

Abstract
The streptococcal promiscuous plasmid pMV158 (5,540 bp) replicates by the rolling-circle mechanism and can be mobilized among a wide number of Gram-positive and -negative bacteria. The plasmid region involved in its conjugative transfer includes the mobM gene, which encodes the MobM relaxase, and the cis-acting origin of transfer (oriT). MobM initiates transfer by cleavage of supercoiled pMV158 DNA at a specific dinucleotide within oriT. In the present work, we have performed a detailed transcriptional analysis to assess the role of MobM in the control of its own gene expression. By in vivo and in vitro approaches, we demonstrated that mobM transcription in Escherichia coli was mostly initiated from a promoter (Pmob2) different from the one (Pmob1) used in Lactococcus lactis. Whereas promoter Pmob1 was embedded within the oriT sequence, promoter Pmob2 was placed apart from but adjacent to oriT. Further, MobM was able to repress the expression of its own gene from both promoters. Given the promiscuity of pMV158, the organization of the mobM promoter region suggests a strategy of the plasmid to cope with different transcription machineries of the hosts it colonizes.
INTRODUCTION
Bacterial horizontal gene transfer (HGT) is mediated by mobile genetic elements, of which plasmids, bacteriophages, and the phage-related chromosomally encoded integrative and conjugative elements (ICEs) constitute the vast majority (14, 53). An increasing number of these mobile elements (the so-called bacterial mobilome) have been found as the number of totally sequenced genomes is increasing. There are nearly 2,000 complete plasmid genomes available at the GenBank, but their transferability has been tested only for a limited number, mostly replicons isolated from Gram-negative (G−) bacteria; much less attention has been given to plasmids from Gram-positive (G+) hosts (16, 41). Initiation of conjugative plasmid transfer involves the relaxation of the supercoiled DNA by a plasmid-encoded protein (the relaxase). This protein cleaves a specific phosphodiester bond (the nick site) of the strand to be transferred and remains covalently bound to its 5′ end. Nicking is followed by a rolling-circle replication-like process in which the relaxase–single-stranded DNA (relaxase-ssDNA) complex is piloted to the cell membrane, where the coupling protein and the type IV secretion system pump the relaxase-ssDNA complex to the recipient cell (28). Once in the recipient, the cell machinery recognizes a single-strand origin of replication generated in the incoming ssDNA to perform the synthesis of the complementary DNA strand by a mechanism of lagging-strand replication (4, 30, 55).
Important features in HGT are the signals that trigger the process, which are largely unknown for many plasmids other than those requiring quorum-sensing signals (13, 16). Accordingly, another key feature is the control of the intracellular levels of the relaxase (and, hence, of the relaxosome formation). In some plasmid transfer systems, this process seems to be exerted by ribbon-helix-helix accessory proteins that play a role in regulating DNA relaxation by inducing bends in the DNA and/or in controlling synthesis of the relaxase by binding to DNA regions close to (or included into) its gene promoter. These are the cases of the TraY of F plasmid (34), TrwA of R388 (33), MbeC of ColE1 (49), and MobC of pC221 (42). In other cases, like TraI of plasmid F, TraA of pIP501, and Mob of pBBR1 (19, 22, 47), it has been shown that expression of the relaxase gene is negatively regulated at the transcriptional level by the activity of the relaxase protein itself. However, the interactions between the relaxase and the host RNA polymerase (RNAP) at the plasmid origin of transfer (oriT) remain to be investigated.
The bacterial RNAP holoenzyme is a complex of six subunits (α2ββ′ωσ). In general, bacterial genomes encode diverse forms of the σ factor, and each of them confers promoter specificity to the RNAP (17, 51). Most transcription in exponentially growing bacterial cells is initiated by RNAPs that carry a housekeeping σ factor similar to the Escherichia coli σ70. Promoters recognized by these RNAPs are characterized by two main sequence elements, the −35 (consensus 5′-TTGACA-3′) and −10 (consensus 5′-TATAAT-3′) hexamers (reviewed in reference 20). Additionally, some of these promoters contain an extended −10 element that is located one nucleotide (nt) upstream of the −10 hexamer. This element is more conserved in G+ bacteria (5′-TRTG-3′ motif) than in E. coli (5′-TG-3′ motif) (32, 38, 50). The two conserved hexamers are separated by a region, termed a “spacer,” which has no consensus sequence but has a structure that is important for σ70 recognition and activity of the promoters (40).
The promiscuous streptococcal plasmid pMV158 (5,540 bp) represents one of the simplest systems for an efficient DNA transfer among different bacterial species, G+ and G−. It has a genetic organization such that all genes that encode proteins are placed in the same orientation (Fig. 1A). In addition to the genes and loci required for its leading-strand rolling-circle replication (repB, copG, and the double-stranded origin, dso), two other cassettes are present in the plasmid, namely, an antibiotic resistance marker (a tetL-type determinant) and a mobilization cassette. The latter includes the mobM gene, which encodes the MobM relaxase protein (494 residues), and the oriT. The MobM protein is the representative of the MOBV family of relaxases, which consists of more than 100 members so far (14). It has been shown that MobM cleaves supercoiled or ssDNA at a specific dinucleotide (coordinates 3595 and 3596; nic) within the oriT sequence (18) (Fig. 1B). The oriT (coordinates 3564 to 3606) is unique in the sense that it has three inverted repeats (IRs) (Fig. 1C, IR1, IR2, and IR3) rather than the common single IR found in most of the studied plasmid oriTs (55). We have shown that IR1 and IR3 are preferentially recognized by MobM protein on ssDNA substrates, at least in vitro (29). The role of IR2, if any, is presently unknown, but the conservation of the oriT sequence among the MOBV plasmid family suggests that any one of the three IRs could be involved in the recognition of oriT by MobM at the initiation of relaxosome formation in the donor cell and/or at the termination reaction to close the transferred strand within the recipient cell. A DNase I footprinting assay showed that MobM binds, although very poorly, to linear double-stranded DNA (dsDNA) fragments containing the oriT sequence (15); the protein is, however, unable to cleave linear dsDNA (18). Specifically, MobM protected a region between coordinates 3582 and 3605, which includes IR2, although the exact upstream border of the footprint remained unclear (Fig. 1B). Recently, using ssDNAs and a truncated MobM protein containing the first 199 residues (MobMN199), the minimal oriT sequence was delimited to a stretch of 26 nucleotides (coordinates 3570 to 3595) that is located just upstream of the nick site. This minimal origin includes IR1 (29).
Fig 1.

(A) Genetic map of pMV158. Only relevant features are indicated. Genes are depicted as arrows pointing in the direction of transcription. copG and repB genes are involved in plasmid DNA replication. The positions of the origins for leading-strand (dso) and lagging-strand (ssoU and ssoA) synthesis are indicated. The tetL gene confers resistance to tetracycline in both G+ and G− bacteria. The origin of transfer (oriT) and the mobM gene are involved in conjugative mobilization. P, PstI; N, NcoI; E, EcoRI. Coordinates are given in parentheses. (B) Nucleotide sequence of the pMV158 region spanning coordinates 3564 and 3650. This region includes the oriT and the promoter region of the mobM gene. The three overlapping inverted repeats (IR1, IR2, and IR3) (29) and the nicking site (nic) (18) constituting the oriT are indicated. The minimal oriT sequence (coordinates 3570 to 3595) on ssDNA is boxed (29). The shadowed sequence (coordinates 3582 to 3605) denotes the MobM binding site defined by DNase I footprinting assays although the precise upstream boundary of protection against DNase I remained unclear (15). The main sequence elements of the Pmob1 (12) and Pmob2 (this work) promoters are underlined in gray and black, respectively. The transcription start site for each promoter is indicated with an arrow. The position of the translation start codon (ATG) of the mobM gene is also shown. (C) Scheme showing the three possible stem-loop structures that could adopt the oriT sequence on ssDNAs. The arrowhead indicates the nic position.
In the present work, we have performed in-depth in vitro and in vivo transcriptional analysis of the pMV158-encoded MobM relaxase. We demonstrate that the major promoter governing transcription of the mobM gene in E. coli is close to, but different from, the one used in the G+ bacteria Lactococcus lactis (12). Whereas the latter promoter is located within the oriT sequence (Fig. 1B), the newly identified promoter is placed just downstream of it. This organization would provide a unique example of the versatility of the transfer system of a plasmid that can be mobilized among many different bacterial species. In addition, we demonstrate that the relaxase MobM is able to negatively regulate its own synthesis, which adds another level of complexity to the compact region of the plasmid spanning its oriT.
MATERIALS AND METHODS
Bacterial strains, plasmids, and oligonucleotides.
The E. coli TOP10 (Invitrogen) and JM109 (54) strains were used as hosts for the plasmids used in this work. E. coli JM109(DE3) (Promega) was employed for the β-galactosidase assays; this strain is a JM109 derivative in which the DE3 lysogen (46) provides the T7 RNA polymerase gene fused to the lacUV5 promoter and the lacIq repressor gene. Thus, expression of the T7 RNA polymerase is induced by isopropyl-β-d-thiogalactopyranoside (IPTG). In addition to the plasmids constructed in this work (see below), we used the following E. coli plasmids: (i) vector pET5 (Novagen); (ii) pLGM2, which is a pET5 derivative that carries the mobM gene under the control of the ϕ10 promoter of phage T7 (18); and (iii) pMP220, which carries a promoterless lacZ gene (44). For small-scale preparations of plasmid DNA, a High Pure Plasmid Isolation Kit (Roche Applied Science) was used. Plasmid DNA from pMV158 (25) was purified from Streptococcus pneumoniae 708 (trt-1 hex-4 end-1 exo-2 malM594) (24) by two consecutive CsCl gradients, as described previously (7). Oligonucleotides used in this work are listed in Table 1.
Table 1.
Oligonucleotides used in this work
| Primer name | Sequence (5′ to 3′)a | Coordinates (nt)b |
|---|---|---|
| DraF | GGTGGAGATTTTTTGAGTG | 3371–3389 |
| DraR | CACGTTCATTATGCTTTAAAGCTCCTCCC | 3807–3779 |
| ssoUF | GGGATCAACTTTGGGAGAGA | 3121–3140 |
| ssoUR | GCGTCTCAAAAACACGTTCA | 3819–3800 |
| ssoAR | TCACAACGCTCACCTCCA | 5495–5478 |
| P−116 | TTATGGTTTTGGTCGGCACT | 3527–3546 |
| P+46 | CACGAGCCGACACAGTCTATT | 3688–3668 |
| ORI | GGCACTGCCGAATTCCTCGCAGAGC | 3541–3565 |
| IR2 | ACACTTTATGAATTCAAAGTATAGTGT | 3568–3594 |
| MR | CAGACGAGCCGCTGCAGTCTATTGCT | 3690–3665 |
| mobM-PE | GCAACCATGTAACTCATAGATTTC | 3748–3725 |
| lacZ-PE | GTGATTTTTTTCTCCATTTTAGC | |
| tetA-PE | GGGGTATGTTGGGTTTCACGTCTG | |
| UC-50 | TTGTGAGCGGATAACAATTTC |
Restriction sites are in bold face, and base changes that generate restriction sites are underlined.
Coordinates are given with respect to pMV158 sequence (accession number NC_010096).
Growth and transformation of bacteria.
E. coli cells were grown in tryptone-yeast extract (TY) medium (Pronadisa) at 37°C. In the case of plasmid-harboring cells, the medium was supplemented with tetracycline (5 μg/ml and 2 μg/ml for pMV158 and pMP220 derivatives, respectively) or 100 μg/ml ampicillin (plasmids pET5 and pLGM2). S. pneumoniae cells harboring pMV158 were grown in medium AGCH (23), supplemented with 0.2% yeast extract, 0.3% sucrose, and 1 μg/ml tetracycline as detailed previously (37). The protocol used to transform E. coli by electroporation was described previously (10).
PCR conditions.
Phusion High-Fidelity DNA Polymerase (Finnzymes) was used for all PCR applications. The reaction mixtures (50 μl) contained 16 mM (NH4)2SO4, 67 mM Tris-HCl, pH 8.8, 1.5 mM MgCl2, 0.2 mM each deoxynucleoside triphosphate ([dNTP] Roche Applied Science), 0.4 μM each primer, 1 ng of template DNA, and 0.65 units of DNA polymerase. An initial denaturation step was performed at 98°C for 30 s, followed by 30 cycles that included the next steps: (i) denaturation at 98°C for 10 s, (ii) annealing at around 55°C (depending on the primer melting temperature [Tm]) for 20 s, and (iii) extension at 72°C for 30 to 60 s. A final extension step was performed at 72°C for 10 min. PCR products were purified with a QIAquick PCR Purification Kit (Qiagen).
RNA isolation and primer extension assays.
JM109 cells harboring pMV158 and JM109(DE3) cells harboring the combination of pLGM2 and pORI-P or the combination of pLGM2 and pIR2-P were exponentially grown at 37°C to an optical density at 600 nm (OD600) of 0.4. The JM109(DE3) cells harboring plasmids were divided into 10-ml aliquots. Each aliquot was supplemented with IPTG (1 mM final concentration) and incubated at the same temperature. As controls, 10-ml cultures incubated without IPTG were used. An Aurum Total RNA Mini Kit (Bio-Rad) was used to isolate total RNA. Cultures were processed as specified by the supplier, and analyses of RNAs and primer extension assays were performed as reported previously (5). Dideoxy-mediated chain termination sequencing reactions were run in the same gel. Labeled products were visualized using the Fujifilm Image Analyzer FLA-3000.
Electrophoretic mobility shift assays (EMSA).
A 437-bp fragment (coordinates 3371 to 3807 of pMV158) was generated by PCR using the DraF and DraR primers (Table 1). The DraR primer included a DraI restriction site. Digestion of the 437-bp DNA with DraI generated a 362-bp DNA fragment (coordinates 3429 to 3790). Reaction mixtures (20 μl) contained 40 mM Tris-HCl, pH 7.5, 150 mM KCl, 10 mM MgCl2, 0.01% Triton X-100, 120 nM E. coli RNAP (Epicentre), and the 362-bp DNA fragment at 5 nM. After 30 min at 37°C, RNAP-DNA complexes were treated with heparin (0.25 to 100 μg/ml final concentration) for 5 min at the same temperature. Electrophoresis conditions and analyses of the retarded bands were performed as reported previously (21).
Electron microscopy.
DNA fragments from pMV158 of two sizes, namely, 2,375 bp (coordinates 3121 to 5495) and 699 bp (coordinates 3121 to 3819), were obtained by PCR using pMV158 DNA as a template and the ssoUF/ssoAR and ssoUF/ssoUR oligonucleotides (Table 1), respectively, as primers. Reaction mixtures (10 μl) contained 40 mM Tris-HCl, pH 7.5, 150 mM KCl, 10 mM MgCl2, 0.01% Triton X-100, 120 nM E. coli RNAP (Epicentre), and the 2,375- or 699-bp DNA fragments at 5 to 20 nM. After 15 min at 37°C, RNAP-DNA complexes were fixed with 0.3% glutaraldehyde for 15 min at the same temperature. Then, reaction mixtures were diluted 10-fold in buffer GA (10 mM triethanolamine chloride, pH 7.5, 10 mM MgCl2), adsorbed onto freshly cleaved mica, positively stained with 2% uranyl acetate, rotary shadowed with Pt/Ir, and covered with a carbon film as described previously (45). Micrographs of the carbon film replica were taken using a Philips CM100 (FEI Company, Hillsboro, Oregon) electron microscope at 100 kV on 35-mm film. The contour length of the RNAP-DNA complexes and the positions on the DNA fragments were measured on projections of 35-mm negatives using a digitizer (LM4; Brühl, Nüremberg, Germany).
DNase I footprinting assays.
A 162-bp DNA fragment (coordinates 3527 to 3688 of pMV158) was generated by PCR using the P−116 and P+46 primers (Table 1). To label this fragment at the 5′ end of a particular strand, the corresponding primer was treated with T4 polynucleotide kinase and [γ-32P]ATP (3,000 Ci/mmol; Hartmann) before the amplification reaction was performed. DNase I footprinting was performed essentially as reported earlier (21), with the concentration of RNAP kept at 10 nM. Dideoxy-mediated chain termination sequencing reactions using pMV158 and either the P−116 or the P+46 oligonucleotide were run in the same gel.
In vitro transcription analysis.
In vitro transcription reactions were carried out under multiple-round conditions. Reaction mixtures (50 μl) contained 45 mM Tris-HCl, pH 7.5, 150 mM KCl, 45 mM NaCl, 10 mM MgCl2, 2 mM dithiothreitol (DTT), 0.01% Triton X-100, 1.5% glycerol, 10 to 30 nM PCR-amplified linear DNA, 250 μM each NTP (Promega), 10 μCi of [α-32P]UTP (3,000 Ci/mmol; GE Healthcare), 10 units of SUPERase-In (Ambion), and 24 nM E. coli RNAP (Epicentre). After incubation at 37°C for 30 min, nonincorporated nucleotide was removed using MicroSpin G-25 columns (GE Healthcare). Samples were then dried in a SpeedVac, dissolved in RNA loading buffer (80% formamide, 10 mM EDTA, pH 8.0, 0.1% bromophenol blue, 0.1% xylene cyanol), heated at 85°C for 5 min, and subjected to electrophoresis in 8 M urea–6% polyacrylamide gels. Dideoxy-mediated chain termination sequencing reactions were run in the same gel. Specifically, the Sequenase Quick-Denature Plasmid Sequencing Kit (USB), pUC19 DNA, the UC-50 primer (Table 1), and [α-32P]dATP (6,000 Ci/mmol, GE Healthcare) were used. Following electrophoresis, the gel was exposed to X-ray films.
Construction of plasmids pORI-P and pIR2-P.
The IncP broad-host-range vector pMP220 has single restriction sites for EcoRI and PstI. These sites are located upstream of the promoterless lacZ gene (44). This plasmid vector was used to evaluate the promoter activity of pMV158 DNA regions by measuring β-galactosidase activity. To construct plasmid pORI-P, a 150-bp region (coordinates 3541 to 3690 of pMV158) was amplified by PCR with the ORI and MR oligonucleotides (Table 1). These oligonucleotides include single EcoRI and PstI restriction sites, respectively. After EcoRI and PstI digestion of the PCR-amplified DNA, the generated product (coordinates 3555 to 3676) was mixed with pMP220 DNA digested with both enzymes. The mixture was then treated with T4 DNA ligase (New England BioLabs). For the construction of plasmid pIR2-P, a 123-bp region of pMV158 (coordinates 3568 to 3690) was amplified using the IR2 and MR primers. The IR2 oligonucleotide included an EcoRI site. After EcoRI and PstI treatment of the PCR-amplified DNA, the digestion product (coordinates 3583 to 3676) was inserted into the pMP220 vector. Ligation mixtures were used to transform E. coli TOP10 cells. Transformants were selected for resistance to 2 μg/ml tetracycline at 37°C. Plasmid DNA was isolated and analyzed by agarose gel mobility. To confirm the constructions, the inserted fragment and the regions of pMP220 that are flanking the insert were sequenced. Dye terminator sequencing was carried out at Secugen (Centro de Investigaciones Biológicas, Madrid, Spain).
β-Galactosidase activity measurements.
E. coli JM109(DE3) cells carrying the indicated plasmids were grown at 37°C, as indicated above, to mid-exponential phase (OD600 of ∼0.4). Then, the cultures were divided in two. IPTG (1 mM final concentration) was added to one half of the cultures to induce the expression of the mobM gene from plasmid pLGM2. As controls, the other half of the cultures did not receive IPTG. Measurement of the activity was performed as reported previously (3), but the samples (200 μl) were dispensed in a 96-well microplate; absorbance data were collected with a VarioskanFlash reader (ThermoScientific). The β-galactosidase-specific activities were calculated in Miller units (MU) (31).
RESULTS
Initiation of mobM transcription in E. coli cells.
The pMV158-mobM cassette is flanked by two lagging-strand origins of replication, ssoU and ssoA (Fig. 1A). It was shown that, whereas either the ssoA or the ssoU could be used for intraspecific transfer in S. pneumoniae, interspecific transfer required an intact ssoU (30). Thus, this origin appeared to be required for the extraordinary promiscuity of pMV158 (8). Plasmid pMV158 has been mobilized between different G+ bacteria, like L. lactis (12) and Enterococcus faecalis (30, 35), using pAMβ1 as an auxiliary plasmid. In the former bacterium, it was shown that transcription of the mobM gene started at coordinate 3609 of pMV158 (12). Thus, the lactococcal RNAP appeared to recognize a promoter sequence, named Pmob1 (Fig. 1B). This promoter has a near-consensus −10 hexamer (5′-TATACT-3′) and a consensus −10 extension (5′-TGTG-3′) and shows a 3/6 match at the −35 element (5′-ATGAAT-3′) (consensus residues are shown in bold). The extension of the −10 sequence, also termed the −15 motif (9), seems to be particularly important for promoters of G+ bacteria (2, 38). Moreover, the −10 and −35 elements of promoter Pmob1 are separated by 16 nucleotides, and the −10 hexamer is located just downstream of the dinucleotide cleaved by MobM (Fig. 1B, nic) (18), placing it within the minimal oriT sequence (29).
In addition to the G+ hosts, pMV158 was shown to replicate in the G− bacterium E. coli (7). Mating experiments showed that the IncPα RP4 or the IncW R388 plasmids were able to mobilize pMV158 between E. coli strains (11). In the case of RP4, it was shown that transfer of pMV158 required the products of the genes traG (coupling protein) (1) and traF (mating pair formation) (27). It was thus interesting to explore in some depth the transcriptional features of the pMV158 mobM gene in the G− host. To identify the transcription initiation site of the mobM gene in E. coli, we carried out primer extension experiments using the mobM-PE oligonucleotide as a primer (coordinates 3748 to 3725) (Table 1) and total RNA isolated from E. coli cells harboring plasmid pMV158. As shown in Fig. 2, two cDNA extension products of 106 (major product) and 140 (minor product) nucleotides were detected. The 140-nt cDNA revealed a transcription initiation event at coordinate 3609 from the Pmob1 promoter, whereas the appearance of the 106-nt cDNA might correspond to (i) a degradation product from the larger 140-nt cDNA, (ii) a premature stop during retrotranscription, or (iii) a transcription initiation event at coordinate 3643 from a different promoter. Sequence analysis of the region located just upstream of coordinate 3643 using the BPROM (Softberry, Mount Kisco, NY) prediction program supported the latter hypothesis. It revealed the existence of an additional promoter sequence, here termed Pmob2 (Fig. 1B). The −10 element (5′-TAAACT-3′) of this putative promoter is located at the proper distance of 7 nucleotides from the transcription start site (coordinate 3643). Moreover, the Pmob2 promoter has a consensus −10 extension (5′-TGTG-3′), and the −10 and −35 (5′-TGGAAG-3′) sequence elements (consensus nucleotides are shown in bold) are separated by 17 nucleotides, which is the optimum spacer length for E. coli (20). Therefore, promoter Pmob2 is placed just downstream of the oriT, and it is close to, but does not overlap with, promoter Pmob1 (Fig. 1B).
Fig 2.
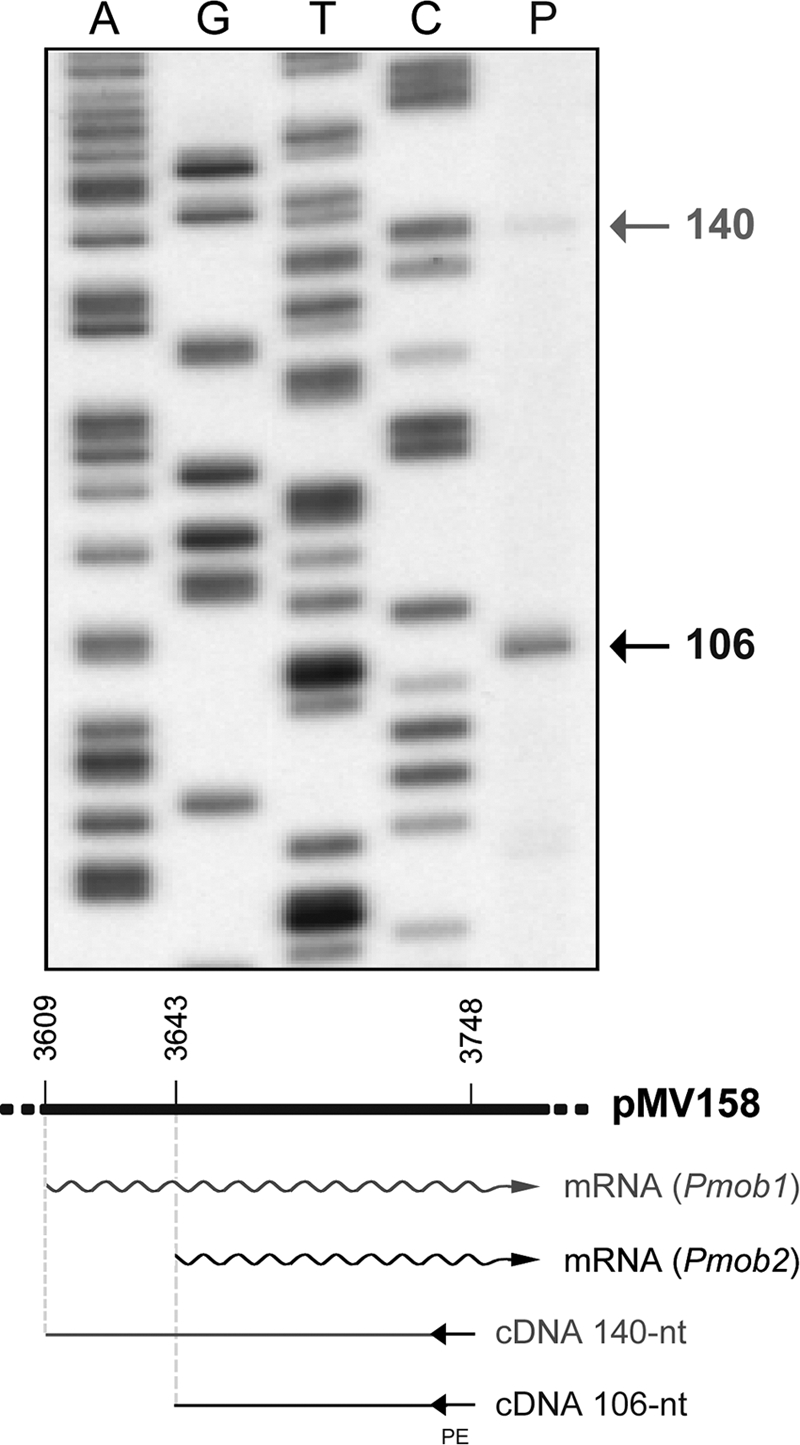
Primer extension on total RNA isolated from E. coli cells carrying plasmid pMV158. The primer (Table 1, mobM-PE) annealed to the transcripts in the region corresponding to coordinates 3725 to 3748 of pMV158. The size expected for the cDNAs if transcription initiation occurs at coordinate 3609 (Pmob1) or 3643 (Pmob2) is indicated below the gel. Primer extension products were resolved on denaturing gels (7 M urea–8% polyacrylamide). As DNA size markers, dideoxy-mediated chain termination sequencing reactions using pMV158 DNA and the mobM-PE primer were run in the same gel (lanes A, G, T, and C). The sizes of the cDNA extension products (lane P) are indicated on the right in nucleotides.
E. coli RNAP binds to the Pmob2 promoter in vitro.
The interaction of the E. coli RNAP with the promoter region of the mobM gene was further studied by several in vitro methods. In a first approach, we performed EMSA using the E. coli RNAP holoenzyme (σ70 factor) and a 362-bp linear DNA fragment (coordinates 3429 to 3790) under conditions that favored generation of open complexes (37°C and in the absence of NTP substrates). The DNA fragment contains the Pmob1 and Pmob2 promoter sequences (Fig. 1B). In these assays, heparin (0.25 to 100 μg/ml) was used as the competitor, and the molar ratio of RNAP to DNA was 24:1 (Fig. 3). Without heparin, aggregates of DNA-bound RNAP molecules, which did not enter the native gel, were observed. After addition of heparin (5 μg/ml), RNAP-DNA complexes with much slower electrophoretic mobility than the free DNA were detected, indicating that nonspecifically bound RNAP molecules were displaced by such a concentration of competitor. RNAP-DNA interactions were disrupted at heparin concentrations above 10 μg/ml (Fig. 3). Hence, the E. coli RNAP was able to interact with the promoter region of the mobM gene, forming complexes that were unstable to heparin challenge. It is worth pointing out that RNAP does not generate stable complexes resistant to competitors in all promoters (for an in-depth discussion, see reference 36).
Fig 3.

EMSA of RNAP-DNA complexes. The E. coli RNAP (120 nM) was incubated with the 362-bp DNA (5 nM) (coordinates 3429 to 3790) at 37°C for 30 min. Then, heparin was added at the indicated concentrations. After 5 min, the reaction mixtures were loaded onto a native gel (5% polyacrylamide). The gel was stained with ethidium bromide. Bands corresponding to free DNA (F) and to specific RNAP-DNA complexes (C) are indicated.
To verify further the specificity of the complexes formed by the E. coli RNAP on the mobM promoter region, we carried out electron microscopy assays. In a first experiment, the E. coli RNAP was incubated with a 2,375-bp linear pMV158 DNA fragment (coordinates 3121 to 5495). This fragment contains the ssoU and ssoA single-strand origins, the oriT sequence, and the mobM gene (Fig. 1A). Then, RNAP-DNA complexes were fixed with glutaraldehyde, prepared for electron microscopy, and visualized as described in Materials and Methods. Electron micrographs of RNAP-DNA complexes are shown in Fig. 4. To determine the RNAP binding site, the contour lengths of the DNA regions between complexes and DNA ends were measured, and the position of the RNAP was determined. Figure 4A shows the distribution of the RNAP positions on the 2,375-bp fragment. Of 174 complexes examined, the majority (73%) showed a RNAP bound to one DNA region located at a maximum distance of 503 bp from one DNA end and of 1,872 bp from the other end. This indicated that the E. coli RNAP binds specifically either around coordinate 3624 (Pmob2 promoter region) or around coordinate 4992 (MobM-coding region) (Fig. 1). No clear indication of complexes of RNAP at the Pmob1 region was observed. To define precisely the RNAP recognition site, we performed similar electron microscopy assays but this time using a 699-bp DNA fragment spanning coordinates 3121 to 3819. The majority (75%) of the 105 complexes analyzed showed RNAP binding in a peak about 187 bp from the nearest DNA end (Fig. 4B). This result positioned the RNAP binding site around coordinate 3632 (Pmob2 promoter region) or 3308. Collectively taken, the above results showed that the E. coli RNAP binds specifically around the coordinates 3624 and 3632, occupying the −10 sequence element of the Pmob2 promoter.
Fig 4.

Electron micrographs of RNAP-DNA complexes. The E. coli RNAP (120 nM) was incubated with the 2375-bp fragment (5 nM) (coordinates 3121 to 5495) (A) or with the 699-bp fragment (20 nM) (coordinates 3121 to 3819) (B). After 15 min at 37°C, complexes were fixed with glutaraldehyde (0.3%) for 15 min at the same temperature. The distribution of the RNAP positions on both DNA fragments is shown. Scale bar, 500 bp.
To define accurately the position of the E. coli RNAP on the mobM promoter region, DNase I footprinting assays were performed using a 162-bp DNA fragment (coordinates 3527 to 3688), which contains the Pmob1 and Pmob2 promoters (Fig. 1). Such a fragment was radioactively labeled either at the 5′ end of the coding strand (Fig. 5A) or at the 5′ end of the noncoding strand (Fig. 5B). On the coding strand, the region spanning the −43 and −15 positions relative to the transcription start site of the Pmob2 promoter was protected against DNase I digestion. Changes in the DNase I sensitivity (diminished cleavages) were also observed at positions of adjacent regions (from −52 to −48 and from −13 to +20). In the case of the noncoding strand, RNAP-mediated protections were observed from −45 to around +21. Therefore, these results demonstrated that the E. coli RNAP recognizes in vitro the Pmob2 promoter rather than the Pmob1 promoter even though the latter promoter was recognized, albeit weakly, in vivo (Fig. 2).
Fig 5.

DNase I footprints of RNAP-DNA complexes. The 162-bp DNA fragment (coordinates 3527 to 3688) was labeled at the 5′ end of either the coding (A) or the noncoding (B) strand. Then, the labeled DNA (2.6 nM) was incubated (lane 2) or not (lane 1) with the RNAP (10 nM). Dideoxy-mediated chain termination sequencing reactions were run in the same gel (lanes A, C, G, and T). The main sequence elements of the Pmob1 and Pmob2 promoters and the RNAP-protected regions are indicated with brackets. The indicated positions are relative to the transcription start point of the Pmob2 promoter.
E. coli RNAP initiates mobM transcription from the Pmob2 promoter in vitro.
We next investigated whether E. coli RNAP was able to transcribe the mobM gene from the Pmob2 promoter in vitro. To this end, in vitro transcription assays under multiple-round conditions were carried out (Fig. 6). Two linear DNA fragments of 362 bp (coordinates 3429 to 3790) and 699 bp (coordinates 3121 to 3819) were used as templates. These fragments, which contain the Pmob1 and Pmob2 promoters, were the same used in the EMSA and electron microscopy studies (see above). Transcription from the Pmob2 promoter should generate runoff transcripts of 148 nt or 177 nt using the 362-bp or 699-bp template, respectively. However, transcription from the Pmob1 promoter should produce runoff transcripts of 182 nt or 211 nt with the 362-bp or 699-bp DNA, respectively. The in vitro transcription products were resolved on denaturing gels, and their sizes were estimated by comparison with the sizes of DNA fragments generated by dideoxy-mediated chain termination sequencing reactions (Fig. 6). When the 362-bp DNA was used as a template (lane 1), an RNA product that comigrated with a 158-nt DNA was detected. In the case of the 699-bp template (lane 2), the main RNA product observed comigrated with a 188-nt DNA. Since RNA runs about 5 to 10% more slowly than DNA of the same size in the sequencing gels (39), we conclude that the major RNA products correspond to runoff transcripts synthesized by recognition of the Pmob2 promoter. Hence, in vitro as well as in vivo, the E. coli RNAP transcribes the mobM gene preferentially from the Pmob2 promoter.
Fig 6.

In vitro transcription assays. Linear DNA fragments of 362 bp (lane 1) and 699 bp (lane 2) were used as templates. The coordinates of both templates are indicated below the gel. The transcription start sites of the Pmob1 and Pmob2 promoters are shown in gray and black, respectively. Reactions were initiated by the addition of E. coli RNAP. A denaturing gel (8 M urea–6% polyacrylamide) was used for resolving transcripts. Dideoxy-mediated chain termination sequencing reactions using pUC19 plasmid DNA (54) and the UC-50 primer (Table 1) were run in the same gel (lanes A, C, G, and T). The sizes of the DNA fragments that comigrate with the runoff transcripts are indicated on both sides of the gel in nucleotides.
Autoregulation of mobM gene expression in E. coli.
Using linear double-stranded DNA fragments from pMV158 and DNase I footprinting techniques, we showed previously that purified MobM protein was able to protect a region spanning coordinates 3582 and 3605 although a vast excess of MobM was needed because of the poor binding of the protein to linear dsDNA (our unpublished observations). Such a region included the IR2 element of oriT (15) (Fig. 1B). Now, we have shown that E. coli RNAP binds preferentially to the Pmob2 promoter both in vivo and in vitro (see above). Specifically, it protected about 66 bp on the noncoding strand, from −45 (coordinate 3598) to about +21 (coordinate 3663) relative to the transcription start point of the Pmob2 promoter (Fig. 5). These results suggested that the binding of MobM to oriT should prevent RNAP from gaining access to the Pmob2 promoter, and, consequently, it should reduce expression of the mobM gene. First, we tested this prediction by performing in vitro transcription experiments with the E. coli RNAP in the presence of MobM. Repression of the Pmob2 promoter was observed, but, as stated above, a large amount of MobM protein was required (data not shown). Subsequently, we designed an in vivo trans-complementation assay based on the use of two compatible plasmids: pLGM2 (18) and pORI-P (this work). Plasmid pLGM2 is a pET5 derivative that carries the mobM gene under the control of the T7 ϕ10 promoter. Plasmid pORI-P is a pMP220 derivative that carries the region of nucleotides 3555 to 3676 of pMV158 inserted upstream of the promoterless lacZ gene. This region, in addition to the Pmob1 and Pmob2 promoters, contains the three IRs of oriT (Fig. 1B). Both plasmids were introduced into JM109(DE3), in which expression of the T7 RNAP is inducible by IPTG. Thus, JM109(DE3) cells harboring pLGM2 and pORI-P synthesize MobM only when they are grown in the presence of IPTG. Measurement of lacZ expression under these conditions showed that β-galactosidase activity decreased nearly 2-fold compared to the activity detected in cells grown in the absence of IPTG (Fig. 7). In contrast, no changes in lacZ expression were detected in JM109(DE3) cells harboring pET5, which lacks the mobM gene, and pORI-P (control strain). The MobM-mediated repression was moderate albeit statistically significant. Similar repression levels were observed for the pMV158-repB gene using a construct similar to the one employed here (6). Whether these effects are due to the employment of the T7 ϕ10 promoter to direct synthesis of the protein or to an intrinsic property of the pneumococcal sequences is not known at present. We can conclude that MobM reduced the activity of the Pmob1 and/or Pmob2 promoters in vivo.
Fig 7.

β-Galactosidase assays. Relevant features of the pORI-P and pIR2-P plasmids are indicated. The Pmob2 (P2) promoter is located just downstream of the right arm of IR2 (Fig. 1B). Unlike pORI-P, pIR2-P lacks the left arm of IR1/IR3 (Fig. 1 B). Thus, pORI-P and pIR2-P differ in the nucleotide sequence just upstream of the −35 element of the Pmob1 (P1) promoter, which was regenerated due to the cloning. Each plasmid was introduced into the E. coli JM109(DE3) strain harboring either pLGM2 (IPTG-inducible expression of the mobM gene) (18) or the pET5 vector (lacking the mobM gene). β-Galactosidase activity (Miller units) was measured in bacteria growing in the absence (−) or in the presence (+) of IPTG. Each result represents the mean of three independent experiments (standard deviation is given).
The above interpretation was further confirmed by primer extension experiments using total RNA isolated from JM109(DE3) cells carrying both plasmids, pLGM2 and pORI-P. As primers, a mixture of the 5′-labeled lacZ-PE and tetA-PE oligonucleotides (Table 1) was used. They anneal to the lacZ and tetA (tetracycline resistance) transcripts of the pORI-P plasmid, respectively. With the primer lacZ-PE and in the absence of IPTG (Fig. 8, lane 3), a minor cDNA product of 131 nt (promoter Pmob1) and a major cDNA product of 97 nt (promoter Pmob2) were detected. The amounts of both products decreased when bacteria were grown in the presence of IPTG for 30 min (lane 4) or 120 min (lane 5). With the primer tetA-PE, used as internal control, a major cDNA product of 85 nt (promoter PtetA) was detected in the absence of IPTG (lane 3). However, the amount of such a product did not change in the presence of IPTG (lanes 4 and 5). We can conclude that MobM was able to repress in vivo not only the minor transcription initiated from promoter Pmob1 but also the major transcription initiated from promoter Pmob2.
Fig 8.

Primer extension on total RNA isolated from JM109(DE3)/pLGM2/pORI-P cells. When the culture reached an OD600 of 0.4, five aliquots (10 ml) were withdrawn and incubated without IPTG for 30 min (lanes 1, 2, and 3) or with IPTG for 30 min (lane 4) or 120 min (lane 5). The lacZ-PE primer (lanes 1, 3, 4, and 5) and/or the tetA-PE primer (lanes 2, 3, 4, and 5) was used. Primer extension products were analyzed by 8 M urea–6% polyacrylamide gel electrophoresis. Dideoxy-mediated chain termination sequencing reactions using pORI-P DNA and the lacZ-PE oligonucleotide were run in the same gel. A longer exposition of the upper part of the gel (lanes 3, 4, and 5) is shown. The sizes of the cDNA extension products are indicated on the right in nucleotides.
We next analyzed whether the IR1/IR3 inverted repeats of the pMV158-oriT were required for such MobM-mediated repression. To this end, we constructed plasmid pIR2-P, which is a pMP220 derivative that carries the region of nt 3583 to 3676 of pMV158 inserted upstream of the promoterless lacZ gene (Fig. 7). The −35 hexamer of the Pmob1 promoter (3575 to 3580 coordinates) was regenerated as a result of the cloning. Thus, plasmid pIR2-P carries an intact IR2 but lacks the left arm of IR1/IR3 (Fig. 1B). Expression of lacZ in JM109(DE3) cells harboring both plasmids, pLGM2 and pIR2-P, decreased nearly 2-fold when they were grown in the presence of IPTG (synthesis of MobM) (Fig. 7). No changes in lacZ expression were observed in the control strain [JM109(DE3) carrying both pET5 and pIR2-P]. MobM-mediated repression was further confirmed by primer extension on total RNA using a mixture of the 5′-labeled oligonucleotides lacZ-PE and tetA-PE as primers (not shown).
Taking all the above results together, we can conclude that MobM is able to repress transcription from the Pmob2 promoter in E. coli and thus regulate its own synthesis. This repression does not require the left arm of IR1/IR3 although we have to consider that the right arm of IR1/IR3 and the left arm of IR2 overlap (Fig. 1B).
DISCUSSION
The promiscuous plasmid pMV158 represents one of the simplest systems for an efficient DNA transfer among different bacterial species, both G+ and G−. Its MobM relaxase is the representative of the MOBV family of relaxases, consisting of more than 100 members (14), representing a wealth of genetic information that merits in-depth exploration. The results presented here demonstrate that the pMV158 mobM gene is transcribed by the E. coli RNAP from a promoter (Pmob2) that is different from the one (Pmob1) previously shown to be used in the G+ bacterium L. lactis (12). The DNA region protected by RNAP extends up to the +20 position relative to the Pmob2 transcription start site (Fig. 5 and 9A). This is indicative of the formation of an open complex, as should be expected under the experimental conditions employed. Inspection of the DNA sequence around the pMV158-oriT showed a high degree of conservation among many different plasmids from G+ bacteria (18). As the number of sequenced replicons increased, we have now found that a few streptococcal plasmids maintain the structure of the two Pmob1 and Pmob2 promoters (Fig. 9B). This is the case of plasmids pER13 and pSMQ172 from Streptococcus thermophilus (43, 48), pVA380-1 from Streptococcus ferus (26), and pRW35 from Streptococcus pyogenes (52). Cross-recognition of the pVA380-1-oriT by the pMV158-MobM was previously demonstrated (15). The genetic structure of the oriT genes and promoter(s) in the mobilization region of these plasmids suggests that it is not by chance that pMV158 harbors two promoters differentially used in G+ and G− bacteria and allows us to postulate that they could also be transferred to E. coli.
Fig 9.

(A) Nucleotide sequence of the pMV158 region that includes the Pmob1 (in red) and Pmob2 (in blue) promoters. Transcription start sites are indicated with arrows. The regions protected by the E. coli RNAP against DNase I digestion (this work) are indicated with brackets. The encircled sequence denotes the MobM binding site defined by DNase I footprinting assays although the precise upstream boundary of protection remained unclear (15). (B) Sequence alignment of the mobM promoter region from streptococcal plasmids closely related to pMV158: pER13 and pSMQ172 from S. thermophilus, pVA380-1 from S. ferus, and pRW35 from S. pyogenes. The nick site (marked by a slash) and the main elements of the Pmob1 and Pmob2 promoters are indicated.
Typically for a small mobilizable plasmid, pMV158 encodes only those conjugative functions required for DNA processing: the cis-acting oriT and the DNA relaxase MobM. Our in vivo transcriptional studies showed the following: (i) that synthesis of the pMV158-relaxase MobM is autoregulated and that regulation of the mobM gene appears to be regulated by MobM solely since we have not found any indication of the presence of a regulatory antisense RNA in the entire mob cassette (our unpublished results); (ii) that the region of oriT involved in autoregulation is represented by IR2; and (iii) that autoregulation does not require the presence of intact IR1/IR3 elements (Fig. 1C). Furthermore, a DNase I footprinting assay using oriT-containing linear double-stranded DNA fragments and a vast excess of purified protein showed that MobM protected IR2 (15). Whether MobM has two modes of binding to oriT, one for relaxosome formation (IR1/IR3) and another for autoregulation (IR2), is presently unknown although it seems likely in light of our present results.
In summary, our data demonstrate that binding of MobM to its cognate oriT is required not only to initiate plasmid transfer but also to control mobM gene expression, as previously hypothesized on the basis of the structure of the DNA region surrounding the pMV158-oriT (29, 30). Autoregulation of the synthesis of a relaxase of the MOBV family was previously shown for the E. coli plasmid pBBR1 although it was based only on transcriptional fusions (47). Similarly, mutational analyses indicated that TraI of plasmid F also self-regulates its own synthesis. However, in neither case, a detailed investigation of the transcriptional structure of the plasmid oriT was performed. To our knowledge, this is the first in-depth transcriptional study performed on a conjugative relaxase from G+ bacteria. Our results show that the mobM gene can be transcribed from two different promoters, which may be used depending upon the host in which the plasmid establishes. While promoter Pmob1 is located within oriT, promoter Pmob2 is adjacent to it (Fig. 1B and 9A), and yet both promoters are subjected to self-regulation. Given the promiscuity of pMV158, such genetic organization suggests a strategy of the plasmid to cope with the different transcription machineries of G+ and G− bacteria.
ACKNOWLEDGMENTS
We thank Lorena Rodríguez for her technical support and help in the purification of protein MobM and DNA from plasmid pMV158.
This research was financed by the Spanish Ministry of Science and Innovation (grants CSD2008-00013, INTERMODS, and BFU2010-19597 to M.E. and BFU2009-11868 to A.B.), by the Consejo Superior de Investigaciones Científicas (grant 201020E030 to A.B.), and by the European Union (grant EU-CP223111, CAREPNEUMO, to M.E.). F.L.-D. was a recipient of a Carlos III Spanish Health Institute fellowship (BF03/00529), and V.S.-C was a recipient of a fellowship from the Spanish Ministry of Science and Innovation (BES-2007-17086).
Footnotes
Published ahead of print 27 January 2012
REFERENCES
- 1. Balzer D, Pansegrau W, Lamka E. 1994. Essential motifs of relaxase (TraI) and TraG proteins involved in conjugative transfer of plasmid RP4. J. Bacteriol. 176: 4285–4295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Camacho A, Salas M. 1999. Effect of mutations in the “extended −10” motif of three Bacillus subtilis sigmaA-RNA polymerase-dependent promoters. J. Mol. Biol. 286: 683–693 [DOI] [PubMed] [Google Scholar]
- 3. Chan WT, et al. 2011. Genetic regulation of the yefM-yoeBSpn toxin-antitoxin locus of Streptococcus pneumoniae. J. Bacteriol. 193: 4612–4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de la Cruz F, Frost LS, Meyer RJ, Zechner EL. 2010. Conjugative DNA metabolism in Gram-negative bacteria. FEMS Microbiol. Rev. 34: 18–40 [DOI] [PubMed] [Google Scholar]
- 5. del Solar G, Acebo P, Espinosa M. 1995. Replication control of plasmid pLS1: efficient regulation of plasmid copy number is exerted by the combined action of two plasmid components, CopG and RNA II. Mol. Microbiol. 18: 913–924 [DOI] [PubMed] [Google Scholar]
- 6. del Solar G, Acebo P, Espinosa M. 1997. Replication control of plasmid pLS1: the antisense RNA II and the compact rnaII region are involved in translational regulation of the initiator RepB synthesis. Mol. Microbiol. 23: 95–108 [DOI] [PubMed] [Google Scholar]
- 7. del Solar G, Díaz R, Espinosa M. 1987. Replication of the streptococcal plasmid pMV158 and derivatives in cell-free extracts of Escherichia coli. Mol. Gen. Genet. 206: 428–435 [DOI] [PubMed] [Google Scholar]
- 8. del Solar G, Giraldo R, Ruiz-Echevarría MJ, Espinosa M, Díaz-Orejas R. 1998. Replication and control of circular bacterial plasmids. Microbiol. Mol. Biol. Rev. 62: 434–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Djordjevic M. 2011. Redefining Escherichia coli σ70 promoter elements: −15 motif as a complement of the −10 motif. J. Bacteriol. 193: 6305–6314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dower WJ, Miller JF, Ragsdale CW. 1988. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16: 6127–6145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Farías ME, Espinosa M. 2000. Conjugal transfer of plasmid pMV158: uncoupling of the pMV158 origin of transfer from the mobilization gene mobM, and modulation of pMV158 transfer in Escherichia coli mediated by IncP plasmids. Microbiology 146: 2259–2265 [DOI] [PubMed] [Google Scholar]
- 12. Farías ME, Grohmann E, Espinosa M. 1999. Expression of the mobM gene of the streptococcal plasmid pMV158 in Lactococcus lactis subsp. lactis. FEMS Microbiol. Lett. 176: 403–410 [DOI] [PubMed] [Google Scholar]
- 13. Fujimoto S, Clewell DW. 1998. Regulation of the pAD1 sex pheromone response of Enterococcus faecalis by direct interaction between cAD1 peptide mating signal and the negatively regulating DNA-binding TraA protein. Proc. Natl. Acad. Sci. U. S. A. 95: 6430–6435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garcillán-Barcia MP, Francia MV, de la Cruz F. 2009. The diversity of conjugative relaxases and its application in plasmid classification. FEMS Microbiol. Rev. 33: 657–687 [DOI] [PubMed] [Google Scholar]
- 15. Grohmann E, Guzmán LM, Espinosa M. 1999. Mobilisation of the streptococcal plasmid pMV158: interactions of MobM protein with its cognate oriT DNA region. Mol. Gen. Genet. 261: 707–715 [DOI] [PubMed] [Google Scholar]
- 16. Grohmann E, Muth G, Espinosa M. 2003. Conjugative plasmid transfer in Gram-positive bacteria. Microbiol. Mol. Biol. Rev. 67: 277–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gruber TM, Gross CA. 2003. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu. Rev. Microbiol. 57: 441–466 [DOI] [PubMed] [Google Scholar]
- 18. Guzmán L, Espinosa M. 1997. The mobilization protein, MobM, of the streptococcal plasmid pMV158 specifically cleaves supercoiled DNA at the plasmid oriT. J. Mol. Biol. 266: 688–702 [DOI] [PubMed] [Google Scholar]
- 19. Haft RJF, et al. 2006. General mutagenesis of F plasmid TraI reveals its role in conjugative regulation. J. Bacteriol. 188: 6346–6353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haugen SP, Ross W, Gourse RL. 2008. Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat. Rev. Microbiol. 6: 507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hernandez-Arriaga AM, Rubio-Lepe TS, Espinosa M, del Solar G. 2009. Repressor CopG prevents access of RNA polymerase to promoter and actively dissociates open complexes. Nucleic Acids Res. 37: 4799–4811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kurenbach B, Grothe D, Farías ME, Grohmann E. 2002. The tra region of the conjugative plasmid pIP501 is organized in an operon with the first gene encoding the relaxase. J. Bacteriol. 184: 1801–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lacks S. 1966. Integration efficiency and genetic recombination in pneumococcal transformation. Genetics 53: 207–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lacks S, Greenberg B. 1977. Complementary specificity of restriction endonucleases of Diplococcus pneumoniae with respect to DNA methylation. J. Mol. Biol. 114: 153–168 [DOI] [PubMed] [Google Scholar]
- 25. Lacks SA, López P, Greenberg B, Espinosa M. 1986. Identification and analysis of genes for tetracycline resistance and replication functions in the broad-host-range plasmid pLS1. J. Mol. Biol. 192: 753–765 [DOI] [PubMed] [Google Scholar]
- 26. LeBlanc DJ, Chen YYM, Lee LN. 1993. Identification and characterization of a mobilization gene in the streptococcal plasmid pVA380-1. Plasmid 30: 296–302 [DOI] [PubMed] [Google Scholar]
- 27. Lessl M, Balzer D, Weyrauch K, Lanka E. 1993. The mating pair formation system of plasmid RP4 defined by RSF1010 mobilization and donor-specific phage propagation. J. Bacteriol. 175: 6415–6425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Llosa M, Gomis-Rüth FX, Coll M, de la Cruz F. 2002. Bacterial conjugation: a two-step mechanism for DNA transport. Mol. Microbiol. 45: 1–8 [DOI] [PubMed] [Google Scholar]
- 29. Lorenzo-Díaz F, et al. 2011. The MobM relaxase domain of plasmid pMV158: thermal stability and activity upon Mn2+ and specific DNA binding. Nucleic Acids Res. 39: 4315–4329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lorenzo-Díaz F, Espinosa M. 2009. Lagging strand DNA replication origins are required for conjugal transfer of the promiscuous plasmid pMV158. J. Bacteriol. 191: 720–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 32. Mitchell JE, Zheng D, Busby SJW, Minchin SD. 2003. Identification and analysis of “extended −10” promoters in Escherichia coli. Nucleic Acids Res. 31: 4689–4695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moncalian G, de la Cruz F. 2004. DNA binding properties of protein TrwA, a possible structural variant of the Arc repressor superfamily. Biochim. Biophys. Acta 1701: 15–23 [DOI] [PubMed] [Google Scholar]
- 34. Nelson WC, Howard MT, Sherman JA, Matson SW. 1995. The traY gene product and integration host factor stimulate Escherichia coli DNA helicase I-catalyzed nicking at the F plasmid oriT. J. Biol. Chem. 270: 28374–28380 [PubMed] [Google Scholar]
- 35. Nieto C, Espinosa M. 2003. Construction of the mobilizable plasmid pMV158GFP, a derivative of pMV158 that carries the gene encoding the green fluorescent protein. Plasmid 49: 281–285 [DOI] [PubMed] [Google Scholar]
- 36. Ross W, Gourse RL. 2009. Analysis of RNA polymerase-promoter complex formation. Methods 47: 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ruiz-Cruz S, Solano-Collado V, Espinosa M, Bravo A. 2010. Novel plasmid-based genetic tools for the study of promoters and terminators in Streptococcus pneumoniae and Enterococcus faecalis. J. Microbiol. Methods 83: 156–163 [DOI] [PubMed] [Google Scholar]
- 38. Sabelnikov AG, Greenberg B, Lacks SA. 1995. An extended −10 promoter alone directs transcription of the DpnII operon of Streptococcus pneumoniae. J. Mol. Biol. 250: 144–155 [DOI] [PubMed] [Google Scholar]
- 39. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 40. Singh SS, Typas A, Hengge R, Grainger DC. 2011. Escherichia coli σ70 senses sequence and conformation of the promoter spacer region. Nucleic Acids Res. 39: 5109–5118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smillie C, Garcillan-Barcia MP, Francia MV, Rocha EPC, de la Cruz F. 2010. Mobility of plasmids. Microbiol. Mol. Biol. Rev. 74: 434–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smith MCA, Thomas CD. 2004. An accessory protein is required for relaxosome formation by small staphylococcal plasmids. J. Bacteriol. 186: 3363–3373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Somkuti GA, Steinbera DH. 2007. Molecular organization of plasmid pER13 in Streptococcus thermophilus. Biotechnol. Lett. 29: 1991–1999 [DOI] [PubMed] [Google Scholar]
- 44. Spaink HP, Okker R, Wijffelman C, Pees E, Lugtengerg B. 1987. Promoters in the nodulation region of the Rhizobium leguminosarum Sym plasmid pRLIJI. Plant Mol. Biol. 9: 27–39 [DOI] [PubMed] [Google Scholar]
- 45. Spiess E, Lurz R. 1988. Electron microscopic analysis of nucleic acids and nucleic acid-protein complexes. Methods Microbiol. 20: 293–323 [Google Scholar]
- 46. Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185: 60–89 [DOI] [PubMed] [Google Scholar]
- 47. Szpirer CY, Faelen M, Couturier M. 2001. Mobilization function of the pBHR1 plasmid, a derivative of the broad-host-range plasmid pBBR1. J. Bacteriol. 183: 2101–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Turgeon N, Moineau S. 2001. Isolation and characterization of a Streptococcus thermophilus plasmid closely related to the pMV158 family. Plasmid 45: 171–183 [DOI] [PubMed] [Google Scholar]
- 49. Varsaki A, Moncalián G, Garcillán-Barcia MP, Drainas C, de la Cruz F. 2009. Analysis of ColE1 MbeC unveils an extended ribbon-helix-helix family of nicking accessory proteins. J. Bacteriol. 191: 1446–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Voskuil MI, Chambliss GH. 1998. The −16 region of Bacillus subtilis and other gram-positive bacterial promoters. Nucleic Acids Res. 26: 3584–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wigneshweraraj S, et al. 2008. Modus operandi of the bacterial RNA polymerase containing the sigma54 promoter-specificity factor. Mol. Microbiol. 68: 538–546 [DOI] [PubMed] [Google Scholar]
- 52. Woodbury RL, et al. 2008. Plasmid-borne erm(T) from invasive, macrolide-resistant Streptococcus pyogenes strains. Antimicrob. Agents Chemother. 52: 1140–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wozniak RAF, Waldor MK. 2010. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 8: 552–563 [DOI] [PubMed] [Google Scholar]
- 54. Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33: 103–109 [DOI] [PubMed] [Google Scholar]
- 55. Zechner EL, et al. 2000. Conjugative-DNA transfer processes, p 87–174 Thomas CM. (ed), The horizontal gene pool. Harwood Academic Publishers, Amsterdam, Netherlands [Google Scholar]