

Abstract
Major histocompatibility complex class I (MHC-I) molecules play an important role in host immunity to infection by presenting antigenic peptides to cytotoxic T lymphocytes (CTLs), which recognize and destroy virus-infected cells. Members of the Herpesviridae have developed multiple mechanisms to avoid CTL recognition by virtue of downregulation of MHC-I on the cell surface. We report here on an immunomodulatory protein involved in this process, pUL56, which is encoded by ORF1 of equine herpesvirus type 1 (EHV-1), an alphaherpesvirus. We show that EHV-1 pUL56 is a phosphorylated early protein which is expressed as different forms and predominantly localizes to Golgi membranes. In addition, the transmembrane (TM) domain of the type II membrane protein was shown to be indispensable for correct subcellular localization and a proper function. pUL56 by itself is not functional with respect to interference with MHC-I and likely needs another unidentified viral protein(s) to perform this action. Surprisingly, pUL49.5, an inhibitor of the transporter associated with antigen processing (TAP) and encoded by EHV-1 and related viruses, appeared not to be required for pUL56-induced early MHC-I downmodulation in infected cells. In conclusion, our data identify a new immunomodulatory protein, pUL56, involved in MHC-I downregulation which is unable to perform its function outside the context of viral infection.
INTRODUCTION
Efficient recognition and destruction of infected cells by antigen-specific cytotoxic CD8+ T lymphocytes (CTLs) are important host defense mechanisms in the control of viral and certain bacterial infections. The CTL-based defense relies on the recognition of pathogen-derived peptides presented on the surface of the infected cells by major histocompatibility complex class I (MHC-I). The MHC-I antigen presentation pathway originates in the cytosol, where viral proteins are processed into small peptides in the proteasome and translocated into the lumen of the endoplasmic reticulum (ER) by the transporters associated with antigen processing (TAPs) 1 and 2. In the ER, a trimolecular complex is formed between the newly synthesized MHC-I heavy chains, the small viral peptides, and β2-microglobulin (β2M) (3, 27). Once the peptides are successfully loaded, the MHC-I complex is released and transported to the cell surface, where a CTL response is triggered. MHC-I molecules that fail to bind peptides are rerouted to the cytosol, where they become degraded by the proteasome (46).
Viruses, especially members of the Herpesviridae, have evolved mechanisms that target multiple steps in the MHC-I antigen presentation pathway to evade immune recognition. A number of viral proteins involved in this process have been identified and studied extensively in the Beta- and Gammaherpesvirinae (14). In the case of the Alphaherpesvirinae, the product of the herpes simplex virus (HSV) US12 gene (ICP47) and the products of the UL49.5 orthologues of several members of the varicelloviruses were shown to interfere with the TAP-mediated peptide transport via different mechanisms (12, 18, 19, 44). In the case of bovine herpesvirus type 1 (BHV-1), pUL49.5 alone is sufficient to downregulate MHC-I expression during virus infection (18). In contrast, the UL49.5 product of human varicella-zoster virus (VZV), while capable of interacting with TAP, is unable to block peptide transport or modulate MHC-I cell surface expression (11, 19). Instead, the VZV ORF66 protein kinase contributes to MHC-I downregulation by delaying MHC-I maturation during its transport from the ER through the Golgi network to the plasma membrane (11). Finally, in the case of a third virus from this group, namely, pseudorabies virus (PRV), it has been reported that MHC-I downregulation is not or only partly dependent on the UL49.5 and US3 gene products, indicating that other, unidentified viral proteins can also be involved in this viral immune evasion process (9).
We here focused on another member of the Varicellovirus genus, namely, equine herpesvirus type 1 (EHV-1). Transmitted by aerosol, EHV-1 is a highly contagious pathogen of horses causing upper respiratory disease, late-stage abortion in mares, and neurological disorders (1). Despite regular vaccinations, EHV-1 remains a constant threat to horses worldwide, mainly because the immune responses induced after both infection and vaccination are not fully protective (8, 26). EHV-1 infection is controlled mainly by the action of CTLs that attack virus-infected cells. The frequency of precursor CTLs specific for EHV-1 antigens is correlated with protection against disease (23). However, EHV-1 has also developed mechanisms to avoid CTL recognition by virtue of a massive downregulation of cell surface MHC-I molecules (2, 29). The UL49.5 product is the only EHV-1 protein identified to modulate the MHC-I-restricted antigen-presenting pathway in E.derm UL49.5EHV-1 cells. The cells stably express EHV-1 pUL49.5, which was shown to block ATP binding to TAP and inhibit transport of proteasome-generated peptides into the ER. This blockade resulted in a downregulation of MHC-I on the cell surface of transfected cells (19). Recently, we observed that infection with EHV-1 strain RacL11 resulted in only mild downregulation of MHC-I, even though it encodes a fully functional pUL49.5 protein (19, 32). These results were in stark contrast to those for another EHV-1 strain, namely, Ab4, which was reported to induce a massive downregulation of MHC-I in infected equine dermal (NBL6) cells (29). This strain-dependent pattern led us to hypothesize that one or more unknown viral proteins participate in downregulation of MHC-I mediated by EHV-1.
In the present study, we demonstrated that the ORF1 gene encodes a novel immune evasion protein that plays a major role in MHC-I downregulation. This ORF1-encoded protein, pUL56, is a phosphorylated type II transmembrane (TM) protein expressed with early kinetics and localized predominantly in Golgi membranes. The TM domain of EHV-1 pUL56 was shown to be indispensable for correct subcellular localization of pUL56 and function.
MATERIALS AND METHODS
Cells and viruses.
Rabbit kidney (RK13) cells, human HeLa cells, and human embryonic kidney 293 (HEKT) cells were maintained in Earle's minimum essential medium (EMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Biochrom AG) and 100 U/ml penicillin and 0.1 mg/ml streptomycin (1% Pen/Strep; Sigma). The equine skin fibroblast cell line NBL6 (ATCC) was maintained in EMEM supplemented with 20% fetal calf serum, 29 mg/ml l-glutamine, 1% nonessential amino acids (Biochrom AG), and 1% Pen/Strep. EHV-1 strains Ab4 and RacL11 were previously constructed as bacterial artificial chromosomes (BACs) by insertion of mini-F sequences containing an egfp marker gene in lieu of gene 71, which encodes glycoprotein gp2 (13, 30). BAC-reconstituted parental viruses Ab4G and RacL11G and all mutant viruses were generated and propagated in RK13 cells.
Antibodies.
Mouse anti-MHC-I monoclonal antibody (MAb) H58A (isotype IgG2a) and anti-CD44 MAb BAT31A (isotype IgG1) were purchased from VMRD and used at a 1/100 dilution. Rabbit anti-EHV-1 UL49.5 polyclonal antibodies (pAbs), mouse anti-EHV-1 gM MAb F6, and mouse anti-EHV-1 gp2 and biotinylated equine polyclonal anti-EHV-1 antibodies have been previously described (32, 42, 45). Rabbit anti-EHV-1 pUL56 pAbs (1/500) were designed and produced by GenScript Corporation (NJ). The β-actin (13E5) rabbit MAbs (1/2,500) were from Cell Signaling Technologies. Alexa Fluor 647-conjugated goat anti-mouse IgG (1/500), Alexa Fluor 488-conjugated goat anti-rabbit IgG (1/500), Texas Red-conjugated goat anti-rabbit (1/100), and streptavidin-phycoerythrin (PE) (1/200) were from Invitrogen, and horseradish peroxidase-labeled goat anti-rabbit and goat anti-mouse IgG (1/10,000) were from Southern Biotech. Goat anti-mouse Cy5 was obtained from Zymed.
Viral mutagenesis.
Two-step Red-mediated (en passant) recombination was used to create mutant and revertant viruses (38). A detailed list of the primers used for generating the mutant viruses is given in Table 1. The genotypes of all virus mutants and revertants were confirmed by restriction enzyme analyses (restriction fragment length polymorphism [RFLP] analysis) and nucleotide sequencing. In addition, the expression of gp2 was restored in the Ab4 parental virus (vAb4), Ab4GΔ1 (vAb4Δ1), Ab4GΔ1_TM (vAb4Δ1_TM) mutants, and the parental RacL11 virus (vRacL11) by cotransfection of RK13 cells with BAC DNA and plasmid DNA containing full-length gene 71, followed by plaque purification (31). Restoration of gp2 was confirmed by Western blot analysis of virus-infected cells using the anti-EHV-1 gp2 MAb 8B6, exactly as previously described (43, 45).
Table 1.
Primers used for generating the different deletion mutant viruses
| Purpose | Descriptiona | Sequence (5′–3′)b |
|---|---|---|
| Deletion of ORF1 | ORF1_F | CGAGGGAGTTTCGCGGGGCCGCGCCTCCTCTGTCTCCATCAAGGAGAAATAAAACGACTGTAGGGATAACAGGGTAATCG |
| ORF1_R | ATTAACCTTTGCGGTACTACAGTCGTTTTATTTCTCCTTGATGGAGACAGAGGAGGCGCGCCAGTGTTACAACCAATTAACC | |
| Deletion of ORF2 | ORF2_F | GTACCGCAAAGGTTAATCGCATTTATTTTTACATGCACTCATTGTGGTAGGGTTTGAGACTAGGGATAACAGGGTAATCG |
| ORF2_R | CGTGGAGGAGGCGCATACACGTCTCAAACCCTACCACAATGAGTGCATGTAAAAATAAATGCCAGTGTTACAACCAATTAACC | |
| Deletion of ORF1 and ORF2 | ORF1&2_F | CGAGGGAGTTTCGCGGGGCCGCGCCTCCTCTGTCTCCATCGGTAGGGTTTGAGACGTGTATAGGGATAACAGGGTAATCG |
| ORF1&2_R | TCCAGCGTGGAGGAGGCGCATACACGTCTCAAACCCTACCGATGGAGACAGAGGAGGCGCGCCAGTGTTACAACCAATTAACC | |
| Deletion of start codon of UL49.5 | UL49.5_F | TGCTCGCGCTCTCCATCTCTCACCACACTCAACCACCACGCTGTCCACGAGATTCGTGACTAGGGATAACAGGGTAATCGATTT |
| UL49.5_R | GGCAGGCGAGAATGGCCAGCGTCACGAATCTCGTGGACAG CGTGGTGGTTGAGTGTGGTGCCAGTGTTACAACCAATTAACC | |
| Deletion of first ATG of ORF1 | ORF11stATG F | TCCACCTGCACCTTTTCCATCTCCTCTCCAACTCGCCGCCAGACCCGAGGGAGTTTCGCGTAGGGATAACAGGGTAATCGATT |
| ORF11stATG R | AGACAGAGGAGGCGCGGCCCCGCGAAACTCCCTCGGGTCTGGCGGCGAGTTGGAGAGGAGCCAGTGTTACAACCAATTAACC | |
| Deletion of second ATG of ORF1 | ORF12ndATG F | GGGAGTTTCGCGGGGCCGCGCCTCCTCTGTCTCCATCTCCTGCCCACCGCCGCCCAATGGTAGGGATAACAGGGTAATCGATT |
| ORF12ndATG R | CCAGCGATGCGCGGCGCGCCCCATTGGGCGGCGGTGGGCAGGAGATGGAGACAGAGGAGGCCAGTGTTACAACCAATTAACC | |
| Deletion of major part (841 bp) of US3 | US3_F | AACCGACTGGTTTTCCACTACGAGCGACGCGTCAGAATCGTAATTTCAACGCTCAAGATAGGATGACGACGATAAGTAGGGATAAC |
| US3_R | TCGAGGAAACTCCTCCGGATTTATCTTGAGCGTTGAAATTACGATTCTGACGCGTCGCTCGGGGTAA TGCCAGTGTTACAACCA | |
| Deletion of predicted catalytic domain of UL13 (aa 221-380) | UL13_F | TACCTTTACAGTGAACCCAGAGATGCACTACAGGCGCGTGTCTATGGCCATGCGGGCGGAGGATGACGACGATAAGTAGGGATAAC |
| UL13_R | CGCCGGTATCAATTTCAAACTCCGCCCGCATGGCCATAGACACGCGCCTGTAGTGCATCTGGGTAATGCCAGTGTTACAACCA | |
| Deletion of transmembrane region (aa 170-192) of ORF1 | ORF1TM_F | CTCCGATCCCCACCAGACCTCTCGAAGGAGACAGAACCCGAGTTCGTTAACAAACGGCAATAGGGATAACAGGGTAATCG |
| ORF1TM_R | AGTCGTTTTATTTCTCCTTCTTGCCGTTTGTTAACGAACTCGGGTTCTGTCTCCTTCGAGGCCAGTGTTACAACCAATTA |
Directionality of the primer: F, forward; R, reverse.
Bold sequence indicates the template binding region of the primers for PCR amplification with pEPkanS.
Construction of the pNTAP-UL56 plasmid and transfection.
To express pUL56, vector pCeMM-NTAP (GS) containing the egfp gene under the control of the human cytomegalovirus immediate-early promoter was used (7). Briefly, the ORF1 gene was PCR amplified from Ab4 DNA using primers NTte (5′-AAAAAAGTCGACAATGAGACCCGAGGGAGTTTCGCGGGGCC-3′) and NTf (5′-AAAAAAGTCGACTTATTTCTCCTTCTTGCCGTTTGTTAAC-3′) (underlined sequence indicates the SalI restriction site) and cloned into the SalI-compatible XhoI restriction site of pCeMM-NTAP (GS). The resulting pNTAP-UL56 construct clones were confirmed by RFLP, using HindIII and NcoI, and sequencing, using NTAP-ORF_F (5′-CCGGTGAGCTGGAGCAGCTA-3′) and NTAP-ORF1_F (5′-CTTCGGCCAGTAACGTTAGG-3′) (data not shown). HEKT or HeLa cells were transfected using the Fugene HD reagent, according to the protocol recommended by the supplier (Promega).
Flow cytometry.
NBL6 cells were mock infected or infected with the various viruses at a multiplicity of infection (MOI) of 5. At different time points (0, 2, 4, 6, 8, 12, and 16 h postinfection [hpi]), mock- and virus-infected cells were trypsinized and resuspended in phosphate-buffered saline (PBS) containing 2.5% FBS and 0.02% sodium azide (FCM buffer). Cells were incubated for 40 min on ice with anti-MHC-I or anti-CD44 MAbs in FCM at a 1/100 dilution. After three times of washing, the cells were incubated with Alexa Fluor 647-conjugated goat anti-mouse IgG (1/500) and analyzed immediately using a FACSCalibur flow cytometer (Becton Dickinson). For each sample, at least 10,000 cells were evaluated, and all data were corrected for unspecific binding using isotype controls. Only enhanced green fluorescent protein (EGFP)-positive cells were analyzed in the case of virus-infected cells. The expression levels of surface MHC-I were presented as mean fluorescence intensities (MFIs).
For the combined transfection-infection experiments, HeLa or HEKT cells were first transfected with the pNTAP-UL56 or empty NTAP vector using Fugene HD and infected with vAb4 or vAb4Δ1 at an MOI of 10. At 24 hpi, cells were collected, washed, and stained for MHC-I expression as described above. To identify the infected cells, a biotinylated anti-EHV-1 antibody was used, followed by streptavidin-PE. Only double-positive cells, with EGFP expression as a marker for successful transfection and PE as a marker for infection, were analyzed for cell surface MHC-I expression, which was presented as MFI.
Western blot analyses.
NBL6 cells were infected with parental or mutant viruses at an MOI of 5, in the presence or absence of the viral DNA synthesis inhibitor phosphonoacetic acid (PAA; 300 μg/ml; Alfa Aesar). The virus was allowed to attach for 1 h at 4°C, followed by a penetration step for 1 h at 37°C. The cells were then treated with citrate-buffered saline (CBS; pH 3.0) for 3 min to remove remaining virus on the cell surface and washed with PBS. At different time points after infection, cells were collected and lysed using radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) with protease inhibitor cocktail (Roche) and benzonase (Novagen). The proteins were separated by SDS-PAGE (10% to 15%) and transferred to polyvinylidene difluoride (PVDF) membranes. To exclude nonspecific binding, membranes were blocked with PBS containing 0.05% Tween 20 (PBST) supplemented with 5% nonfat dry milk overnight at 4°C. After blocking, membranes were incubated with anti-pUL56 (1/500), anti-pUL49.5 (1/500), anti-gM (1/500), or anti-actin (1/2500) antibodies for 1 h at room temperature (RT). After 3 washing steps with PBST, membranes were incubated with peroxidase-conjugated goat anti-rabbit IgG (1/10,000) or goat anti-mouse IgG (1/10,000) for 1 h at RT. Membranes were washed again three times with PBST, and the reactive bands were visualized using an ECL Plus Western blotting detection system (Amersham Pharmacia). To examine pUL56 phosphorylation, cell lysates were treated with lambda protein phosphatase (λ-PPase; New England BioLabs) for 30 min at 30°C before electrophoresis.
Immunofluorescence.
To control for proper pUL56 expression in transfected HEKT cells, cells were incubated with anti-ORF1 pAbs (1/200) in blocking buffer for 1 h at RT and then, after three washing steps, incubated with goat anti-rabbit Texas Red (1/100) for 1 h at RT. Cells were analyzed with a confocal laser scanning microscope (TCS SP2 laser scanning spectral confocal system; Leica Microsystems GmbH, Wetzlar, Germany) and the Leica confocal software.
To evaluate intracellular expression of pUL56 in infected cells, NBL6 cells were grown on coverslips coated with 0.5 mg/ml collagen A (Biochrom AG) in double-distilled H2O (pH 3.5) and infected with parental vAb4, vAb4Δ1, or vAb4Δ1_TM mutant viruses at an MOI of 0.1 for 20 h. To stain the Golgi apparatus, cells were incubated with 5 μM BODIPYTR C5-ceramide complexed to bovine serum albumin (BSA; Invitrogen) in Hank's balanced salt solution with 10 mM HEPES, pH 7.4 (HBSS/HEPES), for 30 min at 4°C, washed once with medium, and incubated in fresh medium for 1 h. To stain the ER, cells were incubated with 1 mM ER-Tracker Red (BODIPYTR glibenclamide; Invitrogen) in HBSS/HEPES for 30 min at 37°C and incubated in fresh medium for 1 h. After 1 h of incubation, cells were fixed with 3.5% PFA for 5 min at RT, followed by permeabilization with 0.1% Tween 20 for 15 min. After blocking with 10% normal goat serum for 1 h, cells were incubated with anti-pUL56 pAbs (1/200) for 1 h at RT. After three washing steps, cells were incubated with secondary Alexa Fluor 488 goat anti-rabbit IgG (1/5,000) for 1 h at RT. Finally, after three washing steps, Vectashield mounting medium with DAPI (4′,6-diamidino-2-phenylindole; Vector Laboratories) was added to cells and the coverslips were inspected under a Zeiss Axio Imager M1 microscope (Zeiss, Germany).
RESULTS
The ability of EHV-1 to downregulate MHC-I is virus strain dependent.
It was reported earlier that the EHV-1 strain Ab4 is able to induce robust downregulation of MHC-I molecules on the surface of infected NBL6 cells (29), a phenomenon confirmed here (Fig. 1A). Expression of the cell surface protein CD44, used as a control, was unaffected during infection (Fig. 1A). In contrast to Ab4, the EHV-1 strain RacL11 resulted in only moderate downregulation of MHC-I surface expression (Fig. 1A and B). This inability could not be attributed to differences in expression of the UL49.5 gene product, since RacL11 encodes a functional pUL49.5 (19) and sequence analyses failed to reveal differences between the UL49.5 genes of Ab4 and RacL11 (data not shown). Hence, these results indicated that another, yet unidentified protein(s) accounts for the difference in MHC-I downregulation capabilities of EHV-1 strains.
Fig 1.

The ability of EHV-1 to downregulate MHC-I is virus strain dependent. (A) NBL6 cells were infected with EHV-1 Ab4G or RacL11G. Cell surface MHC-I expression was analyzed at 16 hpi using flow cytometry. Primary anti-MHC-I MAb H58A and anti-CD44 MAb were used, followed by goat anti-mouse Alexa Fluor 647. (B) Dot plots from a representative experiment depicting MHC-I surface expression in mock- and virus-infected NBL6 cells. Quadrant gates were established using isotype controls and mock-infected cells, respectively. (C) Genomic organization of EHV-1 strains Ab4 and RacL11. A detailed organization of the leftmost ORFs of the UL region of the two virus strains is shown. US, unique short; IR, internal repeat; TR, terminal repeat.
Compared to Ab4, RacL11 specifies a shortened unique long segment (UL) of its genome, and the deletion was mapped to the extreme left genomic terminus (15). When comparing the sequence of this region of RacL11 to that of Ab4 (GenBank accession no. NC_001491.2), we discovered a deletion of 1,283 bp, resulting in the absence of the majority of ORF1 and the complete ORF2 (Fig. 1C). EHV-1 ORF1 is predicted to encode a protein of 202 amino acids (aa) in length which shares homology with pUL56 orthologues of related alphaherpesviruses (4, 36, 37). EHV-1 ORF2 is predicted to encode a 205-aa protein, has a positional counterpart in VZV, and likely arose from the same ancestor as ORF1 (37). We hypothesized that the two open reading frames (ORFs) are plausible candidates to be involved in MHC-I downregulation.
pUL56-mediated MHC-I downregulation is dependent on virus infection but independent of pUL49.5.
To determine whether the proteins encoded by ORF1 and/or ORF2 play a role in MHC-I downregulation, deletion mutants were generated on the basis of the Ab4 strain and cell surface expression levels of MHC-I were evaluated. To this end, the majority of ORF1, the complete ORF2, or a combination of both was deleted, resulting in the recombinant viruses Ab4GΔ1, Ab4GΔ2, and Ab4GΔ1_2, respectively (Fig. 2A). Infection of NBL6 cells with these mutant viruses showed that MHC-I levels on the surface of cells infected with Ab4GΔ1_2 or Ab4GΔ1 were restored (Fig. 2B). In contrast, MHC-I levels remained low after infection with Ab4GΔ2 (Fig. 2B), indicating that the ORF1 but not the ORF2 gene product is involved in MHC-I downregulation. Parental Ab4G and the revertant viruses Ab4GΔ1R and Ab4GΔ1_2R were included as positive controls, and infection of NBL6 cells resulted in a significant decrease of MHC-I expression (Fig. 2B).
Fig 2.

pUL56-mediated MHC-I downregulation is dependent on virus infection. (A) Schematic representation of the structure of ORF1 and ORF2 in wild-type Ab4 as well as in the deletion mutant viruses. (B) NBL6 cells were infected with EHV-1 Ab4G, Ab4GΔ1_2, Ab4GΔ1, Ab4GΔ2, or the revertant viruses Ab4GΔ1_2 R and Ab4GΔ1 R. Cell surface MHC-I expression was analyzed at 16 hpi using flow cytometry. Primary anti-MHC-I MAb H58A was used, followed by goat anti-mouse Alexa Fluor 647. *, P < 0.05. (C) HEKT cells were transfected with pNTAP-UL56 or the empty pNTAP for 48 h. Immunofluorescence stainings of pUL56 expression in transfected cells (EGFP positive) were performed using the rabbit anti-EHV-1 pUL56 pAbs, followed by goat anti-rabbit Texas Red. (D) HEKT and HeLa cells were transfected with pNTAP-UL56 or the empty pNTAP. Cell surface MHC-I expression was analyzed at 48 hpi using flow cytometry. Primary anti-MHC-I MAb H58A was used, followed by goat anti-mouse Cy5. (E) HeLa cells were transfected with pNTAP-UL56 or the empty pNTAP and 24 h later were infected with Ab4GΔ1. Cell surface MHC-I expression was analyzed 24 h later using flow cytometry. Primary anti-MHC-I MAb H58A was used, followed by goat anti-mouse Cy5. To identify infected cells, biotinylated anti-EHV-1 antibodies were used, followed by streptavidin-PE. Mock- and Ab4G-infected cells were included as controls.
In order to analyze whether pUL56 encoded by ORF1 is able to downregulate MHC class I by itself, we constructed a pUL56-expressing plasmid, pNTAP-UL56. Upon transfection of HEKT cells with the plasmid, transfection efficiencies of 55.4% ± 3.7% were achieved and coexpression of pUL56 and EGFP was confirmed by fluorescence microscopy (Fig. 2C). Transfection with the empty vector pNTAP was included as a control, and EGFP-positive cells did not show any pUL56 expression (Fig. 2C). Next, the MHC-I levels in HeLa or HEKT cells were analyzed after transfection with pNTAP-UL56 or the empty pNTAP vector. Surprisingly, no MHC-I downregulation was observed in pUL56-transfected cells (Fig. 2D), despite the fact that infection of the cells with Ab4G, with infection efficiencies of 69.9% ± 5.5%, resulted in massive downregulation of MHC-I (data not shown). This led us to speculate that pUL56 can function properly only in the presence of virus infection. To verify this assumption, HeLa and HEKT cells were transfected with pNTAP-UL56, followed by infection with vAb4GΔ1. The results showed that the combination of transfection and infection led to a significant downregulation of MHC-I in HeLa cells (P = 0.001; Fig. 2E) as well as HEKT cells (P = 0.01; data not shown). In contrast, infection of the same cells with vAb4GΔ1, with infection efficiencies of 80.6% ± 1.5%, alone or after transfection of empty pNTAP vector only slightly downregulated MHC-I in both cell lines (Fig. 2E). These data suggest that pUL56 needs one or more viral partners or cellular partners induced by virus infection to efficiently downregulate cell surface MHC-I.
It was shown previously that EHV-1 pUL49.5 can modulate MHC-I cell surface expression in pUL49.5-expressing equine E.derm cell lines (19). We therefore examined whether the observed pUL56-induced MHC-I downregulation was perhaps dependent on expression of pUL49.5. Recombinant Ab4 viruses were generated where the UL49.5 gene was inactivated by deletion of the start codon, alone or in combination with deletion of ORF1, and the resulting viruses were named Ab4GΔ49.5 and Ab4GΔ1_49.5, respectively. Western blot analyses confirmed that expression of pUL49.5 and/or pUL56 was abrogated in the corresponding mutant viruses (Fig. 3A). Analyses of surface MHC-I expression of NBL6 cells at 16 hpi revealed that the absence of pUL49.5 had no effect on the virus's ability to downregulate MHC-I (Fig. 3B). On the basis of these results, we concluded that pUL56-mediated downregulation of MHC-I during infection is independent of pUL49.5.
Fig 3.

pUL56-mediated downregulation of MHC-I is pUL49.5 independent. (A) Cells were mock infected or infected with parental Ab4G or the mutant viruses Ab4GΔ1, Ab4GΔ49.5, and Ab4GΔ1_49.5, and cell lysates were prepared at 16 hpi. Western blotting was performed to detect pUL49.5 using anti-EHV-1 UL49.5 pAbs, pUL56 using anti-EHV-1 UL56 pAbs, or gM using the MAb F6. The MAb 13E5 was used to detect β-actin, which was included as a loading control. (B) NBL6 cells were mock infected or infected with Ab4G, Ab4GΔ1, Ab4GΔ49.5, or Ab4GΔ1_49.5. At 16 hpi, cell surface expression of MHC-I was analyzed using flow cytometry.
pUL56 is a phosphorylated early protein.
It was previously reported that downregulation of MHC-I by EHV-1 was mediated by an early gene product (29). To examine the kinetics of MHC-I downregulation as well as the expression profile of pUL56, we performed time course studies. NBL6 cells were infected with Ab4G at an MOI of 5, and at different time points after infection, surface MHC-I and pUL56 expression was determined. The results showed that MHC-I downregulation started as early as 2 hpi, was complete by 8 hpi, and was maintained throughout the replication cycle (Fig. 4A). Expression of the surface marker CD44 was not altered at all time points tested. In parallel, we examined the kinetic correlation between MHC-I downregulation and pUL56 expression. At 2 hpi, a single pUL56-specific band of approximately 22 kDa in size was detected, a finding compatible with the expected molecular mass of pUL56 (Fig. 4B). From 4 hpi onwards, a second band with an apparent molecular mass of about 28 kDa appeared, and both moieties reached maximum levels at 8 hpi (Fig. 4B). Besides these two prominent bands, two fainter bands with apparent molecular masses of 20 kDa and 25 kDa were also detected (Fig. 4B). Based on the fact that no specific signal was detected in Ab4GΔ1-infected cells, we concluded that all bands are products of the ORF1 gene and that pUL56 is expressed as different forms. In the presence of PAA, an inhibitor of viral DNA synthesis, expression of pUL56 was only slightly reduced at 16 hpi, indicating that pUL56 expression is independent of DNA synthesis and encoded by a gene with (immediate) early kinetics. As expected, expression of the late viral protein gM, which was used as a control, was significantly inhibited by PAA (Fig. 4B). When infected cell lysates were treated with the phosphatase λ-PPase, all higher-molecular-mass pUL56 moieties disappeared, whereas the 22-kDa protein as well as the smaller proteins remained unaffected, suggesting that the observed 25-kDa and 28-kDa proteins represent phosphorylated forms of pUL56 (Fig. 4B). In contrast, pUL49.5 became expressed only from 4 hpi onwards (Fig. 4B), which is clearly later than the onset of MHC-I downregulation (Fig. 4A). Moreover, PAA treatment partially inhibited pUL49.5 expression, indicating that EHV-1 pUL49.5 is expressed with early-late kinetics. These results were an additional indication that pUL56, but not pUL49.5, is involved in the early downregulation of MHC-I during EHV-1 infection.
Fig 4.

pUL56 is a phosphorylated early protein expressed as different isoforms. (A) Untreated or PAA-treated NBL6 cells were infected with Ab4G parental virus, and at the indicated time points, MHC-I expression on the cell surface was analyzed with anti-MHC-I MAb H58A. CD44 expression was included as a control. (B) NBL6 cells, treated or not treated with PAA, were infected with Ab4G or Ab4GΔ1, and at the indicated time points, cell lysates were prepared. Western blot analyses were performed to detect pUL49.5 using anti-EHV-1 UL49.5 pAbs, pUL56 using anti-EHV-1 pUL56 pAbs, or gM using the MAb F6. The MAb 13E5 was used to detect β-actin, which was included as a loading control.
pUL56 contains an additional, functional in-frame start codon.
It was reported that the UL56 gene product of HSV type 2 (HSV-2) is detectable as several species during infection due to phosphorylation and utilization of an additional in-frame start codon upstream of the canonical AUG (20). Similar to HSV-2, we showed that EHV-1 pUL56 is also phosphorylated and expressed as several protein species that make use of an additional in-frame AUG start codon in the ORF1 gene. This additional in-frame AUG is located 17 codons downstream of the predicted start codon (Fig. 5A), and initiation at this second AUG would theoretically result in a 20-kDa protein, as was observed during the course of infection (Fig. 4B). In order to determine whether this pUL56 form is produced by translation initiated from the second AUG, the first or second in-frame start codon was deleted in the Ab4 virus. pUL56 production and cell surface MHC-I expression were analyzed after infection of NBL6 cells. The pUL56 expression profile (Fig. 5B, lanes 5 and 6) was indistinguishable from that of parental virus (Fig. 5B, lanes 1 and 2) when the second in-frame start codon was deleted, indicating that all its forms are a result of translation initiation from the first AUG. Still, a single unphosphorylated protein with a molecular mass between the 20-kDa and 22-kDa species was detected upon deletion of the first AUG (Fig. 5B, lanes 3 and 4), indicating that the second in-frame AUG can also be used as an alternative initiation site. The expression levels of this unphosphorylated truncated protein, however, were considerably lower than those of the pUL56 species that result from initiation at the first AUG (Fig. 5B). Regardless, this pUL56 moiety was still able to induce significant downregulation of MHC-I (P = 0.009), albeit the downregulation was less effective than the downregulation induced by parental Ab4G virus (Fig. 5C). Based on the results, we concluded that pUL56 can be translated from the second in-frame AUG, which results in a truncated but still functional protein, and that phosphorylation and the N-terminal 17 aa are not required for pUL56 to downregulate MHC-I. Whether production of truncated pUL56 also occurs in vivo remains to be determined.
Fig 5.

pUL56 contains an additional, functional in-frame start codon. (A) The upstream and N-terminal DNA sequence of ORF1 is shown. The start codon of the complete ORF1 and the second in-frame start codon are indicated in shadow. The proposed encoded amino acid sequences are shown as single-letter codes. The arrows indicate the initiation direction from the first or second start codon. (B) NBL6 cells were infected with Ab4G parental virus, Ab4GΔ11stATG, Ab4GΔ12ndATG, or Ab4GΔ1. At 16 hpi, cell lysates were treated or not treated with λ-PPase. Western blot analyses were performed to detect pUL56 using anti-EHV-1 UL56 pAbs or gM using the MAb F6. The MAb 13E5 was used to detect β-actin, which was included as a loading control. (C) NBL6 cells were either mock infected or infected with Ab4G, Ab4GΔ11stATG, Ab4GΔ12ndATG, or Ab4GΔ1, and at 16 hpi, cell surface expression of MHC-I was analyzed using flow cytometry.
pUL56 is not phosphorylated by viral US3 or UL13 kinases.
Alphaherpesviruses have two viral serine/threonine kinases, which are encoded by the US3 and UL13 genes, respectively (10, 28). Given that the US3-encoded kinases of PRV and VZV have been reported to contribute to MHC-I downregulation in certain cell types (9, 11) and EHV-1 pUL56 is a phosphoprotein, we asked if one of these two viral kinases phosphorylated pUL56 and if their expression may affect cell surface MHC-I levels. We therefore generated two kinase mutants, Ab4GΔS3 and Ab4GΔ13, by replacing the majority of the US3 gene (841 bp) or sequences encoding the predicted catalytic domain of UL13 (aa 221 to 380) with the kanamycin resistance gene (aphAI) (Fig. 6A). Infection of NBL6 cells with the generated mutants demonstrated that pUL56 was still phosphorylated (Fig. 6B) and that MHC-I downregulation was not affected by the deletion of US3 or UL13 (Fig. 6C). Taken together, the data indicated that pUL56 is not phosphorylated by either of the two viral kinases and that they do not have detectable contributions to the EHV-1-induced MHC-I downregulation in NBL6 cells.
Fig 6.
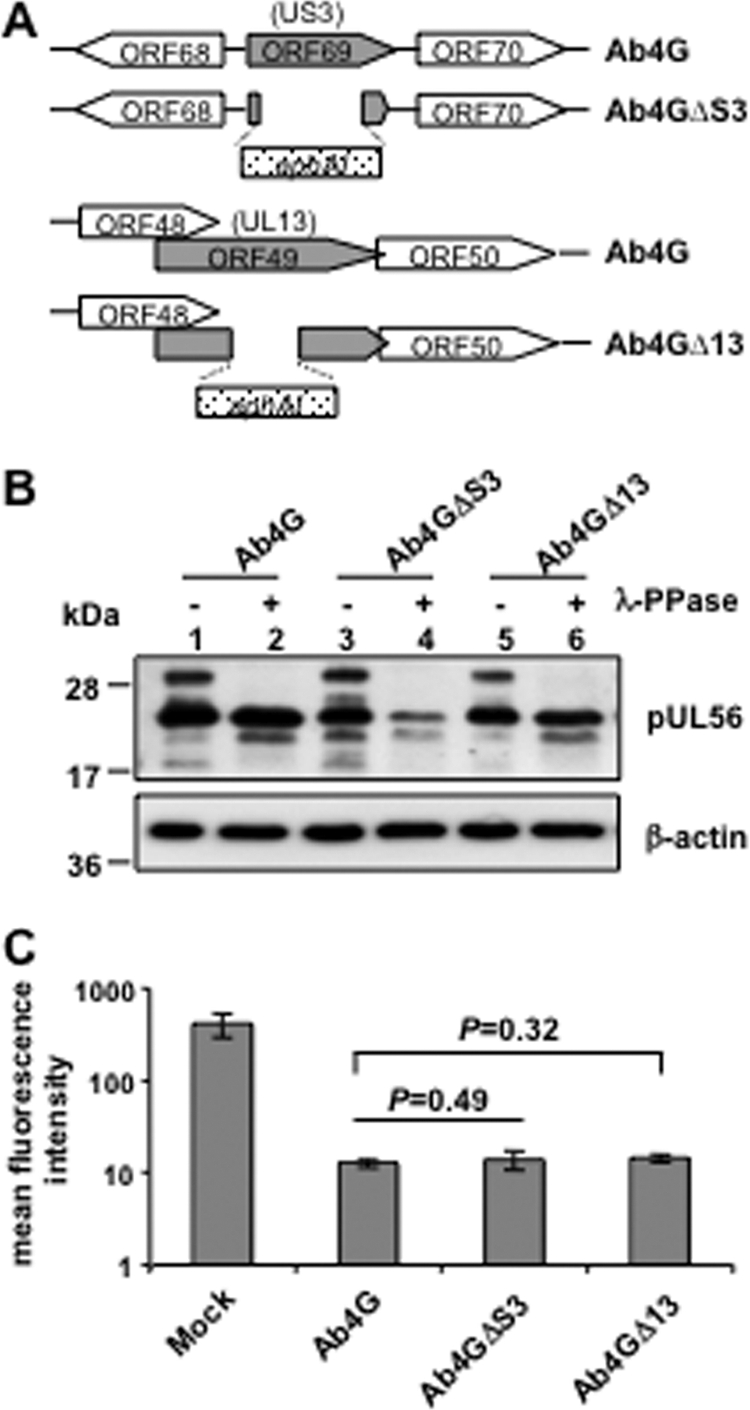
pUL56 is not phosphorylated by the viral protein kinase pUS3 or pUL13. (A) Schematic representation of the structure of US3 and UL13 in wild-type as well as in the deletion mutant viruses. The kanamycin resistance gene is indicated by aphAI (dotted box). (B) NBL6 cells were infected with Ab4G, Ab4GΔS3, or Ab4GΔ13. At 16 hpi, cell lysates were treated or not treated with λ-PPase. Western blotting was performed to detect pUL56 using anti-EHV-1 UL56 pAbs. The MAb 13E5 was used to detect β-actin, which was included as a loading control. (C) NBL6 cells were either mock infected or infected with Ab4G, Ab4GΔS3, or Ab4GΔ13, and at 16 hpi, cell surface expression of MHC-I was analyzed using flow cytometry.
pUL56 is predominantly localized in the Golgi complex, and its transmembrane domain is essential for subcellular localization and function.
The HSV-2 pUL56 homologue has previously been identified to be a type II transmembrane protein (20). Using the TMHMM server (version 2.0) program (http://www.cbs.dtu.dk/services/TMHMM), EHV-1 pUL56 was also predicted to represent a type II transmembrane protein (Fig. 7A). We therefore investigated the intracellular localization of the protein and the importance of the TM domain. To this end, an ORF1 mutant, Ab4GΔ1_TM, that lacked the sequence from aa 170 to aa 192, which was mapped to the TM domain, was created (Fig. 7A). Moreover, parental and mutant viruses were reconstituted such that the gp2 gene was repaired, resulting in egfp-negative viruses vAb4, vAb4Δ1_TM, and vAb4Δ1. NBL6 cells were first infected with vAb4 and fluorescent labels were used to visualize the Golgi complex or the ER. We found that pUL56 accumulated in the Golgi complex but not in the ER in cells infected with wild-type virus (Fig. 7B and C, panels a, b, and c). In contrast, pUL56 exhibited a diffuse appearance in the cytoplasm when the TM domain was deleted (Fig. 7B and C, panels d, e, and f), indicating that the TM domain is required for correct subcellular localization. vAb4Δ1-infected cells were included as a control and showed no pUL56 expression (Fig. 7B and C, panels g). In addition, flow cytometric analyses showed that MHC-I expression levels in cells infected with the TM mutant virus were identical to those in cells infected with Ab4GΔ1 (P = 0.8), the deletion mutant unable to express pUL56 (Fig. 7D). Western blot analyses showed that only unphosphorylated forms of pUL56 were expressed by the Ab4GΔ1_TM mutant (Fig. 7E, lanes 3 and 4). Taken together, our results show that pUL56 is predominantly localized in the Golgi complex and that the TM domain is essential for correct subcellular localization and protein function.
Fig 7.

pUL56 is predominantly localized in the Golgi complex, and its TM domain is essential for subcellular localization and function. (A) The amino acid sequence of the ORF1 gene product pUL56 is shown as single-letter codes. The predicted transmembrane domain (TMD) is indicated in the gray box. The structure of the TM deletion mutant is shown below. wt, wild type. (B) NBL6 cells were infected with the vAb4 parent or the ORF1 mutant vAb4Δ1 or vAb4Δ1_TM for 16 h. Immunofluorescence stainings of the Golgi compartment with BODIPYTR C5-ceramide complexed to BSA or the ER with BODIPYTR glibenclamide were performed. (C) NBL6 cells were either mock infected or infected with Ab4G, Ab4GΔ1, or Ab4GΔ1_TM, and at 16 hpi, cell surface expression of MHC-I was analyzed using flow cytometry. (D) NBL6 cells were infected with Ab4G or Ab4GΔ1_TM, and at 16 hpi, cell lysates were treated or not treated with λ-PPase. Western blot analyses were performed to detect pUL56 using anti-EHV-1 pUL56 pAbs. The MAb 13E5 was used to detect β-actin, which was included as a loading control.
DISCUSSION
Downregulation of cell surface MHC-I is an effective survival strategy for herpesviruses to escape from the host's immune response and helps establish lifelong persistence. For alphaherpesviruses, although they constitute the largest subfamily of the Herpesviridae, the viral proteins involved in MHC-I downregulation are less well understood. We here report that alphaherpesviral pUL56 is a novel immune evasion protein that allows efficient downregulation of cell surface MHC-I expression.
The UL56 gene family encodes tail-anchored membrane proteins and is present in all members of the Alphaherpesvirinae subfamily except BHV-1 and BHV-5 (40), indicating that the encoded proteins may share similar functions. EHV-1 pUL56, like its characterized counterparts, is dispensable for virus growth in vitro but was recently shown to attenuate Ab4 pathogenicity in vivo (35). These findings are in agreement with those in HSV-1 and HSV-2, where pUL56 is also dispensable for virus growth in vitro but contributes to pathogenicity in vivo (5, 17). It is unclear whether pUL56 orthologues in HSV-1 and HSV-2 are also responsible for the downregulation of cell surface MHC-I but may explain the prominent role of pUL56 in HSV-1 and HSV-2 pathogenicity.
It was reported previously that an early EHV-1 gene product, alone or in combination with other proteins, causes MHC-I downregulation (29). We here demonstrate that pUL56 is a phosphorylated early protein that plays a dominant role in the process. Surprisingly, we failed to observe MHC-I downregulation in transfection experiments with a vector expressing pUL56, indicating that pUL56 needs an additional viral early protein or virus-induced cellular partner(s) to completely fulfill its function. HSV-1 and HSV-2 pUL56 are known to be components of the viral envelope (16, 20), and we also demonstrated the presence of pUL56 in purified EHV-1 virions (data not shown). However, based on the fact that UV-inactivated EHV-1 is unable to downregulate MHC-I (29), we exclude the possibility that virion-associated pUL56 or other structural proteins are capable of downregulating MHC-I during virus entry, although MHC-I was shown to be one of the entry receptors for EHV-1 entry (33). Our results seem to suggest that only newly synthesized pUL56 functionally interacts with its partner(s). The pUL56 orthologue of HSV-2 has been reported to interact with pUL11, a viral tegument protein with dynamic membrane-trafficking properties (21, 24); KIF1A, a neuron-specific cellular kinesin (22); and Nedd4, a member of the E3 ubiquitin ligase family (39–41). In future studies, we will address potential interactors of EHV-1 pUL56, including viral and cellular proteins, and the consequences of such interactions.
We discovered that MHC-I downregulation in EHV-1-infected cells occurs independently of pUL49.5. The pUL49.5 orthologues of several members of the genus Varicellovirus, including EHV-1, have been identified to be inhibitors of TAP activity. Using pUL45.9-expressing cell lines, it was shown that pUL49.5 robustly inhibits peptide transport into the ER lumen and precludes migration of peptide-loaded MHC-I complexes to the cell surface (19). In a virus background, EHV-1 pUL49.5 was also shown to inhibit TAP, but the inhibitory effect was much milder and the consequences of pUL49.5-TAP interference on surface MHC-I expression remained elusive (19). It should be noted that a significant population of surface MHC-I in NBL6 cells remains stable for more than 24 h, even when the transport of newly synthesized MHC-I to the cell surface is blocked (29). This finding may explain that the dramatic downregulation of MHC-I at early times of infection is likely not caused by interference of pUL49.5 with TAP. Consistent with this speculation, we here provide evidence that pUL49.5 does not have a readily detectable effect on MHC-I levels on the cell surface during virus infection, at least at early times of infection. A possible explanation for the discrepancy in the results could be that the long-term effect of the pUL49.5-TAP interference is masked by the massive and early effects of pUL56. It is tempting to speculate that removal of MHC-I from the surface of EHV-1-infected cells is the consequence of a coordinated and functional cooperation of pUL56 and pUL49.5: in the early stages of infection, before most viral peptides are loaded onto and presented by MHC-I, pUL56 will remove the majority of the molecules from the cell surface. In a second step, pUL56 would cooperate with pUL49.5 in preventing MHC-I with bound viral peptides to present the viral cargo to CTLs.
It has been reported previously that cell surface MHC-I molecules are not completely removed by EHV-1, as shown with different antibodies used to detect expression, and the downregulation of MHC-I may also be locus or allele specific (29). Although, at first glance, incomplete downregulation may seem to be disadvantageous for the virus in its attempt to escape from CTLs, it will prevent the action of natural killer (NK) cells. Cell surface expression of MHC-I molecules provides inhibitory signals to NK cells, and modulation of MHC-I presentation could sensitize infected cells for NK-mediated cytotoxicity (25). Avoiding NK cell responses by selectively downregulating MHC-I alleles, which do not provide inhibitory signals, or by expressing nonfunctional MHC-I decoys is known for beta- and gammaherpesviruses (14), and similar functions may be at play during alphaherpesvirus infections as well.
Phosphorylation by protein kinases is a common posttranslational modification that regulates protein functions. Combining the facts that EHV-1 pUL56 is a phosphoprotein and is functional only in the virus context, we hypothesized that phosphorylation of pUL56 by a viral protein kinase might be required to fulfill its function. However, we found that even unphosphorylated and truncated pUL56 was still able to efficiently downregulate MHC-I, suggesting that phosphorylation is actually not essential for pUL56. In contrast to what has been reported for PRV and VZV (9, 11), neither of the viral kinases, pUS3 and pUL13, had any detectable effect on the downregulation of surface MHC-I in NBL6 cells. One possibility is that EHV-1 pUS3 might also play a role in delaying MHC-I maturation, as was described for the VZV pUS3 orthologue, the gene 66 product (11), an effect that would likely be undetectable in the early stages of infection. Alternatively, the function of EHV-1 pUS3 may be cell type dependent. Taken together, our data clearly show that pUL56 plays a dominant role in removing surface MHC-I in a fashion that is clearly independent of pUS3, pUL13, and pUL49.5.
We found that pUL56 is predominantly localized in the Golgi compartment and that subcellular localization is dependent on the presence of its TM domain. This intracellular localization is similar to what has been reported for HSV-2 pUL56. Indirect immunofluorescence showed that HSV-2 pUL56 was mainly located to Golgi and other cytoplasmic vesicles in infected cells (20). In contrast, pUL56 was not detected in the ER, which was somewhat anticipated because pUL56 does not contain a double-arginine ER retention motif at its N terminus (34). Furthermore, a significant amount of HSV-2 pUL56 was also shown to be associated with lipid rafts (20), the organizing centers for signal transduction and membrane trafficking (6). These findings indicate that alphaherpesviral pUL56 orthologues may play their role by sorting and trafficking of MHC-I. Rappocciolo et al. (29) suggested that EHV-1 infection may induce enhanced endocytosis of surface MHC-I, based on their observation that EHV-1 infection resulted in a significantly greater loss of surface MHC-I compared to that achieved by treatment of uninfected cells with inhibitors blocking either protein synthesis or transport of newly synthesized protein to the cell surface. Whether this interesting phenomenon is caused by pUL56 is yet to be elucidated.
In conclusion, we have identified a novel immune evasion protein encoded by the UL56 gene family of alphaherpesviruses. We demonstrate that pUL56 is a phosphorylated early protein and plays a dominant role in the downregulation of cell surface MHC-I in the early stages of infection. While independent of pUL49.5, pUS3, and pUL13, pUL56 by itself is unable to downregulate MHC-I. Our current work is focused on elucidating the underlying mechanism of pUL56-mediated MHC-I downregulation.
ACKNOWLEDGMENTS
We declare no conflict of interest.
G.M., S.F., and G.R.V.D.W. performed the experiments. G.M., N.O., and G.R.V.D.W. wrote the paper; G.M., N.O., and G.R.V.D.W. participated in the experimental design.
This work was supported by grant D08-EQ30 from the Morris Animal Foundation. G.R.V.D.W. is a postdoctoral fellow of the FWO Flanders.
We thank Leela Noronha, Cristina Stoyanov, Laura Goodman, Teng Huang, Nora Thormann, and Armando Damiani for their help with preliminary experiments and mutant virus construction.
Footnotes
Published ahead of print 25 January 2012
REFERENCES
- 1. Allen GP, Bryans JT. 1986. Molecular epizootiology, pathogenesis, and prophylaxis of equine herpesvirus-1 infections. Prog. Vet. Microbiol. Immunol. 2:78–144 [PubMed] [Google Scholar]
- 2. Ambagala APN, Gopinath RS, Srikumaran S. 2004. Peptide transport activity of the transporter associated with antigen processing (TAP) is inhibited by an early protein of equine herpesvirus-1. J. Gen. Virol. 85:349–353 [DOI] [PubMed] [Google Scholar]
- 3. Androlewicz MJ, Anderson KS, Cresswell P. 1993. Evidence that transporters associated with antigen processing translocate a major histocompatibility complex class I-binding peptide into the endoplasmic reticulum in an ATP-dependent manner. Proc. Natl. Acad. Sci. U. S. A. 90:9130–9134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baumeister J, Klupp BG, Mettenleiter TC. 1995. Pseudorabies virus and equine herpesvirus 1 share a nonessential gene which is absent in other herpesviruses and located adjacent to a highly conserved gene cluster. J. Virol. 69:5560–5567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berkowitz C, Moyal M, Rosen-Wolff A, Darai G, Becker Y. 1994. Herpes simplex virus type 1 (HSV-1) UL56 gene is involved in viral intraperitoneal pathogenicity to immunocompetent mice. Arch. Virol. 134:73–83 [DOI] [PubMed] [Google Scholar]
- 6. Brown DA, London E. 1998. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14:111–136 [DOI] [PubMed] [Google Scholar]
- 7. Bürckstümmer T, et al. 2006. An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat. Methods 3:1013–1019 [DOI] [PubMed] [Google Scholar]
- 8. Burki F, et al. 1990. Viremia and abortions are not prevented by two commercial equine herpesvirus-1 vaccines after experimental challenge of horses. Vet. Q. 12:80–86 [DOI] [PubMed] [Google Scholar]
- 9. Deruelle MJ, Van den Broeke C, Nauwynck HJ, Mettenleiter TC, Favoreel HW. 2009. Pseudorabies virus US3- and UL49.5-dependent and -independent downregulation of MHC I cell surface expression in different cell types. Virology 395:172–181 [DOI] [PubMed] [Google Scholar]
- 10. de Wind N, Domen J, Berns A. 1992. Herpesviruses encode an unusual protein-serine/threonine kinase which is nonessential for growth in cultured cells. J. Virol. 66:5200–5209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eisfeld AJ, Yee MB, Erazo A, Abendroth A, Kinchington PR. 2007. Downregulation of class I major histocompatibility complex surface expression by varicella-zoster virus involves open reading frame 66 protein kinase-dependent and -independent mechanisms. J. Virol. 81:9034–9049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Früh K, et al. 1995. A viral inhibitor of peptide transporters for antigen presentation. Nature 375:415–418 [DOI] [PubMed] [Google Scholar]
- 13. Goodman LB, et al. 2007. A point mutation in a herpesvirus polymerase determines neuropathogenicity. PLoS Pathog. 3:e160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Griffin BD, Verweij MC, Wiertz EJ. 2010. Herpesviruses and immunity: the art of evasion. Vet. Microbiol. 143:89–100 [DOI] [PubMed] [Google Scholar]
- 15. Hubert PH, Birkenmaier S, Rziha HJ, Osterrieder N. 1996. Alterations in the equine herpesvirus type-1 (EHV-1) strain RacH during attenuation. Zentralbl. Veterinarmed. B 43:1–14 [DOI] [PubMed] [Google Scholar]
- 16. Kehm R, Gelderblom HR, Darai G. 1998. Identification of the UL56 protein of herpes simplex virus type 1 within the virion by immuno electron microscopy. Virus Genes 17:49–53 [DOI] [PubMed] [Google Scholar]
- 17. Kehm R, Rösen-Wolff A, Darai G. 1996. Restitution of the UL56 gene expression of HSV-1 HFEM led to restoration of virulent phenotype; deletion of the amino acids 217 to 234 of the UL56 protein abrogates the virulent phenotype. Virus Res. 40:17–31 [DOI] [PubMed] [Google Scholar]
- 18. Koppers-Lalic D, et al. 2005. Varicelloviruses avoid T cell recognition by UL49.5-mediated inactivation of the transporter associated with antigen processing. Proc. Natl. Acad. Sci. U. S. A. 102:5144–5149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koppers-Lalic D, et al. 2008. Varicellovirus UL 49.5 proteins differentially affect the function of the transporter associated with antigen processing, TAP. PLoS Pathog. 4:e1000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koshizuka T, et al. 2002. Identification and characterization of the UL56 gene product of herpes simplex virus type 2. J. Virol. 76:6718–6728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koshizuka T, Kawaguchi Y, Goshima F, Mori I, Nishiyama Y. 2006. Association of two membrane proteins encoded by herpes simplex virus type 2, UL11 and UL56. Virus Genes 32:153–163 [DOI] [PubMed] [Google Scholar]
- 22. Koshizuka T, Kawaguchi Y, Nishiyama Y. 2005. Herpes simplex virus type 2 membrane protein UL56 associates with the kinesin motor protein KIF1A. J. Gen. Virol. 86:527–533 [DOI] [PubMed] [Google Scholar]
- 23. Kydd JH, Wattrang E, Hannant D. 2003. Pre-infection frequencies of equine herpesvirus-1 specific, cytotoxic T lymphocytes correlate with protection against abortion following experimental infection of pregnant mares. Vet. Immunol. Immunopathol. 96:207–217 [DOI] [PubMed] [Google Scholar]
- 24. Loomis JS, Bowzard JB, Courtney RJ, Wills JW. 2001. Intracellular trafficking of the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 75:12209–12219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marcenaro E, et al. 2011. NK cells and their receptors during viral infections. Immunotherapy 3:1075–1086 [DOI] [PubMed] [Google Scholar]
- 26. Mumford JA, et al. 1987. Serological and virological investigations of an equid herpesvirus 1 (EHV-1) abortion storm on a stud farm in 1985. J. Reprod. Fertil. Suppl. 35:509–518 [PubMed] [Google Scholar]
- 27. Ortmann B, Androlewicz MJ, Cresswell P. 1994. MHC class I/beta 2-microglobulin complexes associate with TAP transporters before peptide binding. Nature 368:864–867 [DOI] [PubMed] [Google Scholar]
- 28. Purves FC, Longnecker RM, Leader DP, Roizman B. 1987. Herpes simplex virus 1 protein kinase is encoded by open reading frame US3 which is not essential for virus growth in cell culture. J. Virol. 61:2896–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rappocciolo G, Birch J, Ellis SA. 2003. Down-regulation of MHC class I expression by equine herpesvirus-1. J. Gen. Virol. 84:293–300 [DOI] [PubMed] [Google Scholar]
- 30. Rudolph J, O'Callaghan DJ, Osterrieder N. 2002. Cloning of genomes of equine herpesvirus type 1 (EHV-1) strains KyA and RacL11 as bacterial artificial chromosomes (BAC). J. Vet. Med. B Infect. Dis. Vet. Public Health 49:31–36 [DOI] [PubMed] [Google Scholar]
- 31. Rudolph J, Osterrieder N. 2002. Equine herpesvirus type 1 devoid of gM and gp2 is severely impaired in virus egress but not direct cell-to-cell spread. Virology 293:356–367 [DOI] [PubMed] [Google Scholar]
- 32. Rudolph J, Seyboldt C, Granzow H, Osterrieder N. 2002. The gene 10 (UL49.5) product of equine herpesvirus 1 is necessary and sufficient for functional processing of glycoprotein M. J. Virol. 76:2952–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sasaki M, et al. 2011. Equine major histocompatibility complex class I molecules act as entry receptors that bind to equine herpesvirus-1 glycoprotein D. Genes Cells 16:343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schutze MP, Peterson PA, Jackson MR. 1994. An N-terminal double-arginine motif maintains type II membrane proteins in the endoplasmic reticulum. EMBO J. 13:1696–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Soboll Hussey G, et al. 2011. Evaluation of immune responses following infection of ponies with an EHV-1 ORF1/2 deletion mutant. Vet. Res. 42:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tai SH, et al. 2010. Complete genomic sequence and an infectious BAC clone of feline herpesvirus-1 (FHV-1). Virology 401:215–227 [DOI] [PubMed] [Google Scholar]
- 37. Telford EA, Watson MS, Perry J, Cullinane AA, Davison AJ. 1998. The DNA sequence of equine herpesvirus-4. J. Gen. Virol. 79(Pt 5):1197–1203 [DOI] [PubMed] [Google Scholar]
- 38. Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197 [DOI] [PubMed] [Google Scholar]
- 39. Ushijima Y, Goshima F, Kimura H, Nishiyama Y. 2009. Herpes simplex virus type 2 tegument protein UL56 relocalizes ubiquitin ligase Nedd4 and has a role in transport and/or release of virions. Virol. J. 6:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ushijima Y, Koshizuka T, Goshima F, Kimura H, Nishiyama Y. 2008. Herpes simplex virus type 2 UL56 interacts with the ubiquitin ligase Nedd4 and increases its ubiquitination. J. Virol. 82:5220–5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ushijima Y, et al. 2010. Herpes simplex virus UL56 interacts with and regulates the Nedd4-family ubiquitin ligase Itch. Virol. J. 7:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. van der Meulen KM, Vercauteren G, Nauwynck HJ, Pensaert MB. 2003. A local epidemic of equine herpesvirus 1-induced neurological disorders in Belgium. Flemish Vet. J. 72:366–372 [Google Scholar]
- 43. Van de Walle GR, et al. 2009. A single-nucleotide polymorphism in a herpesvirus DNA polymerase is sufficient to cause lethal neurological disease. J. Infect. Dis. 200:20–25 [DOI] [PubMed] [Google Scholar]
- 44. Verweij MC, et al. 2011. The capacity of UL49.5 proteins to inhibit TAP is widely distributed among members of the genus Varicellovirus. J. Virol. 85:2351–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. von Einem J, et al. 2004. The truncated form of glycoprotein gp2 of equine herpesvirus 1 (EHV-1) vaccine strain KyA is not functionally equivalent to full-length gp2 encoded by EHV-1 wild-type strain RacL11. J. Virol. 78:3003–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. York IA, Rock KL. 1996. Antigen processing and presentation by the class I major histocompatibility complex. Annu. Rev. Immunol. 14:369–396 [DOI] [PubMed] [Google Scholar]