

Abstract
The efficient transmission of alphaviruses requires the establishment of a persistent infection in the arthropod vector; however, the nature of the virus-arthropod host interaction is not well understood. The phosphatidylinositol 3-kinase (PI3K)-Akt-TOR pathway is a signaling pathway with which viruses interact to manipulate cellular functions. The viral activation of this pathway can enhance translation and inhibit apoptosis, potentially promoting viral replication; conversely, repression can enhance cell death. Using a system to study Sindbis virus RNA replication in Drosophila melanogaster, we found that the overexpression of Akt enhanced Sindbis virus replication. In contrast, a decrease in viral replication was observed for flies hypomorphic for the Akt gene. Infection of cultured Drosophila cells led to the phosphorylation and activation of Akt. The chemical inhibition of PI3K, Akt, and TOR in mosquito cells reduced virus replication, suggesting that this pathway is proviral. Early after infection, there was an increase in the TOR-dependent phosphorylation of 4E-BP1 in mosquito cells and a consequent increase in the translation of a capped reporter mRNA. In contrast, no change in 4E-BP1 phosphorylation was seen in mammalian cells, and the level of translation of the reporter decreased following infection. Finally, we found that the increase in the phosphorylation of 4E-BP1 was stimulated by replicon RNA but not by UV-inactivated virus. Our data indicate that Sindbis virus replication complex formation in mosquito cells activates the PI3K-Akt-TOR pathway, causing the phosphorylation of 4E-BP1 and increasing the formation of eukaryotic initiation factor 4F (eIF4F), which promote cap-dependent translation. This virus-induced increase in cap-dependent translation allows the efficient translation of viral mRNA while minimizing the burden on the cell.
INTRODUCTION
Alphaviruses are arthropod-borne (arboviruses), positive-sense, single-stranded RNA viruses with mosquitoes as their most common vector. Mosquitoes become infected when they feed on vertebrates infected with these viruses. The virus amplifies in the mosquito and can be spread to other vertebrates when they feed. Symptoms of infection range from a mild fever to encephalitis and death, depending on the viral species. Currently, no vaccines are available for the treatment of these viruses, the control measures for prevention are expensive, and no single surveillance measure would be sufficient for all ecological zones and viruses. These factors together illustrate a pressing need for a more thorough understanding of how the virus interacts with its hosts in order to facilitate the development of transmission interventions in the future.
Numerous viruses inhibit host cell translation, usually by interfering with cap-dependent initiation, and employ various mechanisms to promote the translation of their own mRNA. Key eukaryotic initiation factors (eIFs) are either sequestered or modified so as to enable efficient viral mRNA translation and simultaneously curb host translation (7, 39, 45, 48). The cap-binding complex (eIF4F) is required for recruiting ribosomes to mRNAs, and several viruses, such as encephalomyocarditis virus, vesicular stomatitis virus (VSV), and adenovirus, target the eIF4E component of this complex to hinder translation (8, 18, 25). There is growing evidence that alphavirus RNA can be efficiently translated in vertebrate cells in which eIFs are limiting, suggesting that they inhibit host translation and promote viral translation at the initiation step (50). The shutoff of vertebrate host gene expression at the levels of translation and also transcription results in the inhibition of the antiviral response, cytopathology, and, ultimately, cell death (19).
The nature of infection in the arthropod vector, on the other hand, is very different. Once a mosquito ingests a blood meal of an infected vertebrate, the virus spreads in the vector in a stepwise manner. After establishing a productive infection in the midgut epithelium, it escapes to secondary target organs like the salivary glands, where it establishes a persistent infection. A subsequent bite by this mosquito may lead to the transmission of the virus through shedding in mosquito saliva. This persistent infection is recapitulated in cultured mosquito cells, although a degree of variation occurs with regard to cytopathology and viral yield, depending on the cell type (33). In most cases, the virus is produced at high levels in the first 24 to 48 h, after which there is a decline coincident with the establishment of persistence; this is in contrast to the cytolytic infection that occurs in most vertebrate cells (47).
As the virus does not vary genetically in a host-dependent manner, it is apparent that the outcome of infection is determined by the host cell environment and the manner in which the virus interacts with the machinery of the different host cells (1). While specific proteins have been identified to interact with viral components (6, 10, 20, 35, 37), it is still not clear what enables alphaviruses to persistently infect arthropod cells without causing fatal damage.
Many viruses modulate signaling pathways to aid in virus replication (9, 40). The phosphatidylinositol 3-kinase (PI3K)-Akt pathway is a well-characterized signaling pathway that plays an important role in the regulation of cell survival and proliferation (23). This pathway is also highly conserved among different species, including Drosophila melanogaster, Caenorhabditis elegans, and mammals. Signaling through this pathway is initiated by the binding of a hormone or growth factor like insulin or epidermal growth factor (EGF) at the receptor tyrosine kinase (RTK). The activation of RTK leads to the binding and, hence, activation of PI3K, which in turn leads to the conversion of phosphatidyl-inositol-4,5-bisphosphate (PIP2) to its phosphatidylinositol-3,4,5-triphosphate (PIP3) form. PIP3 recruits both Akt and phosphoinositide-dependent protein kinase 1 (PDK1) to the membrane and positions them such that PDK1 can phosphorylate Akt at Thr308, leading to Akt activation (5, 13).
A downstream effect of activated Akt is the activation of the TOR kinase. TOR kinase is a component of the TORC1 protein complex. The TOR signaling pathway has been extensively studied for its role in the control of cap-dependent translation (5, 44). When TOR is active, it phosphorylates eIF4E-binding protein (4E-BP1) (22). The phosphorylation of 4E-BP1 leads to its disassociation from eIF4E, allowing the binding of eIF4G, resulting in a functional eIF4F complex, and facilitating the initiation of translation.
Accumulating evidence indicates that many RNA and DNA viruses coopt the PI3K-Akt signaling pathway, and for some of these viruses, its activity is critical for their propagation (5, 9, 13, 14, 21, 28, 30, 32, 36, 43). However, the role of virus-mediated PI3K-Akt signaling and its connection to alphavirus infection of arthropods are not well understood. In the present study, we take advantage of a system established in our laboratory to study the replication of the type species of the Alphavirus genus, Sindbis virus (SINV), in Drosophila melanogaster (2). Using a transgenic fly line capable of launching/hosting an autonomously replicating SINV replicon RNA, we have employed host genetic analyses to determine the importance of the PI3K-Akt-TOR pathway during virus replication in an arthropod host. The SINV replicon contains the SINV nonstructural protein (nsP) genes along with the 5′-untranslated region (5′-UTR), the 3′-UTR, and a promoter for the production of subgenomic mRNA, which encodes green fluorescent protein (GFP) in place of the structural proteins. This replicon RNA is translated in a cap-dependent fashion to produce the viral nsPs (nsP1, nsP2, nsP3, and nsP4) comprising the viral RNA synthetic complex. This complex is responsible for viral RNA synthesis. During replicon replication, the subgenomic mRNA encoding GFP is produced, which serves as a reporter for replication.
When the SINV replicon fly was crossed with fly lines carrying mutations in the Akt signaling pathway, replication was altered significantly. Mutations inhibiting the pathway reduced replication, whereas the overexpression of Akt led to increased replication. This indicated that this pathway is proviral. Furthermore, a decrease in SINV replication was seen in cultured mosquito cells when PI3K, Akt, and TOR were chemically inhibited. Levels of phosphorylated 4E-BP1 were increased as a consequence of infection in mosquito cells. These findings demonstrated that PI3K-Akt-TOR activation post-SINV infection caused an increased phosphorylation of 4E-BP1, hence freeing eIF4E. The observed 3- to 4-fold increase in cap-dependent translation in infected mosquito cells supports the model that an increase in levels of available eIF4E enables the efficient translation of viral mRNA without a detrimental disruption of cellular function, possibly aiding the establishment of persistence. Finally, we determined that the increase in the phosphorylation of 4E-BP1 could be induced by viral replicon RNA, indicating that the activation of the P13K-Akt-TOR pathway was not dependent on the functions of the viral structural proteins but was probably due to the formation of membrane-associated replication complexes.
MATERIALS AND METHODS
Cells and virus.
Mosquito C6/36 cells (American Type Culture Collection, VA) and BHK-21 cells (American Type Culture Collection, Rockville, MD) were propagated in Eagle minimum essential medium (EMEM) with Earle's balanced salt solution supplemented with 10% fetal bovine serum (FBS) and 0.1% penicillin-streptomycin. Drosophila BG2c2 cells were obtained from the Drosophila Genomics Resource Centre (DGRC), Indiana University, Bloomington, IN. The cells were propagated in M3+Bacto peptone-yeast extract medium supplemented with 10% heat-inactivated FBS and 10 μg/ml insulin. BHK-21 cells were cultured at 37°C in 5% CO2. Mosquito cell cultures were kept under similar conditions, with the exception that the temperature of incubation was 28°C.
SINV was generated by the transfection of BHK-21 cells with RNA transcribed in vitro from pToto1101 (41). The titer of the virus was determined by a plaque assay on BHK cells.
Real-time quantitative reverse transcription-PCR (qRT-PCR) analysis.
RNA was extracted by homogenizing flies or cells in TRIzol reagent (Invitrogen). cDNA was made by using an AffinityScript QPCR cDNA synthesis kit (Stratagene), and PCR amplification was done by using Brilliant II SYBR green QPCR master mix (Stratagene) according to the manufacturer's protocol. Gene expression was normalized to 18S rRNA expression. The comparative threshold cycle (CT) method was used to determine fold changes in levels of transcript present in samples. The oligonucleotides used for the analysis were 5′-GGTTACACACAATAGCGAGGGCTT-3′ (nsP1 forward), 5′-TGGTGTTCCTGTTAGTCCTACCGT-3′ (nsp1 reverse), 5′-CGAAAGTTAGAGGTTCGAAGGCGA-3′ (18S forward-flies), 5′-CCGTGTTGAGTCAAATTAAGCCGC-3′ (18s reverse-flies), 5′-AGCCCAGCTGCTATTACCTTGAAC-3′ (18S forward-C6/36), and 5′-ACGACGGTCTACGAATTTCACCTC-3′ (18s reverse-C6/36).
RNA was harvested from transgenic flies at 3 days posteclosion. To analyze virus infection, flies were intrathoracically injected with 200 PFU of Sindbis virus at 2 days posteclosion, and RNA was harvested at 5 days postinfection.
Western blot analysis.
Samples from whole-cell extracts of uninfected or infected (6 h) C6/36 and BG2c2 cells were separated by SDS-PAGE (8 to 12% polyacrylamide) and transferred onto a nitrocellulose membrane. Blots were blocked and probed in Tris-buffered saline (TBS) with 5% nonfat dry milk and 0.1% Tween 20. Blots were probed with rabbit anti-actin antibody (Sigma), rabbit anti-phospho-Drosophila Akt (Ser505) antibody, rabbit anti-phospho-4E-BP1 (p4E-BP1) (Thr37/46) monoclonal antibody (236B4), rabbit anti-glycogen synthase kinase 3β (GSK-3β) (27C10) monoclonal antibody, rabbit anti-phospho-GSK-3β (pGSK-3β) (Ser9) antibody (Cell Signaling Technologies), and rabbit anti-4E-BP1 antibody (kind gift of Nahum Sonnenberg), followed by an anti-rabbit goat antiserum conjugated to horseradish peroxidase (HRP). Bands were visualized by chemiluminescence (SuperSignal West Pico; Pierce). Quantification was performed by using ImageQuant software.
Inhibitor studies.
Akt inhibitor VIII (isozyme selective, Akt1/2) was obtained from EMD Chemicals, and the PI3K inhibitor (LY-294,002 hydrochloride) was obtained from Sigma. The TOR inhibitors rapamycin and Torin1 were obtained from LC Laboratories and Tocris Biosciences, respectively. For the inhibitor studies, the relevant inhibitor was administered 1 h prior to virus infection, and cells were all harvested at 6 h postinfection (hpi) for RNA extraction. The doses used for the three inhibitors were as follows: the Akt inhibitor at 10 μM, the PI3K inhibitor at 10 μM, and the TOR inhibitors rapamycin and Torin1 at 5 μM.
Plasmids and RNA transcription.
Firefly luciferase (FFluc)-encoding mRNA was produced from a pUC19-derived plasmid possessing a T7 promoter followed by a short nonviral 5′-UTR, the luciferase open reading frame (ORF), a short 3′-UTR, and a poly(A) sequence. The SINrep/GFP plasmid was generated by the removal of lacZ from the pSINrep5LacZ plasmid by XbaI/SphI digestion and replacement with a PCR product encoding GFP (4). Both templates were linearized with XhoI (New England BioLabs [NEB]) and transcribed in the presence of the RNA cap analog mGpppG (NEB) with T7 or Sp6 RNA polymerase (NEB), followed by DNase treatment for 15 min.
Luciferase gene reporter assay.
For the assay described in the legend of Fig. 6A, C6/36 cells were first transfected with the in vitro-transcribed RNA (described above) using Lipofectamine 2000 for 2 h. This step was followed by a rinse with phosphate-buffered saline (PBS) and infection or mock infection with SINV at a multiplicity of infection (MOI) of 10 PFU/cell for 2 h. After infection, the cells were rinsed and lysed in cell culture lysis buffer (Promega), and the luciferase activities were assayed, as suggested by the manufacturer's protocol (Promega), in half-area opaque 96-well plates (Corning). Luciferase assay reagent (Promega) was added just before reading with a BioTek Synergy 2 plate reader.
Fig 6.

Increase in cap-dependent translation in SINV-infected mosquito cells. (A) Translation of a firefly luciferase reporter gene in infected and mock-infected C6/36 cells. Cells were transfected with a capped and polyadenylated RNA encoding firefly luciferase for 2 h and then mock infected or infected with SINV (MOI = 10 PFU/ml) for 2 h prior to harvesting. The value obtained from the control mock-infected cells was set at 100%. (B) Luciferase assay results for control mock-infected cells and cells that were infected for 4 h and then transfected with a capped and polyadenylated RNA encoding firefly luciferase and harvested 4 h later. The value obtained for the control mock-infected cells was set at 100%. (C) Luciferase activity in flies hosting SINV RNA replication and expressing firefly luciferase under the control of the GAL4-UAS system (UAS-Luc;Act5C-GAL4 UAS-SINrep/GFP). Control flies also expressed a nonviral transgene (UAS-Luc;Act5C-GAL4 UAS-GFP). Data shown are representative of three independent experiments. Error bars represent SD. Statistical analysis was performed with Student's t tests. **, P < 0.01 compared to the controls; ***, P < 0.001 compared to the controls.
For the assay described in the legend of Fig. 6B, BHK-21 and C6/36 cells in 6-well dishes were infected at an MOI of 10 PFU/cell. At 4 hpi, cells were rinsed and transfected with 1 μg of in vitro-transcribed luciferase mRNA. At 8 hpi, cells were rinsed and lysed in cell culture lysis buffer (Promega), and the luciferase activities were assayed, as suggested by the manufacturer's protocol (Promega), in half-area opaque 96-well plates (Corning). Luciferase assay reagent (Promega) was added just before reading with a BioTek Synergy 2 plate reader.
UV inactivation of virus.
UV inactivation of SINV was performed by exposing the virus to a germicidal lamp (wavelength, 254 nm) at a distance of 5 cm for 30 min at 4°C as described previously (26). Inactivation was confirmed by performing plaque assays on BHK-21 cells.
Microscopy and imaging of flies.
Live flies were anesthetized with CO2 and viewed under on an Olympus SZX16 dissecting microscope. Photographs were taken by using an Olympus DP72 camera. The software used was cellSens Entry, version 1.3 (build 7990), from Olympus Corporation.
Fly strains.
Fly lines were obtained from the Bloomington Stock Center or were generated previously in our laboratory (2). UAS-Luc flies (second chromosome) were provided by Norbert Perrimon (Harvard Medical School). Fly stocks were raised on standard cornmeal-agar medium at 25°C.
The fly lines were as follows: Akt hypomorph, ry506 P{ry[+t7.2]=PZ}Akt104226/TM3 ry[RK] Sb[1] Ser[1]; Akt overexpression 8191, y1 w1118;P{w[+mC]=UAS-Akt1.Exel}2; Akt overexpression 8192, y1 w1118;P{w[+mC]=UAS-Akt1.Exel}1; GFP expression, w1118 UAS-GFP; Sindbis virus replicon, w1118;Act5C-GAL4,UAS-SINrep/GFP; luciferase expression, y1 w1118;att P40 UAS-Luc.
Nucleotide sequence accession numbers.
The FlyBase (http://flybase.org/) accession numbers for the genes used in this article are CG4006 for the Akt gene and CG4141 for the PI3K gene.
RESULTS
Altered levels of Akt affect SINV RNA replication.
Recent studies have shown that numerous viruses are dependent on the Akt signaling pathway for efficient replication (9). To determine if this pathway played a role in SINV replication, we crossed a fly line hosting SINV replicon (Act5C-GAL4, UAS-SINrep/GFP) with two fly lines overexpressing Akt (UAS-Akt1 8191 and 8192; see Materials and Methods). The overexpression of Akt resulted in significantly increased levels of virus-dependent GFP expression compared to the wild-type fly possessing the SINV replicon, indicating increased viral replication (Fig. 1A). This increase in replication observed by GFP fluorescence was confirmed by qRT-PCR of nsP1-encoding viral genomic RNA (Fig. 1B). The level of viral genomic RNA in the flies overexpressing Akt, 8191 and 8192, were approximately 4- to 6-fold higher than that in the control fly. These flies were analyzed at 3 days posteclosion.
Fig 1.

The level of alphaviral RNA accumulation is higher in flies overexpressing Akt. (A) Bright-field and fluorescent images of wild-type SINV replicon flies, SINV replicon flies with the control transgene (GFP), and SINV replicon flies overexpressing Akt (UAS-Akt1). (B) SINV replication was measured by real-time qRT-PCR analysis of the nsP1 region of the viral genome in control SINV replicon flies with the control transgene (GFP) and flies overexpressing Akt (UAS-Akt1). These values were normalized to 18S rRNA values, and the value obtained for control SINV replicon flies was considered to be 1. Data shown are representative of three independent experiments. Error bars represent standard deviations (SD). Statistical analysis was performed with Student's t tests. *, P < 0.05 compared to SINV replicon flies expressing the control transgene.
In order to control for the enhancer dilution, i.e., the presence of two sets of upstream activation sequences (UASs) in the SINV/Akt-overexpressing fly, the control fly for the qRT-PCR quantification of viral RNA possessed a second set of UAS element upstream of a nonviral transgene. For this fly, the transgene was the GFP gene; however, this in no way complicates the interpretation of the data. As shown in Fig. 1A, the overexpression of Akt (8192) causes an increase in virus-dependent GFP expression compared to expression in the control fly, in which an extra copy of GFP is being expressed (Act5c-GAL4, UAS-SINrep/GFP;UAS-GFP). The detection of increased nsP1-encoding RNA levels further confirms these findings.
To determine if the converse was also true and if levels of SINV replication decreased as a consequence of a reduced level of expression of Akt, the SINV replicon fly was crossed to a fly in which Akt gene expression was disrupted due to a transposon insertion (Akt104226) (46). The reduction of Akt expression levels led to approximately a 50% decrease in viral RNA synthesis compared to the control wild-type SINV replicon fly when measured at 3 days posteclosion (Fig. 2A).
Fig 2.

Decreased levels of viral replication in Akt mutant flies. (A) Real-time qRT-PCR analysis of nsP1-encoding RNA in control SINV replicon flies and SINV replicon flies heterozygous for the Akt1 (akt104226) hypomorphic mutation. These values were normalized to 18S rRNA values, and the value obtained for control SINV replicon flies was considered to be 1. (B) Following the intrathoracic infection of w1118 flies and flies heterozygous for the Akt1 (akt104226) hypomorphic mutation, SINV replication was measured by real-time qRT-PCR analysis of nsP1-encoding RNA. These values were normalized to 18S rRNA values, and the value obtained for control w1118 flies was considered to be 1. Error bars represent SD. Data shown are representative of three independent experiments. Statistical analysis was performed with Student's t tests. *, P < 0.05 compared to control SINV replicon flies; **, P < 0.01 compared to a control w1118 fly.
To confirm the proviral role of Akt during SINV infection in insects, we infected the mutant fly hypomorphic for the Akt gene (Akt104226) intrathoracically with 200 PFU of SINV at 2 days posteclosion. Viral loads were determined by qRT-PCR quantification of viral replicon RNA containing the nsP1 sequence. At 5 days postinfection, viral replication was reduced by approximately 80% in Akt mutant flies compared to flies of the wild type for Akt (Fig. 2B). These data demonstrate a consistent proviral role for Akt using multiple alleles in multiple fly lines.
SINV infection leads to increased levels of phosphorylated Akt in Drosophila cells.
The results described above suggested that Akt is proviral. To test whether virus infection led to Akt activation, infected and uninfected Drosophila cells (BG2c2) were harvested at 6 hpi, and using anti-phospho-Drosophila Akt antibody, the levels of phosphorylated Akt in the whole-cell lysate were examined by Western blotting. Levels of phosphorylated Akt were increased in the infected cells compared to the levels in uninfected cells, indicating the activation of Akt by 6 h following infection with SINV (Fig. 3A). This site of activation (Ser505) is homologous to mammalian Ser473, and the phosphorylation of this residue was previously shown to be important for activation (12, 24).
Fig 3.
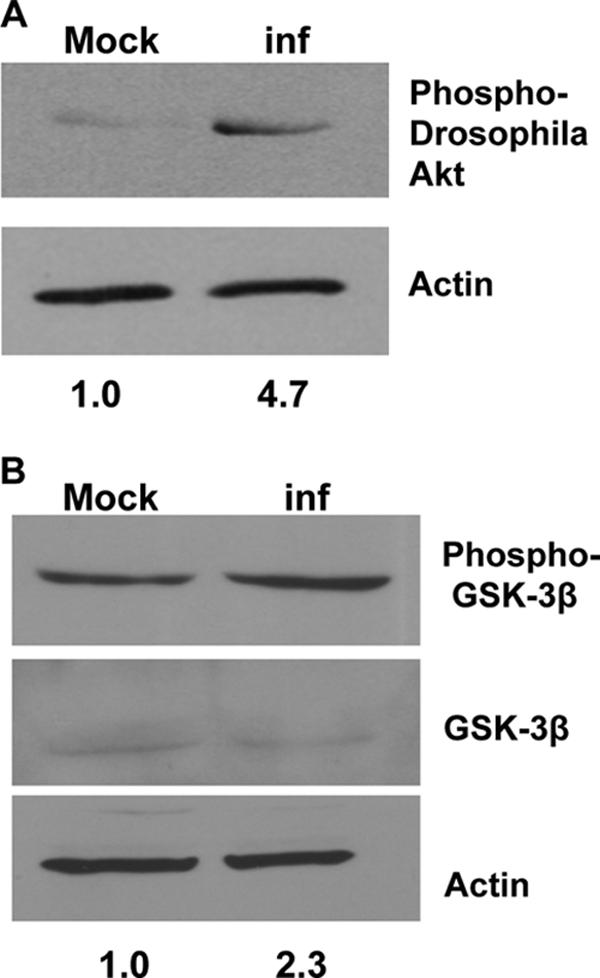
SINV infection activates Akt and GSK-3 phosphorylation in Drosophila cells. (A) Western blot with anti-phospho-Drosophila Akt. Relative quantities of phospho-Akt are given standardized to the actin loading control. (B) Anti-phospho-GSK3 and anti-GSK-3 antibodies on whole-cell extracts of BG2c2 cells infected with SINV. Relative quantities of phospho-GSK-3 are shown standardized to the total amount of GSK-3. BG2c2 cells were mock infected or infected (inf) at an MOI of 10 PFU/cell for 6 h, and whole-cell lysates were made and probed with the relevant antibody. β-Actin was used as the loading control.
To confirm the increase in levels of activated Akt, we examined the phosphorylation of glycogen synthase kinase 3β (GSK-3β) on serine 9; this site is a direct target of Akt (12). GSK-3β is a known downstream substrate for Akt, and its phosphorylation would verify Akt activation. We observed that the increase in levels of phospho-Akt correlated with an increase in levels of phospho-GSK-3β, confirming that Akt activity is increased in cells infected with SINV (Fig. 3B).
PI3K-Akt-TOR pathway inhibition in mosquito cells.
Results from transgenic flies showed that a reduction in the level of Akt inhibited viral RNA synthesis and that the infection of Drosophila cells led to Akt activation. To extend these studies and examine the effects of the pathway on virus production in a natural host cell, growth curves were performed for mosquito cells (C6/36) in which Akt was chemically inhibited. Cells were treated with Akt inhibitor VIII (3, 11) 1 h prior to infection. At each time point, the medium was removed from the cells for viral titer estimations, and fresh medium supplemented with the inhibitor was added back to the cells. Viral titers for each time point were determined. Viral growth kinetics (Fig. 4A) indicated that the inhibition of Akt led to a decrease in virus production compared to growth in control cells. This trend began at approximately 8 h postinfection and was most prominent at around 24 to 32 h of infection. Significant cell death was not observed when the cells were monitored visually throughout infection in the absence of the inhibitor; however, a cytopathic effect became apparent in infected cells treated with the inhibitor (Fig. 4B). The observation of the cytopathic effect following the infection of mosquito cells in which the pathway was inhibited provides evidence that the pathway is important not only for virus replication but possibly also for cell survival after infection. The inhibitors did not appear to be toxic to the cells, as shown in Fig. 4B. Following 48 h of treatment with the inhibitor, uninfected cells did not display obvious signs of cytotoxicity. While the cell number seemed to be reduced in the presence of the inhibitor, this was probably a consequence of a reduction in cell division.
Fig 4.

PI3K-Akt-TOR inhibition leads to decreased viral replication in mosquito cells. (A) Growth curve indicating viral titers over time from C6/36 cells infected with SINV at an MOI of 10 PFU/cell compared to cells infected with SINV and undergoing Akt inhibitor VIII treatment. Cells were treated with the inhibitor (inh) for 1 h prior to infection. Medium was removed at each time point thereafter for viral titer estimations, and fresh medium containing the inhibitor was added to ensure the constant presence of the inhibitor. The viral titers are expressed as PFU or PFU/ml. Titers are average titers from two independent experiments. (B) Phase-contrast microscopy analysis of C6/36 cells mock infected, infected with SINV, mock infected undergoing Akt inhibitor treatment, and infected with SINV and undergoing Akt inhibitor treatment. All these images were taken at 48 h postinfection. (C) Real-time qRT-PCR analysis of nsP1 mRNA in control C6/36 cells infected with SINV for 6 h and C6/36 cells treated with Akt inhibitor VIII, the PI3K inhibitor, and the TOR inhibitor for 1 h, followed by SINV infection for 6 h. These values were normalized to 18S rRNA values, and the value obtained for control C6/36 cells infected for 6 h was considered to be 1. Data are representative of three independent experiments. Error bars represent SD. Statistical analysis was performed with Student's t tests. *, P < 0.05 compared to infected cells without the inhibitor; ***, P < 0.001 compared to infected cells without the inhibitor. (D) Virus growth in C6/36 cells treated with the TOR inhibitor rapamycin and control dimethyl sulfoxide (DMSO) treatment, performed as described above for panel A.
Further examinations of the effect of Akt inhibition on viral replication in mosquito cells were done by qRT-PCR to detect nsP1-encoding RNA in mosquito cells infected with SINV for 6 h, treated with and without the Akt inhibitor. The results indicated a 60 to 70% drop in the levels of viral replication in the cells treated with the Akt inhibitor (Fig. 4C). Hence, the chemical inhibition of Akt in mosquito cells led to a decrease in viral replication as well as a decrease in viral titers, and this seemed to begin early in infection.
Similar inhibition experiments with PI3K and TOR, signaling proteins upstream and downstream of Akt, indicated a drop of 40 to 50% in the levels of viral RNA in cells treated with the inhibitors compared to the control cells (Fig. 4C). A growth curve for C6/36 cells with the TOR inhibitor indicated a decrease of 2 logs in viral titers at around 24 to 32 h (Fig. 4D), with the decrease in virus production beginning early in infection. From these data, we concluded that the PI3K-Akt-TOR pathway is an important regulator of viral replication in mosquito cells as well as Drosophila.
As a reduction in viral RNA levels was observed early (6 h) in infection when the pathway was inhibited, we believe that it is unlikely that the cell death observed later in infection following pathway inhibition accounts entirely for the decrease in virus production shown in Fig. 4A and D. Rather, we conclude that since viral genome levels are reduced by pathway inhibition, the reduction in virus production is independent of cell death. Hence, the PI3K-Akt-TOR pathway is an important factor for efficient virus replication.
4E-BP1 is phosphorylated following SINV infection.
TOR lies downstream of Akt and upstream of 4E-BP1 and regulates its phosphorylation. If TOR is activated during SINV infection through the action of PI3K-Akt, it is probable that 4E-BP1 will be phosphorylated (22). The phosphorylation of 4E-BP1 leads to its release from eukaryotic translation initiation factor 4E (eIF4E) and, hence, increased cap-dependent translation.
Western blotting with anti-p4E-BP1 antibody showed that the levels of phosphorylated 4E-BP1 (Thr37/Thr46) were increased in SINV-infected C6/36 cells compared to levels in mock-infected control cells, with the levels of total 4E-BP1 remaining constant in both (Fig. 5A). Multiple sequential phosphorylation events are required to release 4E-BP1 from eIF4E, and the phosphorylation of 4E-BP1 at Thr37 and Thr46 is a necessary priming event (17). In the presence of rapamycin, a TOR inhibitor, the levels of phosphorylated 4E-BP1 were decreased in both infected and mock-infected cells (Fig. 5B). The inhibitor treatment was initiated at 1 h preinfection. These data are consistent with SINV infection resulting in the phosphorylation of 4E-BP1 via the activation of the PI3K-AKT signaling pathway in concert with TOR in arthropod cells.
Fig 5.

Differential 4E-BP1 phosphorylation post-SINV infection in mosquito and vertebrate cells. (A) Western blot with anti-p4E-BP1 antibody and anti-4E-BP1 antibody on whole-cell extracts of C6/36 cells. Cells were either mock infected or infected with SINV for 6 h before being harvested for analysis. (B) Western blot with anti-p4E-BP1 antibody on whole-cell extracts of C6/36 cells. Cells were infected or mock infected in the presence and absence of the TOR inhibitor. (C and D) Western blot with anti-p4E-BP1 and anti-4E-BP1 antibody on whole-cell extracts of BHK-21 (C) and 293 (D) cells. Cells were mock infected, infected with SINV for 6 h, and infected with SINV for 6 h and treated with the TOR inhibitor before being harvested for analysis. Relative quantities of p4E-BP1 standardized to the total 4E-BP1 signal are shown. β-Actin was used as the loading control.
In contrast, there was no effect on the levels of phosphorylated 4E-BP1 in BHK-21 and 293 cells at 6 hpi (Fig. 5C and D). In 293 cells, there appeared to be a small decrease in the overall amounts of 4E-BP1 present in the cells, but the change in the levels of total and phosphorylated 4E-BP1 in 293 cells was proportional. The phosphorylation of 4E-BP1 in BHK-21 cells caused a shift in migration in SDS-PAGE gels. The inhibition of phosphorylation by Torin resulted in an unphosphorylated form of 4E-BP1 that migrated faster than the phosphorylated forms (Fig. 5C). It was previously reported by Mohankumar et al. that Torin and not rapamycin inhibits the TOR-mediated phosphorylation of 4E-BP1 in some mammalian cell lines, including BHK-21 (34).
These contrasting results for the virus-induced phosphorylation of 4E-BP1 in arthropod and mammalian cells suggest that the PI3K-Akt-TOR signaling pathway could be differentially regulated in mammalian and arthropod cells, perhaps playing a role in the different outcomes of virus infection.
SINV infection of mosquito cells causes increased cap-dependent translation.
4E-BP1 interacts with eIF4E, which is a limiting component of the multisubunit eIF4F complex that recruits 40S ribosomal subunits to the 5′ ends of mRNAs. The interaction of the 4E-BP1 protein with eIF4E inhibits eIF4F complex assembly and hence represses cap-dependent translation (29, 38). TOR activation leads to the phosphorylation of 4E-BP1, resulting in its dissociation from eIF4E and the activation of mRNA translation (29, 38). As infection led to increased levels of phosphorylated 4E-BP1, we examined whether the level of cap-dependent translation was also affected. C6/36 cells were transfected with a capped reporter mRNA encoding firefly luciferase (FFluc). At 2 h posttransfection, the cells were infected with SINV at an MOI of 10. Cells were harvested at 2 hpi or after a total of 4 h posttransfection, and luciferase activity levels were determined to measure translation in the cells. Mock-infected C6/36 cells transfected with the reporter RNA were used as a control. We observed a 3.5-fold increase in the luciferase activity in infected mosquito cells, demonstrating that infection caused an increase in cap-dependent translation early in infection, consistent with the increase in levels of phosphorylated 4E-BP1 (Fig. 6A).
To determine whether the changes in translation levels persisted later into infection, both C6/36 and BHK-21 cells were infected with SINV for 4 h, followed by 4 h of transfection (total of 8 h of infection). This timing was chosen to ensure that measurements of luciferase activity were taken at a consistent time after the transfection of reporter RNA. It was found that at 8 h postinfection, the cap-dependent translation of luciferase was increased by 20 to 30% in arthropod cells and was decreased by 90% in mammalian cells compared to uninfected cells (Fig. 6B). An increase in the levels of luciferase activity of 30 to 40% was also seen when we examined flies hosting the SINV replicon and expressing FFluc (Fig. 6C), further substantiating that SINV causes an increase in cap-dependent translation in arthropod cells. The finding that the 3- to 4-fold increase in the translation of the reporter, observed very early in infection, was not maintained as infection progressed is not surprising. Viral mRNA levels will increase during infection, effectively competing for the available translation factors and reducing the translation of the reporter at later times postinfection. The decrease in reporter translation in BHK-21 cells is consistent with previous reports of the shutoff of host cell translation following an infection of vertebrate cells (19, 42).
The observations described above strongly suggest that the activation of the PI3K-Akt-TOR pathway occurs early in infection in mosquito cells, leading to 4E-BP1 phosphorylation and an increase in cellular translation. This may play a role in repressing cytopathic effects and facilitating the establishment of persistence in mosquito cells. It is also apparent that while cellular translation is severely inhibited in vertebrate cells, 4E-BP1 phosphorylation is unaffected, implying that virus-induced translational repression is not mediated through this pathway.
SINV replication leads to 4E-BP1 phosphorylation in mosquito cells.
Since the activation of the pathway occurs early in infection, it could be a consequence of attachment, entry, the translation of nonstructural proteins, or early events in RNA replication. Utilizing a SINV replicon launched from the genome of Drosophila, we observed changes in viral genome replication in flies possessing defects in this pathway. These changes were seen with SINV replicon RNA that does not encode the viral structural proteins; hence, it is capable of self-replication but not of subsequent infection. This suggests that the interaction between the virus and the PI3K-Akt-TOR pathway does not require infection or viral structural proteins. On this basis, we think that it is probable that the pathway is activated by a component or formation of the plasma membrane-associated viral RNA synthetic complex (15).
To examine this hypothesis, C6/36 cells were transfected with SINV replicon RNA expressing GFP from the subgenomic RNA but not encoding structural proteins. Levels of phosphorylated 4E-BP1 in the whole-cell extracts were determined at 6 and 12 h posttransfection. As shown in Fig. 7A, the levels of phosphorylated 4E-BP1 were higher in the cells transfected with the replicon RNA than in the mock-transfected cells at both 6 and 12 h posttransfection.
Fig 7.
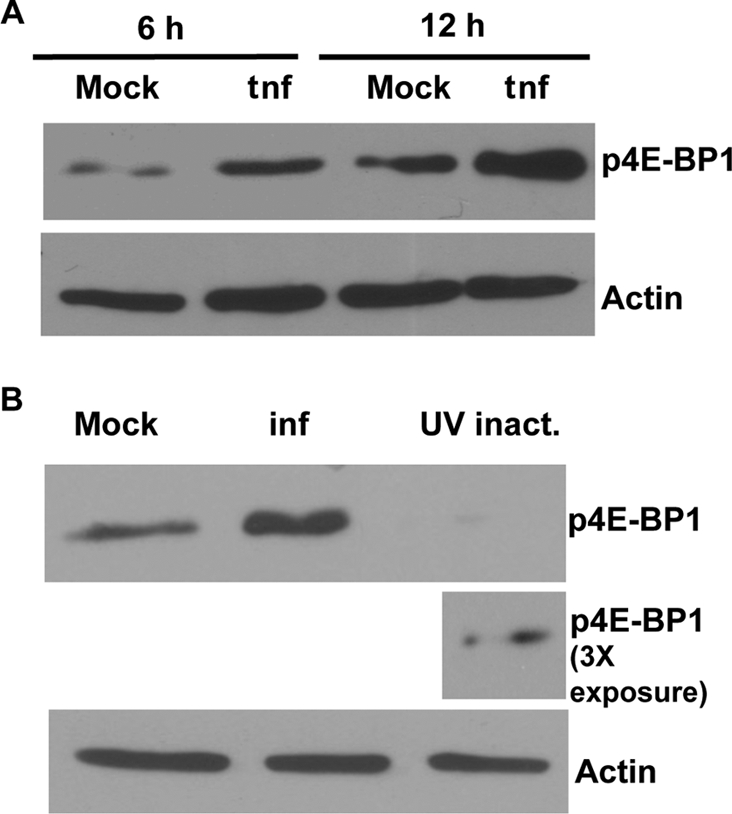
SINV replication is essential for the increase in levels of phosphorylation of 4E-BP1. (A) Western blot with anti-p4E-BP1 antibody on whole-cell extracts of C6/36 cells. Cells were either mock transfected or transfected with SINrep/GFP RNA for 6 h or 12 h before being harvested for analysis. (B) Western blot with anti-p4E-BP1 antibody on whole-cell extracts of C6/36 cells either mock infected or infected with replication-competent SINV or UV-inactivated SINV for 6 h before harvesting for analysis. β-Actin was used as the loading control. Two exposures of the blot for p4E-BP1 in the UV-inactivated treated sample are shown.
To further determine the importance of viral RNA replication, cells were infected with SINV or UV-inactivated SINV (equivalent of an MOI of 10 PFU/cell). UV-inactivated virus is capable of attaching to and entering cells (26). The UV inactivation of the virus was confirmed by checking virus titers on BHK-21 cells (data not shown). The UV inactivation of SINV (Fig. 7B) prevented the virus from causing the phosphorylation of 4E-BP1, further emphasizing the requirement for virus replication or an associated complex to activate this pathway and cause an increase in overall translation.
DISCUSSION
The patterns of infection established by alphaviruses in vertebrate and arthropod hosts clearly demonstrate that the virus interacts with host cells in distinctly different ways. In a vertebrate host, infection results in cytolysis and disease, whereas in a mosquito host, infection results in a life-long persistent infection with minimal fitness cost to the host. As the same virus infects both these different cell types, it is apparent that it is the difference in the host cell environment that leads to this difference in the outcome of infection.
SINV infection of vertebrate cells causes a shutoff of host cell gene expression at the levels of both transcription and translation (19). This facilitates the sequestration of host cell machinery by the virus and inhibits the antiviral response, resulting in high levels of virus replication (19). The shutoff of host cell gene expression also contributes to cell death. Virus production at the expense of cell survival in vertebrates has no detrimental consequence for virus transmission and in fact results in high-level viremia, facilitating uptake by, and infection of, the mosquito vector (27). During the acute phase of SINV infection in arthropod cells, the virus replicates to the same levels as in vertebrate cells, but it does so without detrimental effects on the host (47). This pattern of infection is essential for virus transmission. A severe fitness cost to the vector would be detrimental to transmission, whereas persistent, noncytolytic infection facilitates transmission. In order for this pattern of infection to be established, the virus cannot shut off vector host gene expression; nevertheless, the virus still needs to utilize host cell machinery, particularly for the translation of viral proteins, in order to replicate to high levels. The data presented in this study describe how the virus can, through the activation of a cellular signaling pathway, increase the translational capacity of the arthropod host, thus allowing an enhanced translation of viral messages while not depleting the cell of translational machinery to the point at which cytopathology occurs.
The PI3K-Akt pathway is an important signaling pathway that can be regulated by viruses (9). The activation of this pathway can lead to TOR activation and increased phosphorylation of the eIF4E-binding protein 4E-BP1. This is a very important point of control in cap-dependent translation, as the phosphorylation of 4E-BP1 leads to the release and increased availability of eIF4E, which can then form the eIF4F complex on capped mRNA. The data described above indicate that the PI3K-Akt-TOR pathway is activated early following SINV infection of arthropod cells as a consequence of viral replication complex formation or activity. This activation results in the phosphorylation of 4E-BP1, which in turn results in an increase in overall translation within the infected cell. Interestingly, the infection of vertebrate cells does appear to result in a change in 4E-BP1 phosphorylation early in SINV infection. It is well documented that host translation is inhibited following infection in vertebrate cells; however, the mechanism of translational inhibition is not understood (19, 42, 47). From previous studies, it seems likely that multiple viral factors play a role in translational shutoff. nsP2, nsP4, and viral subgenomic mRNA transcription have been implicated in this process (31, 42, 49). While there may be multiple, possibly redundant, means by which shutoff is achieved, it is apparent from our studies that the virus-mediated shutoff of vertebrate cell translation does not correlate with a change in the 4E-BP1 phosphorylation state.
In contrast to the situation in vertebrate cells, cellular translation in mosquito cells is not inhibited during infection. Based on our observations of two arthropod systems, mosquito cells and transgenic Drosophila, we hypothesize that SINV infection activates the PI3K-Akt-TOR pathway in arthropod cells, which in turn leads to the phosphorylation of 4E-BP1 and the consequent release of eIF4E, enhancing cap-dependent translation (Fig. 8). As both viral genomic and subgenomic RNAs are capped, an increased availability of factors necessary for cap-dependent translation would allow the efficient translation of viral proteins and, hence, virus replication without the need to inhibit host processes in order to acquire the necessary machinery. Therefore, the upregulation of this pathway facilitates viral replication while limiting the impact of infection on host cell gene expression. Another RNA virus, cricket paralysis virus, is known to modulate this pathway differently in Drosophila cells by causing a dissociation of eIF4G from eIF4F, resulting in the inhibition of host mRNA translation, but allowing viral mRNA translation through an internal ribosome entry mechanism (16).
Fig 8.

Activation of the PI3K-Akt-TOR pathway and viral persistence in arthropod cells. The activation of the PI3K-Akt-TOR pathway following virus infection leads to the phosphorylation of 4E-BP1 and the release of sequestered eIF4E. The availability of eIF4E leads to the increased translation of both viral and cellular mRNAs, allowing efficient virus replication and cell survival following infection. Pathway inhibitors are shown at the left of the pathway.
We recognize that increased translation is just one outcome of PI3K-Akt-TOR activation, and other consequences may also influence virus replication and host survival (23). Further investigations of the outcomes of pathway activation are necessary. However, the identification of a pathway activated by infection that functions to promote virus replication and that may enhance arthropod host cell survival provides an obvious target for the development of transmission intervention strategies.
ACKNOWLEDGMENTS
We thank Suchetana Mukhopadhyay and Pranav Danthi for their invaluable suggestions and guidance throughout the project; Kevin Sokoloski for critical reading of the manuscript; and members of the Hardy, Mukhopadhyay, and Danthi laboratories for discussions of the data. We thank Norbert Perrimon (Harvard Medical School) for the UAS-Luc flies and Nahum Sonnenberg (Cold Spring Harbor Laboratory) for the 4E-BP1 antibody.
This work was supported by grant R01 AI0090077 from the National Institutes of Health to R.W.H.
Footnotes
Published ahead of print 18 January 2012
REFERENCES
- 1. Arrigo NC, Adams AP, Weaver SC. 2010. Evolutionary patterns of eastern equine encephalitis virus in North versus South America suggest ecological differences and taxonomic revision. J. Virol. 84:1014–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Avadhanula V, Weasner BP, Hardy GG, Kumar JP, Hardy RW. 2009. A novel system for the launch of alphavirus RNA synthesis reveals a role for the Imd pathway in arthropod antiviral response. PLoS Pathog. 5:e1000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barnett SF, et al. 2005. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem. J. 385:399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bredenbeek PJ, Frolov I, Rice CM, Schlesinger S. 1993. Sindbis virus expression vectors: packaging of RNA replicons by using defective helper RNAs. J. Virol. 67:6439–6446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. 2008. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat. Rev. Microbiol. 6:266–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burnham AJ, Gong L, Hardy RW. 2007. Heterogeneous nuclear ribonuclear protein K interacts with Sindbis virus nonstructural proteins and viral subgenomic mRNA. Virology 367:212–221 [DOI] [PubMed] [Google Scholar]
- 7. Bushell M, Sarnow P. 2002. Hijacking the translation apparatus by RNA viruses. J. Cell Biol. 158:395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Connor JH, Lyles DS. 2002. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1. J. Virol. 76:10177–10187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cooray S. 2004. The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J. Gen. Virol. 85:1065–1076 [DOI] [PubMed] [Google Scholar]
- 10. Cristea IM, et al. 2010. Host factors associated with the Sindbis virus RNA-dependent RNA polymerase: role for G3BP1 and G3BP2 in virus replication. J. Virol. 84:6720–6732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Degtyarev M, et al. 2008. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J. Cell Biol. 183:101–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dionne MS, Pham LN, Shirasu-Hiza M, Schneider DS. 2006. Akt and FOXO dysregulation contribute to infection-induced wasting in Drosophila. Curr. Biol. 16:1977–1985 [DOI] [PubMed] [Google Scholar]
- 13. Dunn EF, Connor JH. 2011. Dominant inhibition of Akt/protein kinase B signaling by the matrix protein of a negative-strand RNA virus. J. Virol. 85:422–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fothergill T, McMillan NA. 2006. Papillomavirus virus-like particles activate the PI3-kinase pathway via alpha-6 beta-4 integrin upon binding. Virology 352:319–328 [DOI] [PubMed] [Google Scholar]
- 15. Frolova EI, Gorchakov R, Pereboeva L, Atasheva S, Frolov I. 2010. Functional Sindbis virus replicative complexes are formed at the plasma membrane. J. Virol. 84:11679–11695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garrey JL, Lee YY, Au HH, Bushell M, Jan E. 2010. Host and viral translational mechanisms during cricket paralysis virus infection. J. Virol. 84:1124–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gingras AC, et al. 2001. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 15:2852–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gingras AC, Svitkin Y, Belsham GJ, Pause A, Sonenberg N. 1996. Activation of the translational suppressor 4E-BP1 following infection with encephalomyocarditis virus and poliovirus. Proc. Natl. Acad. Sci. U. S. A. 93:5578–5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gorchakov R, Frolova E, Frolov I. 2005. Inhibition of transcription and translation in Sindbis virus-infected cells. J. Virol. 79:9397–9409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gorchakov R, Garmashova N, Frolova E, Frolov I. 2008. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J. Virol. 82:10088–10101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guo H, et al. 2007. Regulation of hepatitis B virus replication by the phosphatidylinositol 3-kinase-Akt signal transduction pathway. J. Virol. 81:10072–10080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hara K, et al. 1997. Regulation of eIF-4E BP1 phosphorylation by mTOR. J. Biol. Chem. 272:26457–26463 [DOI] [PubMed] [Google Scholar]
- 23. Hawkins PT, Anderson KE, Davidson K, Stephens LR. 2006. Signalling through class I PI3Ks in mammalian cells. Biochem. Soc. Trans. 34:647–662 [DOI] [PubMed] [Google Scholar]
- 24. Howlett E, Lin CC, Lavery W, Stern M. 2008. A PI3-kinase-mediated negative feedback regulates neuronal excitability. PLoS Genet. 4:e1000277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang JT, Schneider RJ. 1991. Adenovirus inhibition of cellular protein synthesis involves inactivation of cap-binding protein. Cell 65:271–280 [DOI] [PubMed] [Google Scholar]
- 26. Jan JT, Griffin DE. 1999. Induction of apoptosis by Sindbis virus occurs at cell entry and does not require virus replication. J. Virol. 73:10296–10302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Griffin DE. 2001. Alphaviruses, p 917–962 In Knipe DM, et al. (ed), Fields virology, 4th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 28. Li E, Stupack D, Bokoch GM, Nemerow GR. 1998. Adenovirus endocytosis requires actin cytoskeleton reorganization mediated by Rho family GTPases. J. Virol. 72:8806–8812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin TA, et al. 1994. PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science 266:653–656 [DOI] [PubMed] [Google Scholar]
- 30. Mannova P, Beretta L. 2005. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: control of cell survival and viral replication. J. Virol. 79:8742–8749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mayuri Geders TW, Smith JL, Kuhn RJ. 2008. Role for conserved residues of Sindbis virus nonstructural protein 2 methyltransferase-like domain in regulation of minus-strand synthesis and development of cytopathic infection. J. Virol. 82:7284–7297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Menges CW, Baglia LA, Lapoint R, McCance DJ. 2006. Human papillomavirus type 16 E7 up-regulates AKT activity through the retinoblastoma protein. Cancer Res. 66:5555–5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miller ML, Brown DT. 1992. Morphogenesis of Sindbis virus in three subclones of Aedes albopictus (mosquito) cells. J. Virol. 66:4180–4190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mohankumar V, Dhanushkodi NR, Raju R. 2011. Sindbis virus replication, is insensitive to rapamycin and torin1, and suppresses Akt/mTOR pathway late during infection in HEK cells. Biochem. Biophys. Res. Commun. 406:262–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Montgomery SA, Berglund P, Beard CW, Johnston RE. 2006. Ribosomal protein S6 associates with alphavirus nonstructural protein 2 and mediates expression from alphavirus messages. J. Virol. 80:7729–7739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Norman KL, Sarnow P. 2010. Herpes simplex virus is Akt-ing in translational control. Genes Dev. 24:2583–2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pardigon N, Strauss JH. 1996. Mosquito homolog of the La autoantigen binds to Sindbis virus RNA. J. Virol. 70:1173–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pause A, et al. 1994. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 371:762–767 [DOI] [PubMed] [Google Scholar]
- 39. Pestova TV, et al. 2001. Molecular mechanisms of translation initiation in eukaryotes. Proc. Natl. Acad. Sci. U. S. A. 98:7029–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47 [DOI] [PubMed] [Google Scholar]
- 41. Rice CM, Levis R, Strauss JH, Huang HV. 1987. Production of infectious RNA transcripts from Sindbis virus cDNA clones: mapping of lethal mutations, rescue of a temperature-sensitive marker, and in vitro mutagenesis to generate defined mutants. J. Virol. 61:3809–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rupp JC, Jundt N, Hardy RW. 2011. Requirement for the amino-terminal domain of Sindbis virus nsP4 during virus infection. J. Virol. 85:3449–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saeed MF, Kolokoltsov AA, Freiberg AN, Holbrook MR, Davey RA. 2008. Phosphoinositide-3 kinase-Akt pathway controls cellular entry of Ebola virus. PLoS Pathog. 4:e1000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sarbassov DD, Ali SM, Sabatini DM. 2005. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 17:596–603 [DOI] [PubMed] [Google Scholar]
- 45. Schneider RJ, Mohr I. 2003. Translation initiation and viral tricks. Trends Biochem. Sci. 28:130–136 [DOI] [PubMed] [Google Scholar]
- 46. Spradling AC, et al. 1999. The Berkeley Drosophila genome project gene disruption project: single P-element insertions mutating 25% of vital Drosophila genes. Genetics 153:135–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58:491–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thompson SR, Sarnow P. 2000. Regulation of host cell translation by viruses and effects on cell function. Curr. Opin. Microbiol. 3:366–370 [DOI] [PubMed] [Google Scholar]
- 49. van Steeg H, et al. 1981. Shutoff of neuroblastoma cell protein synthesis by Semliki Forest virus: loss of ability of crude initiation factors to recognize early Semliki Forest virus and host mRNA's. J. Virol. 38:728–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ventoso I, et al. 2006. Translational resistance of late alphavirus mRNA to eIF2alpha phosphorylation: a strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev. 20:87–100 [DOI] [PMC free article] [PubMed] [Google Scholar]