼10−18 M, suggesting that Cu2+ is avidly associated with Mek1. Since the Cu2+ binding stoichiometry is ∼2.5 mol equiv, from these experiments, we cannot resolve whether the Cu2+ binding sites are equivalent.
Cu stimulates Mek1-mediated phosphorylation of Erk1 in vitro.
The metal pulldown and binding studies support the identification of Mek1 as a Cu-binding protein. Additionally, Cu deficiency imposed by loss of Ctr1, or through Cu chelating agents or competitors of Cu uptake, resulted in impaired Mek1-mediated phosphorylation of Erk1. To further explore the role Cu plays in the stimulation of Mek1-dependent phosphorylation of Erk1/2, a series of in vitro experiments were carried out. For these experiments, purified human Mek1 protein was incubated with a kinase-dead isoform of hErk2 so that assessments of Erk2 phosphorylation mediated by Mek1 could be made in the absence of Erk2 autophosphorylation (56). The data shown in Fig. 8A are representative results of 3 in vitro Mek1 kinase activity assays that yielded similar trends for Mek1 activity. When recombinant Mek1 was incubated with kinase-dead Erk2, an ∼2-fold increase in Erk2 phosphorylation was observed that may have been due to residual Cu that copurified with recombinant Mek1 protein (Fig. 7A, as-isolated sample) compared to kinase-dead Erk2 alone, which in and of itself still retains some autophosphorylation ability. However, Mek1 kinase activity was greatly enhanced by Cu addition in a dose-dependent manner, with Mek1 activity ∼20 times greater in the presence of 2.5 μM CuSO4. Furthermore, Mek1 activity in the presence of 2.5 μM CuSO4 was blunted by the addition of TTM, a Cu-chelating agent. Similar in vitro kinase assays were performed with recombinant wild-type Erk2 protein, and no effect of Cu addition on Erk2 phosphorylation of MBP, a commonly used substrate for Erk kinase assay, was observed (Fig. 8B).
Fig 8.

Mek1 kinase activity and association with Erk are stimulated by Cu. (A) Recombinant, GST-tagged human kinase-dead Erk2 and recombinant, GST-tagged human Mek1 were incubated with increasing amounts of CuSO4 with or without TTM or Mek inhibitor 1. Mek1 phosphorylation of Erk2 was assessed by Western blotting with Erk1/2 phosphospecific antibody (n = 3). (B) Erk kinase activity in not enhanced by the addition of Cu. Recombinant GST-hErk2 and recombinant MBP were incubated with increasing amounts of CuSO4. Erk2 phosphorylation of MBP was assessed by Western blotting with MBP phosphospecific antibody (n = 2). (C) Coimmunoprecipitation of Mek1 and Erk1/2 in Ctr1+/+ and Ctr1−/− MEFs. Mek1 immunoprecipitation (IP) from Ctr1+/+ and Ctr1−/− and binding of Erk1/2 were assessed by Western blotting with Mek1 and Erk1/2 antibodies. RalB immunoprecipitation was used as a negative control. Cu deficiency was assessed by immunoblot analysis of CCS protein levels in whole-cell extract (WCE). Immunoblot analyses of total Mek1, Erk1/2, and β-tubulin served as loading controls (n = 3).
One possible mechanism of a role for Cu in stimulating Mek1 phosphorylation of Erk is enhancement of the association of the two proteins. We examined the interaction between endogenous Mek1 and Erk1/2 under Cu-replete (Ctr1+/+ MEFs) or Cu-deficient (Ctr1−/− MEFs) conditions using coimmunoprecipitation experiments. While a fraction of Mek1 and Erk1/2 can be coimmunoprecipitated in Ctr1+/+ MEFs, this interaction was significantly reduced in Ctr1−/− MEFs (Fig. 8C). These results suggest that Cu stimulates the physical association of Mek1 with Erk1/2, which could augment Mek1 phosphorylation of Erk1/2.
Physiological role for Ctr1 in Erk activation in mice.
The results presented here demonstrate a strong role for Cu in the Ras/Raf/Mek/Erk signal transduction pathway in flies and cultured cells and support direct Cu binding to Mek1. Moreover, our results suggest that a Cu transport-competent form of Ctr1 is required to provide Cu to Mek1. To test for a potential physiological requirement for Ctr1 in Mek1 function in animals, mice were generated with cardiac-tissue-specific ablation of Ctr1 expression (Ctr1hrt/hrt mice) as previously described (29). Protein extracts from two control (C) and two Ctr1hrt/hrt mutant (M) littermates were evaluated for Erk1/2 phosphorylation by immunoblotting. As shown in Fig. 9, hearts from the two Ctr1hrt/hrt mice were Cu deficient, as evidenced by the increased steady-state levels of CCS compared to those of wild-type control littermates. Moreover, cardiac tissue protein extracts from Ctr1hrt/hrt mice showed a clear reduction in Erk1/2 phosphorylation and a concomitant increase in phospho-Mek1/2 levels compared to those of wild-type littermates. The results from tissue-specific ablation of Ctr1 in mice parallel those observed comparing cultured Ctr1+/+ and Ctr1−/− MEFs and demonstrate a clear physiological role for Ctr1 in Mek-mediated Erk phosphorylation in mammalian tissues.
Fig 9.
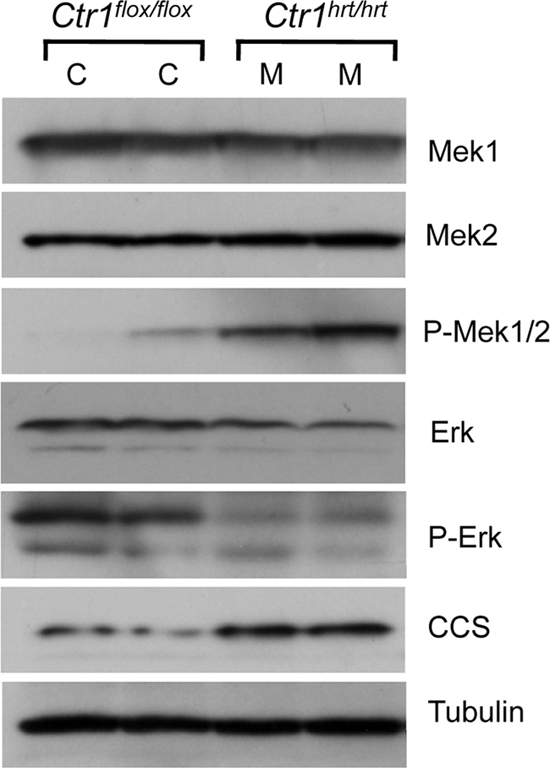
Ras/MAPK signaling from mice with Ctr1-deficient hearts recapitulates cell culture observations. Heart lysates from two Ctr1 wild-type (Ctr1flox/flox) animals were analyzed by immunoblotting for Mek1, Mek2, phospho-Mek1/1, Erk, phospho-Erk, CCS, and tubulin as a loading control. C indicates an individual Ctr1flox/flox mouse, and M indicates an individual Ctr1hrt/hrt mouse analyzed.
DISCUSSION
Cu is essential for normal cell proliferation and differentiation, yet the constellation of biochemical mechanisms in which it participates in these processes is not well understood. While traditionally viewed as a structural or catalytic cofactor that stabilizes protein folds or drives redox chemistry, Cu ions also participate in signaling events through the activation of metalloregulatory transcription factors in bacteria, fungi, and mammals (54, 58) and in other canonical signaling pathways (13, 38). In this work, we provide strong evidence that the Ctr1 high-affinity Cu+ transporter and intracellular Cu play important roles in Ras/MAPK pathway activation. In both flies and mice and in cultured cells, we find that reduction of intracellular Cu accumulation imposed by loss of Ctr1 Cu transporter protein levels or Ctr1 function, results in impaired insulin/FGF-stimulated activation of Ras/MAPK signaling, as evidenced by a reduction in Erk phosphorylation. Although the downstream Erk-dependent functions that may be altered in response to changes in bioavailable Cu have not been evaluated in this work, previous studies suggest that transcript levels known to be dependent on Mek signaling are significantly increased in Cu-treated mammalian cells (41).
Additional evidence for the stimulation of Ras/MAPK signaling by Cu comes from our finding that adequate Cu levels are important for Mek1/2-dependent phosphorylation of Erk1/2. Moreover, the observations that purified recombinant Mek1 binds ∼2 Cu atoms with high affinity and specificity and in vitro experiments that demonstrate a Cu dose-dependent stimulation of Mek1 kinase activity provide strong evidence to support a direct role for Cu ions in Mek1 action. A similar observation of reduced phosphorylation of Erk2 despite activating phosphorylation of Mek1 has been made in cells expressing Mek1 mutants where the amino terminus of the protein, a domain important for Erk2 interactions, was deleted (63). Hence, one possible mechanistic explanation for the reduced Erk phosphorylation observed in Cu-deficient cells and tissues could be reduced interactions between Mek1 and Erk. A corollary to this is that Cu binding may stimulate the Mek1-Erk interaction, perhaps by stabilizing Mek1 in a conformation compatible with productive Erk interactions. Our studies reported here demonstrate that Cu stimulates the kinase activity of recombinant Mek1 toward Erk and promotes the physical association of Mek1 with Erk in cultured cells. Further studies are under way to identify Cu ligands within Mek1 and determine whether the integrity of these ligands is essential for Cu-stimulated Erk1 phosphorylation.
The stimulation of Mek-dependent Erk phosphorylation and the demonstration that Mek1 binds Cu with high specificity and affinity are reminiscent of bacterial Cu-responsive two-component gene regulatory systems in which elevated Cu levels activate the expression of bacterial genes encoding Cu+-transporting efflux pumps. Recently, potential histidine ligands have been proposed for the periplasmic Cu-responsive CinS histidine kinase of Pseudomonas putida, suggesting direct binding of Cu to the Cu-sensing domain of the kinase, which, in turn, would phosphorylate and activate the CinR regulatory protein (51). The nature of potential Cu ligands in Mek1 is currently unknown, as are the mechanisms by which Cu is delivered to Mek1. While both the CCS and Atox1 Cu chaperones deliver Cu+ to cytosolic Cu+-binding domains, low-molecular-weight intracellular Cu ligands could also provide Cu to Mek1 (5). It is notable that Mek activity in vitro is stimulated by Cu, but Cu may not be an absolute requirement for Mek phosphorylation of Erk. In this view, flux of intracellular Cu accumulation or mobilization may provide a stimulatory signal for this pathway in response to other proliferative signals.
The equilibrium binding studies with Mek1 only address the thermodynamics of the binding reaction. These studies indicate that Mek1 directly binds ∼2 atoms of Cu2+ but do not address the kinetic off rate of Cu binding. Kinetic lability in metal binding is one basis for metalloregulation. One example is the Cu-responsive CueR transcriptional regulator in E. coli that binds Cu+ with an affinity of 10−21 M (10). A second example is the kinetic lability in a zinc finger pair motif in the zinc metalloregulatory factor Zap1 in yeast (4). The Zap1 zinc finger pair exhibits the same thermodynamic stability of zinc binding as a related zinc finger pair in the nonregulatory DNA-binding motif in Zap1, but the two differ markedly in kinetic lability, with the metalloregulatory finger pair exhibiting a fast off rate in metal exchange studies. Metalloregulation can be achieved either through a low-affinity metal-binding site (for example, calcium regulation of calmodulin) or through a kinetically labile but thermodynamically stable site. Cu modulation of Mek1 function likely arises from the kinetic lability of the metal site(s).
Our studies are in partial contrast to a previous report which showed reduced Erk phosphorylation in FGF-stimulated Xenopus embryos treated with morpholinos against the Xenopus Ctr1, Xctr1 (20). The authors proposed that this occurs in a Cu+ transport-independent fashion and that Ctr1 serves as a scaffold to regulate Mek signaling. Consistent with this report, we demonstrate in cultured Drosophila and mouse cells, and in fly and mouse models, that the Ctr1 high-affinity Cu+ transporter plays a significant role in signaling through growth factor receptors to the MAPK pathway. However, our studies strongly suggest that this Ctr1-mediated signaling occurs in a Cu-dependent manner. First, Cu chelation, but not chelation of Fe, greatly reduced Erk phosphorylation. Second, Ag, a metal that is isoelectric to Cu+ and competes for entry across the plasma membrane via Ctr1, also diminished Erk phosphorylation. Third, while insulin-stimulated Erk phosphorylation was strongly diminished in Ctr1−/− MEFs compared to wild-type MEFs, exogenous Cu largely restored insulin-stimulated Erk phosphorylation. Since this was observed in the absence of Ctr1, it is likely that Cu entered Ctr1−/− MEFs via a Ctr1-independent Cu uptake mechanism that has been previously described (34). Fourth, the analysis of cells expressing a Cu+ transport-incompetent Ctr1 protein strongly suggests that the Cu+ transport activity of Ctr1 is inextricably linked to the stimulation of Mek activity. Finally, we show that purified recombinant Mek1 directly and specifically binds two atoms of Cu in vitro.
Whereas the present studies demonstrate a regulatory role for Cu ions in Mek1 activity, previous X-ray crystallographic studies did not identify Cu as a bound cofactor. Two considerations may account for this discrepancy. First, the published crystal structures of recombinant Mek1 were solved from proteins with amino-terminal truncations (16, 24, 48, 59). The extents of these truncations varied, but all removed the Erk-binding/docking domain of Mek1. Removal of this domain within Mek1 is reported to reduce its ability to phosphorylate Erk2 in vitro and to interact with and stimulate activation of Erk2 in cells, while still being competent for binding by the upstream activator Raf-1 (63). Similarly, we have found that Mek1 from Cu-deficient cells can still be activated by Raf but has a severely diminished capacity to phosphorylate Erk1/2. Second, recombinant truncated Mek1 protein used in crystallography studies was expressed and purified, with the exception of one study, from bacteria. E. coli does not contain any cuproenzymes in the cytoplasm, and Cu ions are efficiently pumped from the cytoplasm to the periplasm, thereby restricting the available Cu concentration in the cytoplasm (62). Thus, cuproenzymes expressed in E. coli are often recovered as apoproteins. Future studies will be aimed at identifying those amino acid residues required for Cu binding and will include more-detailed spectroscopic analyses, such as X-ray absorption spectroscopy, that can provide information about the coordination chemistry of the metal within the protein (11).
We present data to specifically support Mek1 as a Cu-binding protein, but the data presented in this study do not address whether Mek2 can bind Cu and how this binding might affect Mek2 activity and Erk1/2 phosphorylation. Future studies should investigate whether Mek2 binds Cu directly and, if so, whether apo-Mek2 has a similar effect on Erk1/2 phosphorylation and whether there may be residues conserved between Mek1 and Mek2 that mediate coordination of the metal. Furthermore, while Mek2 can also phosphorylate Erk1/2, data have emerged that support a complex interactive relationship between Mek1 and Mek2 and suggest that Mek1 functions as the critical kinase modulator of Erk signaling (9). In the absence of Mek1, Mek2 demonstrates prolonged phosphorylation and activation that, in turn, results in prolonged phosphorylation of Erk1/2, supporting the idea that Mek1 exerts a negative regulatory effect on Mek2 phosphorylation and activation. Thus, while data regarding the Cu-binding competency of Mek2 are currently unavailable, a model of the role of Cu in regulating Erk1/2 signaling via Mek1/2 proteins can be proposed that is not mutually exclusive of a potential role for Cu in the regulation of Mek2 activity: in the case where Cu could regulate Mek2 activity, Cu deficiency may have an effect on Mek2 activity similar to that reported here for Mek1, or it may be that Cu deficiency, while diminishing Mek1 activity toward Erk1/2, does not affect Mek1 negative regulation of Mek2.
As Cu ions play an important role in stimulating Ras-Mek-Erk pathway activation, the results described here have implications for understanding the connections between Cu availability and cancer cell proliferation and metastasis. Cancer cells and serum from cancer patients have been shown to have alterations in Cu accumulation (19). Moreover, tumor growth and metastasis are strongly dependent on angiogenesis, a process that has been demonstrated to be stimulated by Cu (6, 18). For example, treatment of endothelial cells with Cu stimulates cell migration (40) and Cu-deficient rabbits were shown to be unable to mount strong angiogenic responses in corneal angiogenesis assays (49, 52, 65). Recently, Cu chelators have been investigated for efficacy in clinical trials for the treatment of a range of cancers (18, 25, 27). While the specific roles of Cu in tumor cell proliferation have been suggested to involve the secretion or expression of proangiogenic growth factors, Cu, Zn, superoxide dismutase activity, and other targets (6, 18, 39), the precise Cu-dependent mechanisms that drive cancer cell proliferation and angiogenesis and the targets for Cu chelating anticancer therapies have not been clearly elucidated. As the MAPK pathway plays important roles in both cell proliferation and angiogenesis (15, 30), Mek protein kinases may be critical targets for Cu chelators as chemotherapeutic agents. In support of this possibility, the Mek kinase inhibitor U0126 was recently identified in a screening for compounds that affect Cu metabolism in zebrafish (23).
ACKNOWLEDGMENTS
The Phantom Gal4, UAS mCD8::GFP/TM6, Tb flies were provided by M. O'Connor (University of Minnesota), and plasmid pCMV-HA-Mek1 was provided by A. Catling (LSU Health Science Center). We thank Eric Spana for expert advice on Drosophila experiments and Jamie Roebuck for Drosophila embryo injections at the Duke University Model Systems Genomics Core Facility.
We gratefully acknowledge funding support from an American Heart Association postdoctoral fellowship to B.-E. Kim (09POST2251047) and National Institutes of Health grants DK074192 to D.J.T., CA123031 to C.M.C., and GM083292 to D.R.W. M.L.T. was a Ph.D. student in the NIH-funded Duke University Program in Genetics and Genomics. H.K. was supported by training grant T32 NIH DK07115. D.C.B. was supported by training grant T32 NIH CA059365-14.
Footnotes
Published ahead of print 30 January 2012
REFERENCES
- 1. Alessi DR, et al. 1996. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 15:6541–6551 [PMC free article] [PubMed] [Google Scholar]
- 2. Aoki Y, Niihori T, Narumi Y, Kure S, Matsubara Y. 2008. The RAS/MAPK syndromes: novel roles of the RAS pathway in human genetic disorders. Hum. Mutat. 29:992–1006 [DOI] [PubMed] [Google Scholar]
- 3. Baker NE, Rubin GM. 1992. Ellipse mutations in the Drosophila homologue of the EGF receptor affect pattern formation, cell division, and cell death in eye imaginal discs. Dev. Biol. 150:381–396 [DOI] [PubMed] [Google Scholar]
- 4. Bird AJ, et al. 2003. Zinc fingers can act as Zn2+ sensors to regulate transcriptional activation domain function. EMBO J. 22:5137–5146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boal AK, Rosenzweig AC. 2009. Structural biology of copper trafficking. Chem. Rev. 109:4760–4779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brewer GJ. 2005. Anticopper therapy against cancer and diseases of inflammation and fibrosis. Drug Discov. Today 10:1103–1109 [DOI] [PubMed] [Google Scholar]
- 7. Caldwell PE, Walkiewicz M, Stern M. 2005. Ras activity in the Drosophila prothoracic gland regulates body size and developmental rate via ecdysone release. Curr. Biol. 15:1785–1795 [DOI] [PubMed] [Google Scholar]
- 8. Caruano-Yzermans AL, Bartnikas TB, Gitlin JD. 2006. Mechanisms of the copper-dependent turnover of the copper chaperone for superoxide dismutase. J. Biol. Chem. 281:13581–13587 [DOI] [PubMed] [Google Scholar]
- 9. Catalanotti F, et al. 2009. A Mek1-Mek2 heterodimer determines the strength and duration of the Erk signal. Nat. Struct. Mol. Biol. 16:294–303 [DOI] [PubMed] [Google Scholar]
- 10. Changela A, et al. 2003. Molecular basis of metal-ion selectivity and zeptomolar sensitivity by CueR. Science 301:1383–1387 [DOI] [PubMed] [Google Scholar]
- 11. Chen X, et al. 2008. Copper sensing function of Drosophila metal-responsive transcription factor-1 is mediated by a tetranuclear Cu(I) cluster. Nucleic Acids Res. 36:3128–3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dhillon AS, Hagan S, Rath O, Kolch W. 2007. MAP kinase signalling pathways in cancer. Oncogene 26:3279–3290 [DOI] [PubMed] [Google Scholar]
- 13. Donnelly PS, et al. 2008. Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-beta peptide. J. Biol. Chem. 283:4568–4577 [DOI] [PubMed] [Google Scholar]
- 14. Dougherty MK, et al. 2005. Regulation of Raf-1 by direct feedback phosphorylation. Mol. Cell 17:215–224 [DOI] [PubMed] [Google Scholar]
- 15. Eliceiri BP, Klemke R, Stromblad S, Cheresh DA. 1998. Integrin alphavbeta3 requirement for sustained mitogen-activated protein kinase activity during angiogenesis. J. Cell Biol. 140:1255–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fischmann TO, et al. 2009. Crystal structures of MEK1 binary and ternary complexes with nucleotides and inhibitors. Biochemistry 48:2661–2674 [DOI] [PubMed] [Google Scholar]
- 17. Friedman A, Perrimon N. 2006. A functional RNAi screen for regulators of receptor tyrosine kinase and ERK signalling. Nature 444:230–234 [DOI] [PubMed] [Google Scholar]
- 18. Goodman VL, Brewer GJ, Merajver SD. 2004. Copper deficiency as an anti-cancer strategy. Endocr. Relat. Cancer 11:255–263 [DOI] [PubMed] [Google Scholar]
- 19. Gupte A, Mumper RJ. 2009. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 35:32–46 [DOI] [PubMed] [Google Scholar]
- 20. Haremaki T, Fraser ST, Kuo YM, Baron MH, Weinstein DC. 2007. Vertebrate Ctr1 coordinates morphogenesis and progenitor cell fate and regulates embryonic stem cell differentiation. Proc. Natl. Acad. Sci. U. S. A. 104:12029–12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heise CJ, Cobb MH. 2006. Expression and characterization of MAP kinases in bacteria. Methods 40:209–212 [DOI] [PubMed] [Google Scholar]
- 22. Horng YC, et al. 2005. Human Sco1 and Sco2 function as copper-binding proteins. J. Biol. Chem. 280:34113–34122 [DOI] [PubMed] [Google Scholar]
- 23. Ishizaki H, et al. 2010. Combined zebrafish-yeast chemical-genetic screens reveal gene-copper-nutrition interactions that modulate melanocyte pigmentation. Dis. Model Mech. 3:639–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iverson C, et al. 2009. RDEA119/BAY 869766: a potent, selective, allosteric inhibitor of MEK1/2 for the treatment of cancer. Cancer Res. 69:6839–6847 [DOI] [PubMed] [Google Scholar]
- 25. Juarez JC, et al. 2006. Copper binding by tetrathiomolybdate attenuates angiogenesis and tumor cell proliferation through the inhibition of superoxide dismutase 1. Clin. Cancer Res. 12:4974–4982 [DOI] [PubMed] [Google Scholar]
- 26. Karnoub AE, Weinberg RA. 2008. Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 9:517–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khan G, Merajver S. 2009. Copper chelation in cancer therapy using tetrathiomolybdate: an evolving paradigm. Expert Opin. Invest. Drugs 18:541–548 [DOI] [PubMed] [Google Scholar]
- 28. Kim BE, Nevitt T, Thiele DJ. 2008. Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 4:176–185 [DOI] [PubMed] [Google Scholar]
- 29. Kim BE, et al. 2010. Cardiac copper deficiency activates a systemic signaling mechanism that communicates with the copper acquisition and storage organs. Cell Metab. 11:353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kolch W. 2005. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 6:827–837 [DOI] [PubMed] [Google Scholar]
- 31. Kolch W. 2000. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 351(Pt. 2):289–305 [PMC free article] [PubMed] [Google Scholar]
- 32. Kubota Y, O'Grady P, Saito H, Takekawa M. 2011. Oncogenic Ras abrogates MEK SUMOylation that suppresses the ERK pathway and cell transformation. Nat. Cell Biol. 13:282–291 [DOI] [PubMed] [Google Scholar]
- 33. Kuo YM, Zhou B, Cosco D, Gitschier J. 2001. The copper transporter CTR1 provides an essential function in mammalian embryonic development. Proc. Natl. Acad. Sci. U. S. A. 98:6836–6841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee J, Petris MJ, Thiele DJ. 2002. Characterization of mouse embryonic cells deficient in the ctr1 high affinity copper transporter. Identification of a Ctr1-independent copper transport system. J. Biol. Chem. 277:40253–40259 [DOI] [PubMed] [Google Scholar]
- 35. Lee J, Prohaska JR, Thiele DJ. 2001. Essential role for mammalian copper transporter Ctr1 in copper homeostasis and embryonic development. Proc. Natl. Acad. Sci. U. S. A. 98:6842–6847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee YS, Carthew RW. 2003. Making a better RNAi vector for Drosophila: use of intron spacers. Methods 30:322–329 [DOI] [PubMed] [Google Scholar]
- 37. Levin-Salomon V, Kogan K, Ahn NG, Livnah O, Engelberg D. 2008. Isolation of intrinsically active (MEK-independent) variants of the ERK family of mitogen-activated protein (MAP) kinases. J. Biol. Chem. 283:34500–34510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lim S, et al. 2010. Copper and zinc bis(thiosemicarbazonato) complexes with a fluorescent tag: synthesis, radiolabelling with copper-64, cell uptake and fluorescence studies. J. Biol. Inorg. Chem. 15:225–235 [DOI] [PubMed] [Google Scholar]
- 39. Lowndes SA, Sheldon HV, Cai S, Taylor JM, Harris AL. 2009. Copper chelator ATN-224 inhibits endothelial function by multiple mechanisms. Microvasc. Res. 77:314–326 [DOI] [PubMed] [Google Scholar]
- 40. McAuslan BR, Reilly WG, Hannan GN, Gole GA. 1983. Angiogenic factors and their assay: activity of formyl methionyl leucyl phenylalanine, adenosine diphosphate, heparin, copper, and bovine endothelium stimulating factor. Microvasc. Res. 26:323–338 [DOI] [PubMed] [Google Scholar]
- 41. McElwee MK, Song MO, Freedman JH. 2009. Copper activation of NF-kappaB signaling in HepG2 cells. J. Mol. Biol. 393:1013–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mercer JF. 2001. The molecular basis of copper transport diseases. Trends Mol. Med. 7:64–69 [DOI] [PubMed] [Google Scholar]
- 43. Mercer JF, Llanos RM. 2003. Molecular and cellular aspects of copper transport in developing mammals. J. Nutr. 133:1481S–1484S [DOI] [PubMed] [Google Scholar]
- 44. Mirth C, Truman JW, Riddiford LM. 2005. The role of the prothoracic gland in determining critical weight for metamorphosis in Drosophila melanogaster. Curr. Biol. 15:1796–1807 [DOI] [PubMed] [Google Scholar]
- 45. Mirth CK, Riddiford LM. 2007. Size assessment and growth control: how adult size is determined in insects. Bioessays 29:344–355 [DOI] [PubMed] [Google Scholar]
- 46. Mufti AR, et al. 2006. XIAP is a copper binding protein deregulated in Wilson's disease and other copper toxicosis disorders. Mol. Cell 21:775–785 [DOI] [PubMed] [Google Scholar]
- 47. Nose Y, Kim BE, Thiele DJ. 2006. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 4:235–244 [DOI] [PubMed] [Google Scholar]
- 48. Ohren JF, et al. 2004. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat. Struct. Mol. Biol. 11:1192–1197 [DOI] [PubMed] [Google Scholar]
- 49. Parke A, Bhattacherjee P, Palmer RM, Lazarus NR. 1988. Characterization and quantification of copper sulfate-induced vascularization of the rabbit cornea. Am. J. Pathol. 130:173–178 [PMC free article] [PubMed] [Google Scholar]
- 50. Puig S, Lee J, Lau M, Thiele DJ. 2002. Biochemical and genetic analyses of yeast and human high affinity copper transporters suggest a conserved mechanism for copper uptake. J. Biol. Chem. 277:26021–26030 [DOI] [PubMed] [Google Scholar]
- 51. Quaranta D, McEvoy MM, Rensing C. 2009. Site-directed mutagenesis identifies a molecular switch involved in copper sensing by the histidine kinase CinS in Pseudomonas putida KT2440. J. Bacteriol. 191:5304–5311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Raju KS, Alessandri G, Ziche M, Gullino PM. 1982. Ceruloplasmin, copper ions, and angiogenesis. JNCI 69:1183–1188 [PubMed] [Google Scholar]
- 53. Roberts DB. 1998. Drosophila: a practical approach, 2nd ed IRL Press at Oxford University Press, Oxford, England [Google Scholar]
- 53a. Rushworth L. K., et al. 2006. Regulation and role of Raf-1/B-Raf heterodimerization. Mol. Cell. Biol. 26:2262–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rutherford JC, Bird AJ. 2004. Metal-responsive transcription factors that regulate iron, zinc, and copper homeostasis in eukaryotic cells. Eukaryot. Cell 3:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schubbert S, Shannon K, Bollag G. 2007. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 7:295–308 [DOI] [PubMed] [Google Scholar]
- 56. Seger R, et al. 1991. Microtubule-associated protein 2 kinases, ERK1 and ERK2, undergo autophosphorylation on both tyrosine and threonine residues: implications for their mechanism of activation. Proc. Natl. Acad. Sci. U. S. A. 88:6142–6146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shaul YD, Seger R. 2007. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim. Biophys. Acta 1773:1213–1226 [DOI] [PubMed] [Google Scholar]
- 58. Solioz M, Stoyanov JV. 2003. Copper homeostasis in Enterococcus hirae. FEMS Microbiol. Rev. 27:183–195 [DOI] [PubMed] [Google Scholar]
- 59. Tecle H, et al. 2009. Beyond the MEK-pocket: can current MEK kinase inhibitors be utilized to synthesize novel type III NCKIs? Does the MEK-pocket exist in kinases other than MEK? Bioorg. Med. Chem. Lett. 19:226–229 [DOI] [PubMed] [Google Scholar]
- 60. Turski ML, Thiele DJ. 2007. Drosophila Ctr1A functions as a copper transporter essential for development. J. Biol. Chem. 282:24017–24026 [DOI] [PubMed] [Google Scholar]
- 61. Turski ML, Thiele DJ. 2009. New roles for copper metabolism in cell proliferation, signaling, and disease. J. Biol. Chem. 284:717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Waldron KJ, Robinson NJ. 2009. How do bacterial cells ensure that metalloproteins get the correct metal? Nat. Rev. Microbiol. 7:25–35 [DOI] [PubMed] [Google Scholar]
- 63. Xu B, Wilsbacher JL, Collisson T, Cobb MH. 1999. The N-terminal ERK-binding site of MEK1 is required for efficient feedback phosphorylation by ERK2 in vitro and ERK activation in vivo. J. Biol. Chem. 274:34029–34035 [DOI] [PubMed] [Google Scholar]
- 64. Zhang BH, Guan KL. 2000. Activation of B-Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. EMBO J. 19:5429–5439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ziche M, Jones J, Gullino PM. 1982. Role of prostaglandin E1 and copper in angiogenesis. JNCI 69:475–482 [PubMed] [Google Scholar]
- 66. Zimmermann M, et al. 2009. Metal binding affinities of Arabidopsis zinc and copper transporters: selectivities match the relative, but not the absolute, affinities of their amino-terminal domains. Biochemistry 48:11640–11654 [DOI] [PubMed] [Google Scholar]