

Abstract
Vaccinia mature virus enters cells through either endocytosis or plasma membrane fusion, depending on virus strain and cell type. Our previous results showed that vaccinia virus mature virions containing viral A26 protein enter HeLa cells preferentially through endocytosis, whereas mature virions lacking A26 protein enter through plasma membrane fusion, leading us to propose that A26 acts as an acid-sensitive fusion suppressor for mature virus (S. J. Chang, Y. X. Chang, R. Izmailyan R, Y. L. Tang, and W. Chang, J. Virol. 84:8422–8432, 2010). In the present study, we investigated the fusion suppression mechanism of A26 protein. We found that A26 protein was coimmunoprecipitated with multiple components of the viral entry-fusion complex (EFC) in infected HeLa cells. Transient expression of viral EFC components in HeLa cells revealed that vaccinia virus A26 protein interacted directly with A16 and G9 but not with G3, L5 and H2 proteins of the EFC components. Consistently, a glutathione S-transferase (GST)-A26 fusion protein, but not GST, pulled down A16 and G9 proteins individually in vitro. Together, our results supported the idea that A26 protein binds to A16 and G9 protein at neutral pH contributing to suppression of vaccinia virus-triggered membrane fusion from without. Since vaccinia virus extracellular envelope proteins A56/K2 were recently shown to bind to the A16/G9 subcomplex to suppress virus-induced fusion from within, our results also highlight an evolutionary convergence in which vaccinia viral fusion suppressor proteins regulate membrane fusion by targeting the A16 and G9 components of the viral EFC complex. Finally, we provide evidence that acid (pH 4.7) treatment induced A26 protein and A26-A27 protein complexes of 70 kDa and 90 kDa to dissociate from mature virions, suggesting that the structure of A26 protein is acid sensitive.
INTRODUCTION
Vaccinia virus belongs to the genus Orthopoxvirus of the family Poxviridae and has a wide host range in vitro and in vivo (35). Vaccinia virus replicates in the cytoplasm of infected cells and produces two infectious forms of virus particles, the mature virion (MV) and extracellular enveloped virion (EV) (14). MVs are the most abundant constituent of vaccinia virus in the infected cells and can be readily purified, with stable biochemical properties, for further analyses. MVs contain about 76 to 80 viral proteins, including more than 20 envelope proteins (12, 43). Four proteins are known to play a role in MV attachment. Among them, viral envelope H3 (32), A27 (13), and D8 (22) proteins bind to cell surface glycosaminoglycans (GAGs), while the fourth protein, A26, binds to the extracellular matrix protein laminin (11). Furthermore, a virus entry-fusion complex (EFC) consisting of 12 proteins, A16 (40), A21 (51), A28 (53), F9 (5), G3 (26), G9 (39), H2 (46), I2 (37), J5 (62), L1 (3), L5 (50), and O3 (44), plays an essential role in postattachment membrane fusion although the fusion mechanism remains unknown at the moment.
After cell attachment vaccinia virus MVs penetrate into host cells through either endocytosis (19, 28) or plasma membrane fusion (6, 17, 33) pathways, depending on virus strain (2) and cell type (60). Although phenotypic entry differences were examined via electron microscopy (EM) and described in early literature (1, 7, 15, 42), the molecular mechanisms were not investigated until recent years. It has been shown that vaccinia virus MV entry is sensitive to cytoskeleton inhibitors that block actin polymerization, and dominant negative forms of small GTPases and various kinase inhibitors also blocked vaccinia virus MV entry (33). Entry after endocytosis of vaccinia virus MVs is dependent on low pH (4.5 to 5.0) and is sensitive to chemicals such as NaF and cytochalasin B (15, 42), as well as bafilomycin (BFLA), which blocks acidification of endosomes (52). Exposure of MVs to low pH in the range of 4.5 to 5.0 during infection forces the MV membrane to fuse with the plasma membrane, thus bypassing the need for endosomal acidification (19). The endocytic pathway of MV infection in HeLa cells was reported by Mercer and Helenius as dynamin-independent macropinocytosis (34) and by Huang et al. as a dynamin-dependent, VPEF-dependent fluid-phase endocytosis (23). Although vaccinia virus MVs are rich in phosphatidylserine (PS) (25), reconstitution of the MV membrane with other lipids rescued virus infectivity (29), demonstrating that apoptotic mimicry (34) is not essential for MV entry.
Despite the fact that virus strain-related variations of MV entry pathways were well documented, the reason behind this phenomenon was not known. Using several vaccinia virus strains, we recently demonstrated that A26 protein in MVs is the major determinant of endocytic choice since virus strains containing A26 protein, such as WR and IHD-J, enter cells through an endocytic pathway, whereas other virus strains lacking A26 protein, such as IHD-W, MVA, and Copenhagen, entered HeLa cells through plasma membrane fusion (8). Indeed, deletion of MV envelope protein A26 from the vaccinia virus Western Reserve (WR) strain generated WRΔA26L MV particles and triggered massive fusion from without (FFWO) without low-pH treatment, suggesting that viral A26 protein functions as a fusion suppressor of MVs at neutral pH (8). To understand how A26 protein suppresses membrane fusion, we hypothesized that it interacts with subcomponents of viral EFC to block fusion activity of the latter. In the present study, we identify specific components of EFC that physically interact with A26 protein and investigate how acidic pH affects A26 protein.
MATERIALS AND METHODS
Cell culture and viruses.
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen). The Western Reserve (WR) strain of vaccinia virus was prepared and purified by CsCl gradient centrifugation as previously described (18, 41). VTF7-3 was obtained from ATCC. The vA28i virus, in which the A28L open reading frame (ORF) is under isopropyl-β-d-thiogalactopyranoside (IPTG) regulation (48), was obtained from Bernard Moss. WRΔA56R and WRΔK2L were described previously (54, 56) and obtained from Amy L. MacNeill with consent of Richard W. Moyer. Anti-vaccinia virus MV antibody (Ab) was generated previously in rabbits by injecting purified vaccinia virus MVs of the WR strain (21). Anti-A26 (11), anti-A27 (13), anti-G3 (26), anti-D8 (22), anti-H3 (32), and anti-L1 (10) rabbit antibodies were all previously described. Anti-vaccinia virus A56 mouse monoclonal antibody ([MAb] B2D10) was obtained from Yasuo Ichihashi (38, 45). Anti-G9 antibody was generated in New Zealand White (NZW) rabbits by injecting recombinant G9 protein containing amino acids 1 to 340 produced in bacteria. Anti-A25, -A16, and -L5 antibodies were raised in NZW rabbits against synthetic peptides containing sequences of A25 (amino acids [aa] 10 to 35), A16 (aa 273 to 292), and L5 (aa 56 to 75), respectively. Anti-Myc (Abcam), anti-V5 (Serotec), anti-green fluorescent protein ([GFP] BD Biosciences), anti-Flag (Sigma), and anti-hemagglutinin ([HA] Covance) antibodies were purchased.
Construction of expression plasmids.
For the expression of vaccinia viral proteins in HEK293T cells, vaccinia viral ORFs of the WR strain were codon optimized (Gene Script, Inc.) and subsequently cloned into various mammalian expression vectors as described below. Vaccinia virus H2R, L5R, and G3L genes were cloned into pLKO_AS3.1.EGFP, pLKO_AS3w.neo, and pLKO_AS3w.puro, respectively, without any tag sequences. The A56R ORF was cloned into pcDNA3.1/CT-GFP-TOPO and fused in frame with GFP at the 3′ end without any tag sequences. The HA and Myc tags were added to the N terminus of A16L and G9R, respectively, by PCR prior to cloning into pcDNA3.1. A Flag tag was added to the C terminus of the K2L gene by PCR prior to cloning into pcDNA3.1. The A26L ORF was cloned into pcDNA3.1/V5-His TOPO and fused in frame with the C-terminal V5-His sequences of the vector.
Generation of the recombinant virus, WR-Flag-A26. (i) Plasmid construction.
To obtain the Flag-A26/LacZ fragment, overlapping PCR was performed using two sets of primers. First, the Flag-A26 fragment driven by synthetic late promoter was amplified from the WR genomic DNA using the following primers (SalI restriction site is underlined): a, 5′-GGGGTCGACAATTGGATCAGCTTTTTTTTTTTTTTTTTTGGCATATAAATGGATTACAAGGATGACGACGATAAGATGGCGAACATTATAAAT-3′ (this primer also contains a synthetic late promoter, a Flag tag and first 18mer of A26 ORF); and b, 5′-AAAAAAACAAAATGAAATTCGTTATAAAATCGTAGATCTCCC-3′. The LacZ fragment driven by the viral p11k late promoter was then amplified from p11k-LacZ-containing plasmid (pSC11-360-luciferase/p11k-LacZ) using the following primers (SalI restriction site is underlined): c, 5′-GGGAGATCTACGATTTTATAACGAATTTCATTTTGTTTTTTT-3′; and d, 5′-AAAGTCGACTTATTATTATTTTTGACA-3′. After the first PCR, these two fragments were mixed in a molar ratio of 1:1 for overlapping PCR with primers a and d again, and the resulting Flag-A26/LacZ fragment was digested with SalI and ligated with SalI-digested pBluescript II KS-A25/Luc/GPT/A27/A28 (8), resulting in pBluescript II KS-A25/Flag-A26/LacZ/Luc/A27/A28. The plasmids were sequenced to ensure that there was no mutation.
(ii) Isolation of the recombinant virus, WR-Flag-A26.
CV-1 cells were infected with WRΔA26L virus (8) at a multiplicity of infection (MOI) of 1 PFU per cell and transfected with (1 μg) the pBluescript II KS-A25/Flag-A26/LacZ/Luc/A27/A28 plasmid. The cells were cultured for another 3 days and harvested for recombinant virus isolation through three rounds of plaque purification in agar containing 150 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) as described previously (9).
Immuno-electron microscopy.
Immunogold labeling of viral protein in the infected cells was based on the established methods, as previously described (52). In brief, HeLa cells were infected with CsCl gradient-purified wild-type WR or WRΔA26L virus at an MOI of 100 PFU per cell at 4°C for 60 min and washed with cold phosphate-buffered saline (PBS; pH 7.4), and the cells were shifted to 37°C for 10 min and fixed for immunogold-conjugated antibody staining with anti-vaccinia virus MV primary Ab (1:50 dilution) and goat anti-rabbit Ab conjugated to 6-nm gold particles (1:40 dilution). After cells were embedded and sectioned, samples in thin sections were stained with 7% uranyl acetate in 50% ethanol and then with 0.01% lead citrate and photographed under a Zeiss 902 transmission electron microscope. For quantification, sections of infected cells (>30) were scored for the presence of endocytosed MVs enclosed in vesicles, which were scored as endocytosis. Alternatively, cells containing viral cores associated with immunogold labeled plasma membrane were scored as plasma membrane fusion (PM). The percentage of endocytosis was calculated as follows: (number of cells containing endocytosed MVs/total number of cells) × 100%. The percentage of PM fusion was calculated as follows: (number of cells containing viral cores with immunogold-labeled PM/total number of cells) × 100%. The experiments were repeated twice.
Coimmunoprecipitation.
Cell lysates used for coimmunoprecipitation and GST pulldown were prepared from cells under three different conditions. (i) HeLa cells were infected with wild-type WR, WRΔA26L, WRΔA56R, WRΔK2L, WR-Flag-A26 or VA28i virus at an MOI of 5 PFU per cell and harvested at 24 h postinfection (p.i.). (ii) 293T cells (8 × 106 cells/100 mm) were transiently transfected with 3 μg each of pMyc-G9R, pHA-A16L, pA26L-V5, pGFP-A56R, and pFlag-K2L using a calcium phosphate transfection protocol and harvested after 24 h. (iii) 293T cells in 60-mm dishes were infected with a recombinant virus VTF7-3 at an MOI of 5 PFU per cell, subsequently transfected with 3 μg of pGFP-A56R, pFlag-K2L, and pA26L-V5 using a calcium phosphate transfection protocol, and harvested after 24 h. Cells were then lysed in a lysis buffer consisting of 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 20 mM N-ethylmaleimide (NEM; Sigma), and protease inhibitor cocktail (Roche). Lysates were cleared by low-speed centrifugation, incubated with anti-Flag, anti-GFP, anti-V5, or anti-G9 antibody that was conjugated to agarose beads (Sigma) or to protein A beads (GE Healthcare) at 4°C for 2 h, washed four times, and centrifuged. The immunoprecipitates were then resuspended in SDS-containing sample buffer, separated by SDS-PAGE, and analyzed by immunoblot analyses as previously described (8). The experiments were repeated three times.
GST pulldown assays.
GST and GST-A26 were overexpressed in the Escherichia coli BL21(DE3) strain and purified on glutathione-Sepharose 4B beads (Amersham Biosciences) as previously described (20). Cell lysates were prepared from virus-infected HeLa cells or transiently transfected 293T cells as described in the previous paragraph (experiments described in i and ii). For GST pulldown assays, lysates were first precleaned with 30 μg of GST bound to glutathione-Sepharose 4B beads at 4°C for 2 h and centrifuged again. The supernatant was subsequently incubated with 30 μg of GST or GST-A26 protein bound to the glutathione-Sepharose 4B beads at 4°C for 2 h. After centrifugation, samples were washed, separated by 10% SDS-PAGE, and transferred for immunoblot analyses as previously described (8). The experiments were repeated three times.
Triton X-114 partitioning.
The protein partition during phase separation in solutions of Triton X-114 was based on experimental procedures as previously described (4, 16). In brief, 8 μg of purified vaccinia virus WR MV particles was solubilized in 2% Triton X-114 in buffer containing 10 mM Tris-HCl and 150 mM NaCl at pH 7.4 or 4.7 at 4°C for 10 min. These samples were further incubated at 37°C for 10 min and then centrifuged at 300 × g at 25°C for 3 min to separate the solution into aqueous and detergent phases. The aqueous (top) phase and the detergent (bottom) phases were both collected and analyzed by immunoblot analysis as described above. The experiments were repeated twice.
RESULTS
EM revealed vaccinia virus MV membrane fused with plasma membrane at neutral pH in the absence of A26 protein.
Our previous results showed that the wild-type vaccinia virus WR strain MV entered HeLa cells through bafilomycin-sensitive endocytosis, whereas WRΔA26L entered through bafilomycin-resistant plasma membrane fusion (8). To provide additional evidence, we performed electron microscopic (EM) analyses. HeLa cells were infected with either wild-type WR or WRΔA26L virus at an MOI of 100 PFU per cell at 4°C for 60 min, washed, and fixed at 10 min p.i. with anti-vaccinia virus MV antibody in immunogold labeling analyses. HeLa cells infected with the vaccinia virus WR strain contained multiple vesicles with intact MV enclosed in cytoplasmic vesicles (Fig. 1A) (23). In contrast, HeLa cells infected with WRΔA26L virus contained intracellular viral cores with viral membrane, which was decorated with anti-vaccinia virus MV antibody, already fused with plasma membrane (Fig. 1B). This result shows that WRΔA26L enters HeLa cells through plasma membrane fusion. Bafilomycin, previously shown to block WR strain endocytosis, did not inhibit WRΔA26L entry, and the intracellular viral core release appeared normal by EM (data not shown). These quantification results provide new and direct evidence that the vaccinia virus WR strain is endocytosed into HeLa cells, whereas WRΔA26L virus fused with the plasma membrane (Fig. 1C). Since removal of A26 protein allowed plasma membrane fusion at neutral pH, we concluded that A26 protein acts as an MV fusion suppressor for MV-mediated membrane fusion (8).
Fig 1.
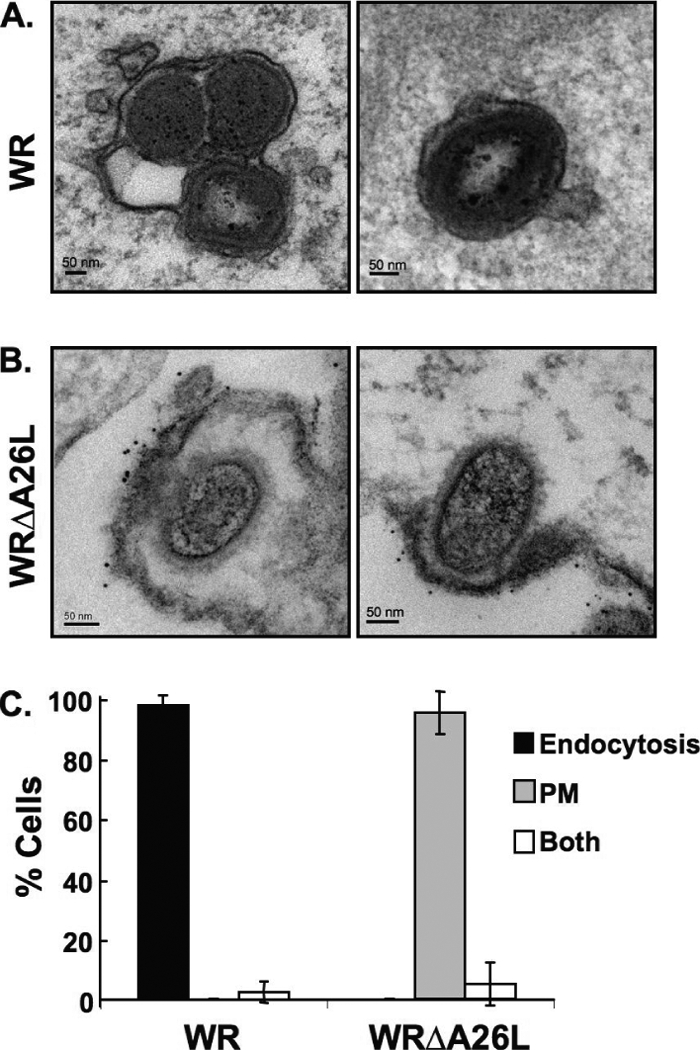
Immunogold electron microscopy analyses of HeLa cells infected with vaccinia virus MVs. (A) HeLa cells were infected with vaccinia virus wild-type WR and WRΔA26L MVs as described in Materials and Methods. After infection, cells were fixed and stained with anti-vaccinia virus MV primary antibody and goat anti-rabbit antibody conjugated to 6-nm gold particles and analyzed by EM. MV particles of wild-type strain WR are enclosed within intracellular vesicles. (B) HeLa cells were infected with WRΔA26L MVs and analyzed by EM as described in panel A. MV particles of WRΔA26L fused with the cell plasma membrane that were decorated with the immunogold-labeled antibody. (C) Quantification of cells (>30) infected with vaccinia virus MVs through different entry routes. Cells containing MV entry through endocytosis or plasma membrane fusion pathways were quantified as described in Materials and Methods. Data represent percentages of cells that contain MVs within intracellular vesicles (endocytosis), MVs fused with the plasma membrane (PM), or both.
A26 interacts with multiple protein components of the viral EFC in virus-infected cells.
Previous results demonstrated that 12 components of the viral entry-fusion complex (EFC) are required for vaccinia virus membrane fusion (3, 5, 26, 37, 39, 40, 44, 46, 50, 51, 53). Thus, we addressed the relationship between A26 and the EFC by testing whether A26 interacts with components of the EFC. We constructed a recombinant WR virus, WR-Flag-A26, expressing A26 protein fused with a Flag tag at the N terminus. HeLa cells were infected with MVs of the wild-type WR strain or WR-Flag-A26 at an MOI of 5 PFU per cell and harvested at 24 h p.i. for coimmunoprecipitation analyses. Multiple components of the EFC, including G9, A16, G3, and L5, were detected in the immunoprecipitates, whereas non-EFC components, such as envelope protein D8 and core protein A4, were not (Fig. 2A). These data indicate that A26 interacts specifically with multiple components of the EFC. To test which EFC components directly interact with the A26 protein, we took advantage of the recombinant virus vA28i that expresses an HA-tagged A28 protein only in the presence of IPTG (48). When A28 is not expressed (in the absence of IPTG), other EFC components fail to form the EFC, despite the formation of small subcomplexes such as A16-G9 (59) and G3-L5 (61). HeLa cells were infected with WR or vA28i, cultured in medium with or without IPTG, and harvested at 24 h p.i. In the presence of IPTG, anti-G9 antibody immunoprecipitated G9 and other associated EFC components as expected (Fig. 2B). Anti-G9 antibody also brought down A26 protein but not D8 or A4 protein, consistent with the data shown in Fig. 2A. In the absence of IPTG, anti-G9 antibody coimmunoprecipitated only A16 and A26 proteins but not other EFC components. These results suggested that the interaction of A26 protein with G9 and A16 proteins is direct and not mediated through other EFC components. We noticed a reduced amount of A26 protein in the infected cells when A28 expression was repressed, resulting in less A26 protein coimmunoprecipitated with anti-G9 antibody. Although the A26 reduction was reproducible, the reason behind this observation is currently unknown.
Fig 2.

Coimmunoprecipitation of A26 protein with multiple components of the viral EFC in virus-infected cells. (A) Coimmunoprecipitation of A26 with EFC components in virus-infected cells. HeLa cells were either mock infected or infected with WR or WR-Flag-A26 virus at an MOI of 5 PFU/cell, harvested at 24 h p.i. for immunoprecipitation (IP) with anti-Flag-agarose, and analyzed by immunoblotting with various antibodies, shown at the right side of the gel. (B) Coimmunoprecipitation of G9 protein with A26 protein in virus-infected cells. HeLa cells were either mock infected or infected with wild-type WR or vA28i virus at an MOI of 5 PFU/cell, incubated in medium with or without 100 μM IPTG, harvested at 24 h p.i. for immunoprecipitation with anti-G9 (1:50) antibody, and analyzed as described for panel A. Input represents 1% of total cell lysates.
A26 interacts individually with G9 and A16 proteins in vitro.
To further demonstrate that A26 interacts with the A16 and G9 proteins, GST and GST-A26 fusion proteins (Fig. 3A) were prepared from E. coli, as described previously (20), for in vitro pulldown analyses. Recombinant GST or GST-A26 proteins were incubated for 2 h at 4°C with cell lysates prepared from mock-infected or WR-infected (Fig. 3A, M or V, respectively) cells, and the GST pulldown pellets were analyzed by immunoblot analyses. As shown in Fig. 3B, the GST-A26 fusion protein (but not the control GST protein) pulled down the A16 and G9 proteins but none of the other EFC components, such as G3 and L5 proteins, nor the control D8 and A4 proteins from virus-infected cells (Fig. 3B). To eliminate the possibility that other viral late proteins contributed to the interactions among A26 and A16/G9 proteins, HA-tagged A16 and Myc-tagged G9 proteins were transiently expressed individually or in combination in 293T cells. Cell lysates were then prepared for pulldown analyses as described above. Consistent with the above results, GST-A26 fusion protein specifically pulled down A16 and G9 proteins (Fig. 4A) but not the other EFC subunit proteins, G3 and L5 (Fig. 4B). Furthermore, although A16 and G9 proteins were known to form a complex, GST-A26 protein could interact with A16 or G9 protein individually (Fig. 4A), suggesting that A26 protein may contain distinct binding sites to A16 and G9 proteins in vitro.
Fig 3.

In vitro interaction of A26 protein with G9 and A16 proteins expressed from virus-infected cells. (A) Coomassie blue staining of recombinant GST and GST-A26 proteins purified from E. coli. The asterisk represents the full-length GST-A26 protein, and the arrowhead represents the GST protein. (B) GST-A26 pulled down A16 and G9 proteins from virus-infected cells. Recombinant GST and GST-A26 protein (30 μg) were incubated with cell lysates prepared from either mock-infected (M) or vaccinia virus WR-infected (V) HeLa cells, as described in Materials and Methods, harvested at 24 h p.i., and analyzed by immunoblotting with various antibodies, as shown in Fig. 2. Input represents 1% of total cell lysates.
Fig 4.

A26 interacts individually with A16 and G9 but not with G3 and L5 proteins in vitro. (A) GST-A26 pulled down A16 and G9 individually. HEK293T cells were transfected with plasmids expressing HA-tagged A16, Myc-tagged G9, or both and harvested 24 h later. The lysates were subjected to GST pulldown analyses using 30 μg of recombinant GST and GST-A26 proteins as described in Materials and Methods and analyzed by immunoblotting with anti-HA (A16) (1:1,000) and anti-Myc (G9) (1:1,000) antibodies. (B) GST-A26 did not pull down G3 and L5. HEK293T cells were transfected with plasmids expressing vaccinia G3 and L5 proteins and harvested 24 h later, and the lysates were subjected to GST pulldown analyses as described above using anti-G3 (1:1,000) and anti-L5 (1:1,000) antibodies. Input represents 1% of total cell lysates.
A26 interacts with A16 and G9 proteins individually in transfected HEK293T cells.
Next, to confirm that A26 could interact with A16 and G9 proteins individually in vivo, 293T cells were transiently transfected, individually or in different combinations, with expression plasmids encoding V5-tagged A26, A16, and G9 proteins, and lysates were harvested for immunoprecipitation analyses. As shown in Fig. 5, anti-V5 antibody immunoprecipitated A26 protein from cells coexpressing A16, G9, or both. Immunoblot analyses showed that A26 protein brought down A16 and G9 proteins from cells expressing either one or both proteins. Notably, without A26 protein expression, anti-V5 antibody did not cross-react with either the A16 or G9 protein, providing strong evidence that the G9 and A16 proteins were coimmunoprecipitated through interaction with A26 protein. The results are consistent with the above in vitro pulldown showing that A26 interacts with the A16 and G9 proteins individually in vivo. We noticed that an obvious increase of A26 protein level in cells coexpressing A16 and G9 proteins led to more A26 protein immunoprecipitated by anti-V5 antibody and consequently more A16 and G9 proteins coimmunoprecipitated, implying that A16 and G9 proteins may stabilize A26 protein in vivo.
Fig 5.

A26 protein interacts with A16 and G9 proteins individually in transfected HEK293T cells. HEK293T cells were transiently transfected with plasmids expressing A26-V5, A16, or G9 protein individually or in combination. Coimmunoprecipitations were performed with anti-V5 antibody-conjugated agarose beads and analyzed by immunoblotting with anti-V5 (1:4,000), anti-A16 (1:1,000), and anti-G9 (1:5,000) antibodies. Input represents 1% of total cell lysates.
To further support that A26 interactions with A16 and G9 are specific, we engineered codon-optimized viral ORFs to express other components of vaccinia EFC in transiently transfected 293T cells. Some of the EFC components such as A28 and A21 were expressed poorly in 293T cells (data not shown). As shown in Fig. 6, A26 associated with A16 and G9 in coimmunoprecipitation analyses (Fig. 6A) but not with G3 and L5 (Fig. 6B) although the latter two were recently shown to form a subcomponent complex (61). A26 protein also did not associate with the H2 protein (Fig. 6C). In summary, our results showed a specific interaction among the A26, A16, and G9 proteins not shared by other EFC component proteins or subcomponent complexes.
Fig 6.
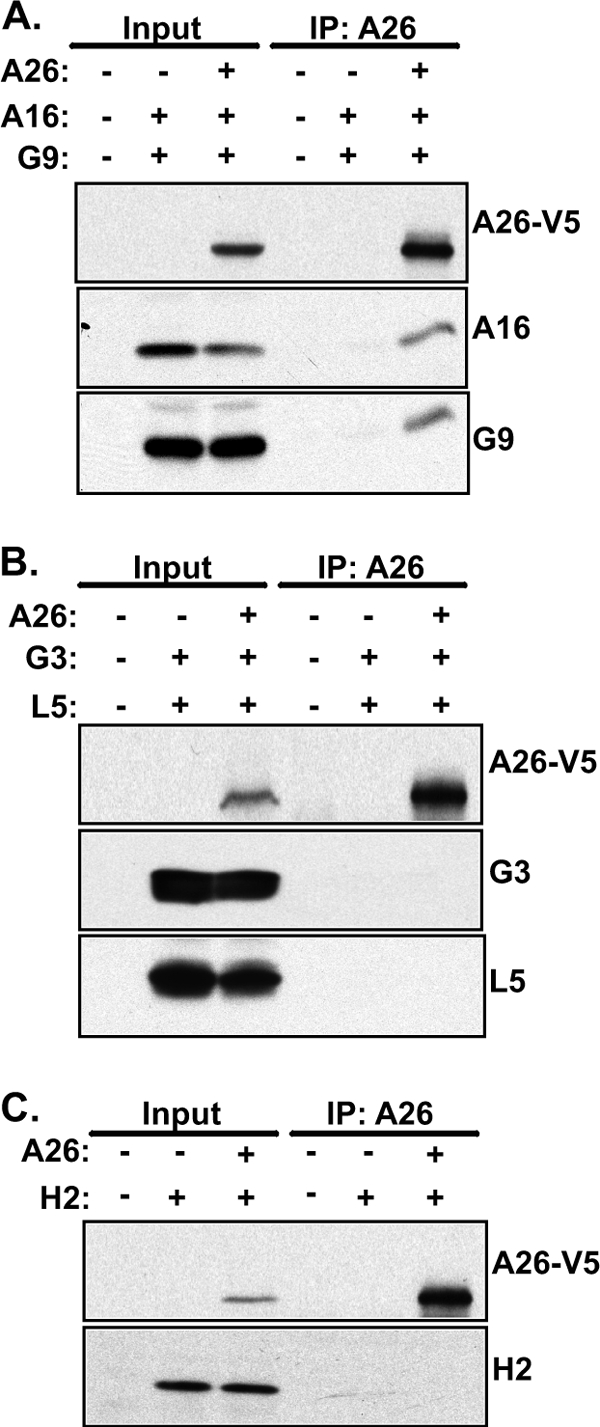
A26 does not interact with EFC component proteins H2, G3, and L5 in transfected HEK293T cells. HEK293T cells were transiently transfected with individual plasmids expressing A26-V5 along with A16 and G9 (A) or G3 and L5 (B) or H2 (C) protein, harvested for coimmunoprecipitations with anti-V5 agarose beads, and analyzed by immunoblotting with anti-V5 (1:4,000), anti-A16 (1:1,000), anti-G9 (1:5,000), anti-G3 (1:1,000), anti-L5 (1:1,000), and anti-H2 (1:1,000) antibodies. Input represents 1% of total cell lysates.
A26 and A56/K2 proteins do not interact with the same A16 and G9 proteins in vitro and in vivo.
Previous studies showed that A56 and K2 are fusion regulators on the infected cell surface and bind to A16 and G9 proteins of the progeny virus to prevent cell-to-cell fusion (57–59). Therefore, we investigated whether A26 and A56/K2 interact with the same A16 and G9 in the virus-infected cells. To address this issue, we first tested whether the presence of A56/K2 proteins in the infected cells will reduce the availability of A16 and G9 proteins interacting with exogenous GST-A26 protein in vitro. HeLa cells were infected with WR, WRΔA56R, and WRΔK2L virus at an MOI of 5 PFU per cell and harvested at 24 h p.i. for GST pulldown analyses. As shown in Fig. 7A, GST-A26 protein specifically pulled down comparable levels of A16 and G9 proteins from cells infected with the wild-type WR, WRΔA56R, and WRΔK2L viruses, suggesting that A56/K2 proteins in cells did not interfere with the ability of A16 and G9 proteins to bind to GST-A26 protein. In addition, GST-A26 did not pull down A56 protein in vitro. Due to the lack of antibody recognizing the endogenous K2 protein, we repeated the GST pulldown assays using cell lysates from transiently transfected 293T cells expressing Flag-tagged K2, HA-tagged A16, Myc-tagged G9, and A56-fused GFP proteins. Consistently, GST-A26, but not GST, pulled down A16 and G9 but not the A56 and K2 proteins (Fig. 7B). The data suggest that A26 protein binds to the A16 and G9 proteins that are not in complex with the A56/K2 proteins in vitro.
Fig 7.
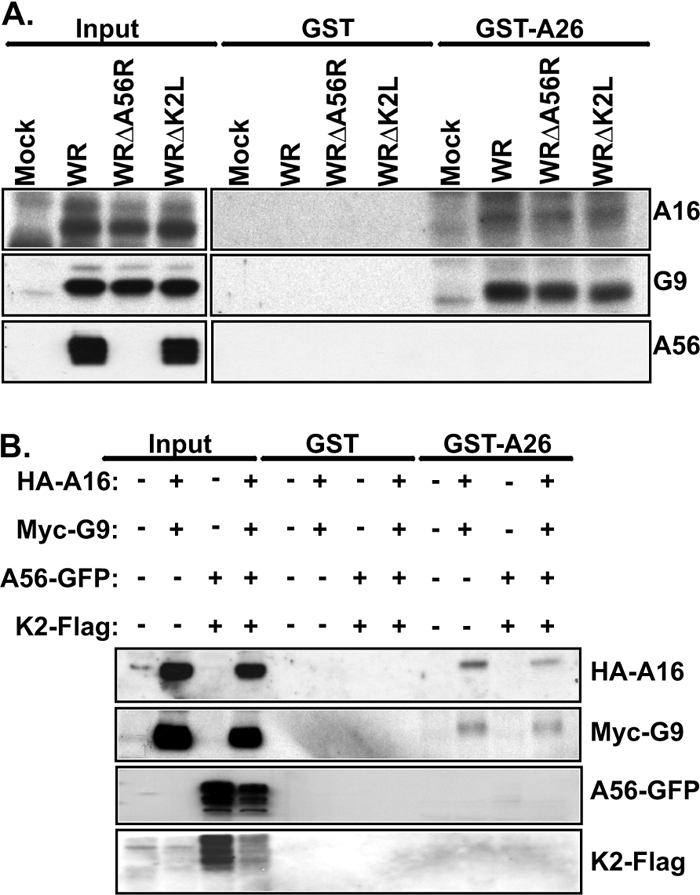
A26 protein in vitro binds to A16 and G9 proteins that are not in complex with A56/K2. (A) GST-A26 pulled down A16 and G9 proteins but not A56 protein. HeLa cells were infected with WR, WRΔA56R, and WRΔK2L at an MOI of 5 PFU/cell; cell lysates were harvested at 24 h p.i. for GST pulldown analyses with 30 μg of recombinant GST and GST-A26 proteins and analyzed by immunoblotting with anti-A16 (1:1,000), anti-G9 (1:5,000), and anti-A56 MAb (B2D10) (1:500). (B) GST-A26 did not pull down K2 protein. HEK293T cells were transiently transfected with plasmids expressing HA-A16, Myc-G9, A56-GFP, and K2-Flag proteins individually or in combination. The lysates were harvested for GST pulldown and analyzed by immunoblotting as described for panel A with anti-HA (1:1,000), anti-Myc (1:1,000), anti-GFP (1:4,000), and anti-Flag (1:1,000) antibodies. Input represents 1% of total cell lysates.
Next, to verify whether the A26 and A56/K2 proteins could interact with the same A16 and G9 proteins in vivo, 293T cells were transiently transfected, individually or in different combinations, with plasmids encoding V5-tagged A26, GFP-fused A56, and Flag-tagged K2 proteins. The cells were subsequently infected with VTF7-3 and harvested at 24 h p.i. for immunoprecipitation analyses. As shown in Fig. 8A, the anti-V5 antibody immunoprecipitated A26 protein associated with viral G9 and A16 proteins in the infected cells; however, neither the A56 nor K2 protein was detected in the immunoprecipitate using anti-GFP and anti-Flag antibodies, indicating that A26 protein brought down a subset of G9 and A16 proteins that were not bound by A56/K2 proteins in the infected cells. To reinforce the concept that A26 and A56/K2 proteins did not interact with the same A16/G9 proteins, we then immunoprecipitated A56 protein using an anti-GFP antibody and found that not only A56 protein but also K2, A16, and G9 were brought down from the infected cells (Fig. 8B). However, no A26 protein was detected in the immunoprecipitates. Finally, we immunoprecipitated K2 protein using an anti-Flag antibody. Again, immunoblot analyses confirmed that K2 proteins were associated with A56, A16, and G9 proteins in the infected cells but not with A26 protein (Fig. 8C). Together, the above results showed that the A26 and A56/K2 proteins do not bind to same A16 and G9 proteins in vitro and in vivo.
Fig 8.
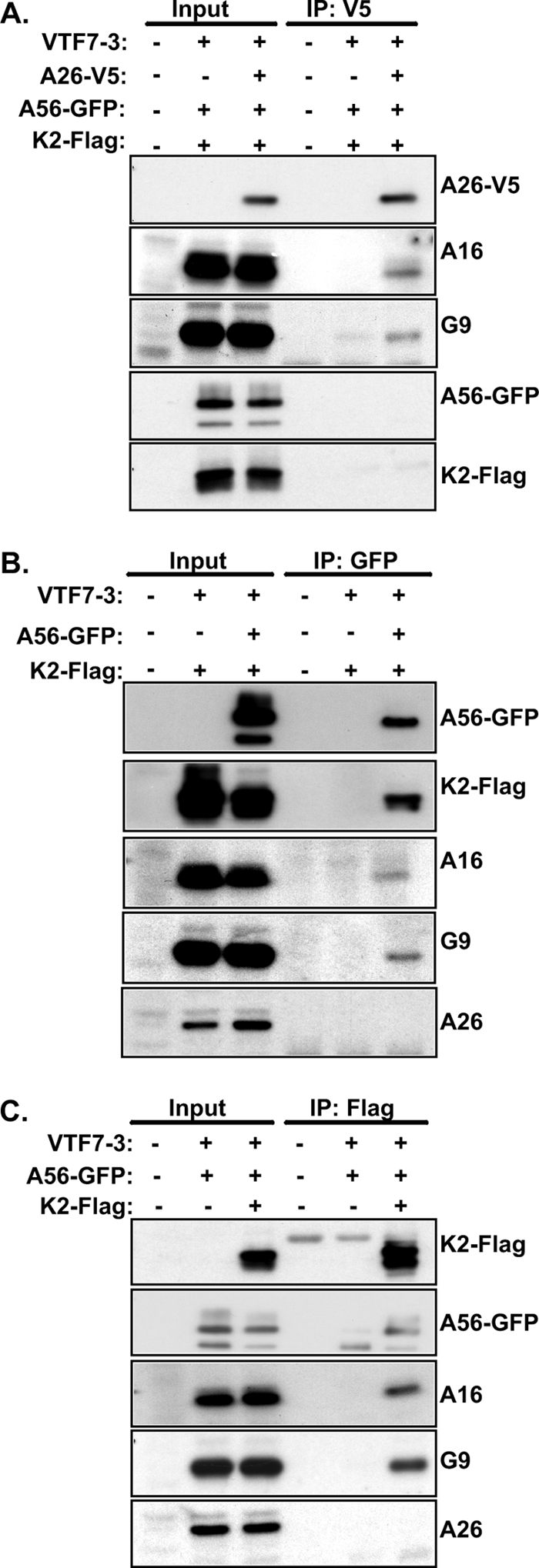
A26 protein binds to A16 and G9 proteins in vivo that are not in complex with A56/K2. (A) HEK293T cells were infected with VTF7-3 at an MOI of 5 PFU/cell and transfected with plasmids expressing A26-V5, A56-GFP, and K2-Flag proteins; cells were harvested at 24 h p.i. for coimmunoprecipitations with anti-V5 antibody conjugated to agarose beads and analyzed by anti-V5 (1:4,000), anti-A16(1:1,000), anti-G9 (1:5,000), anti-GFP (1:4,000), and anti-Flag (1:1,000) antibodies. (B) HEK293T cells were infected with VTF7-3 at an MOI of 5 PFU/cell, transfected with plasmids expressing A56-GFP and K2-Flag proteins, harvested at 24 h p.i. for coimmunoprecipitations with anti-GFP antibody, and analyzed by immunoblotting as described for panel A. (C) HEK293T cells were infected with VTF7-3 at an MOI of 5 PFU/cell, transfected with plasmids expressing A56-GFP and K2-Flag proteins as described for panel B for coimmunoprecipitations with anti-Flag antibody conjugated to agarose beads, and analyzed by immunoblotting as described for panel A. Input represents 1% of total cell lysates.
Low pH increases the hydrophilicity of A26 and A27 proteins.
The above-described experiments supported our hypothesis that vaccinia virus A26 protein acts as a fusion suppressor of the wild-type vaccinia virus WR MV particles by binding to A16 and G9 proteins so that the MV particles do not fuse with the plasma membrane of cells at neutral pH. Upon infecting cells, these MV particles are endocytosed into HeLa cells in which acidification of the endosomal environment is required to initiate membrane fusion (23, 49), implying that the low-pH environment may provoke conformational changes of A26 protein and reduce its association with A16 and G9 proteins. In order to investigate this, we took advantage of the nonionic detergent Triton X-114 that allows partitioning of proteins into hydrophilic and hydrophobic phases at room temperature upon centrifugation (4). Purified WR MV particles were treated with 2% Triton X-114 in a solution of pH 7.4 or 4.7, as described in Materials and Methods, and centrifuged, and both aqueous and detergent phases were collected for immunoblot analyses (Fig. 9). At neutral pH, viral envelope proteins such as A25, A26, and A27 and EFC components A16, G9, G3, and L5 were hydrophobic and only partitioned in the detergent (bottom) phase. Envelope L1, D8, and A4 core proteins as well as H3 protein were more hydrophilic and became solubilized into the aqueous (top) phase. Interestingly, low-pH treatment of MVs at pH 4.7 significantly increased partition of A26 and A27 proteins into the aqueous phase, suggesting that a low-pH environment may cause structural alteration of A26 and A27 proteins. In contrast, low-pH treatment did not alter the aqueous partition of other above-mentioned proteins, including A25, A16, G9, G3, and L5, all of which remained in the hydrophobic phase (bottom fraction), nor did it increase partition of L1, D8, H3, and A4 into aqueous phase. The increased hydrophilicity of A26 and A27 proteins in the Triton X-114 partition experiments may imply specific conformational changes of both A26 and A27 proteins induced by low pH.
Fig 9.
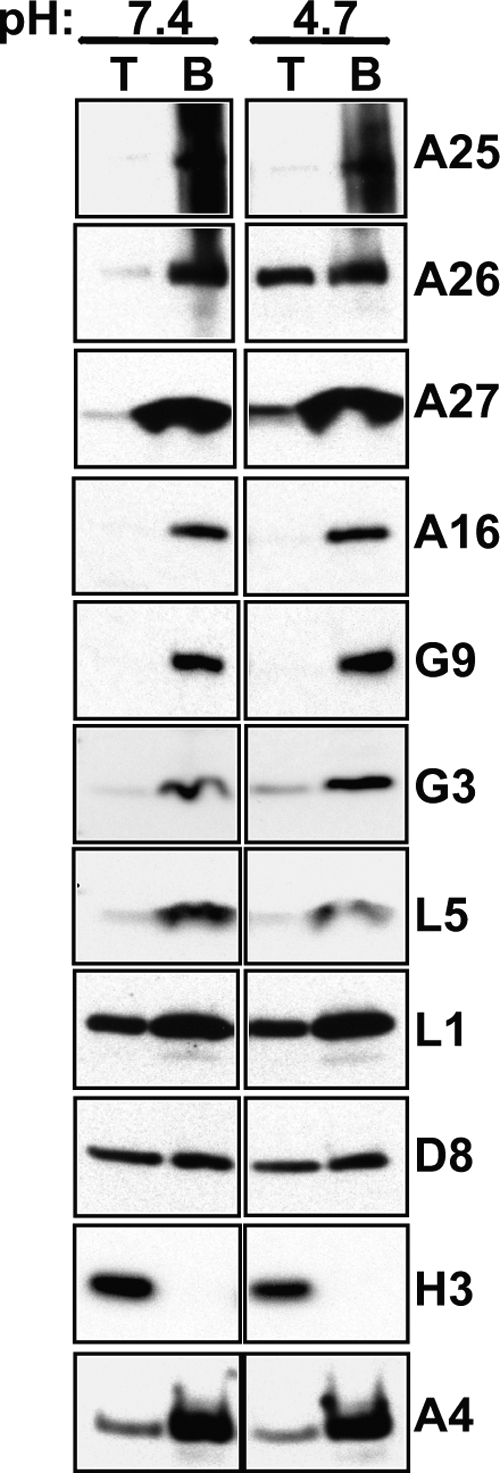
Effect of low-pH treatment on the hydrophilicity/hydrophobicity of A26 and A27 proteins. Vaccinia MV particles (8 μg) were solubilized in 2% Triton X-114 in buffer of pH 7.4 or pH 4.7, incubated at 37°C for 10 min, and then centrifuged at 300 × g for 3 min, as described in Materials and Methods. The aqueous (top fraction, T) and detergent (bottom fraction, B) phases were collected and analyzed by immunoblot analysis with various antibodies as described in Materials and Methods.
Viral A26 protein, alone or in complex with A27 protein, dissociated from vaccinia virus MV particles upon low-pH treatment in vitro.
The above results in the Triton X-114 experiments prompted us to test whether the structural changes of viral A26 and A27 proteins induced by low pH could lead to protein dissociation from MV particles. We therefore treated purified WR MV particles with either neutral (pH 7.4) or acidic (pH 4.7) PBS at 37°C for 3 min, followed by centrifugation to separate supernatant and pellets for immunoblot analysis (Fig. 10A). Neutral buffer (pH 7.4) treatment did not affect the integrity of MV particles, and both A26 and A27 proteins were detected in the pellets (Fig. 10A, lane 4), with only weak signals in the supernatant fractions (Fig. 10A, lane 1). Interestingly, after treatment of MV particles with acidic buffer (pH 4.7) for 3 min, the amounts of A26 and A27 proteins released into the supernatant increased (Fig. 10A, lane 2). Prolonged acidic buffer treatment (>3 min) did not further increase the amounts of A26 and A27 proteins in the supernatant (Fig. 10A, lane 3). Other envelope proteins, such as EFC components G9, A16, and control H3, remained in the pellets regardless of the pH treatment, showing that acid triggered specific dissociation of A26 and A27 proteins from MV particles.
Fig 10.
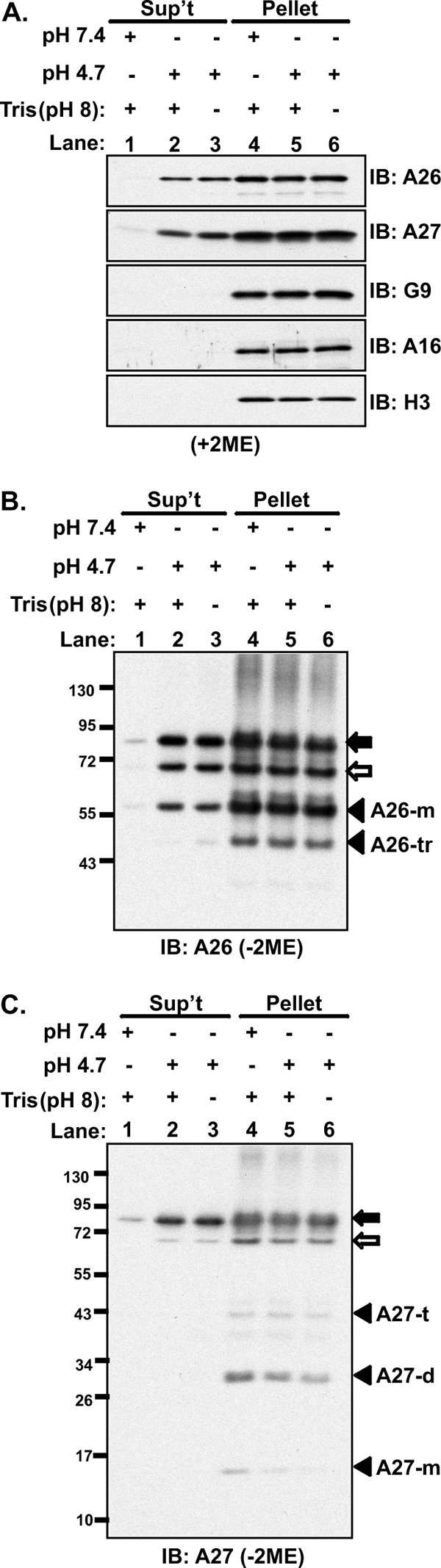
Low pH dissociates vaccinia A26 and A26-A27 protein complexes from MVs. (A). Purified vaccinia virus MV particles (24 μg) were treated with PBS at pH 7.4 or PBS at pH 4.7 at 37°C for 3 min with or without neutralization with 15 mM Tris-HCl (pH 8), followed by centrifugation. The supernatant (Sup't) and pellet fractions were collected, separated on SDS-PAGE gels under reducing (with 2-mercaptoethanol [+2ME]) conditions, and analyzed by immunoblotting (IB) using anti-A26 (1:1,000), anti-A27 (1:5,000), anti-G9 (1:5,000), anti-A16 (1:1,000), and anti-H3 (1:1,000) antibodies. (B and C) The samples described in panel A were separated under nonreducing (−2ME) conditions on 4 to 12% SDS-PAGE gels and analyzed by immunoblotting with anti-A26 (1:1,000) (B) and anti-A27 (1:5,000) (C) antibodies. The m, d, t, and tr suffixes with the A26 and A27 proteins represent monomer, dimer, trimer, and truncated, respectively.
Previous studies showed that A26 protein is incorporated into MV particles in three different forms, A26 alone and A26-A27 protein complexes of 70 and 90 kDa formed by disulfide bonding (10). We thus performed SDS-PAGE under the nonreducing (without with 2-mercaptoethanol [2ME]) condition and analyzed immunoblots using anti-A26 antibody. The results showed that A26 monomer (∼58 kDa) as well as A26-A27 protein complexes of 70 kDa and 90 kDa were detected in supernatant fractions upon acid treatment (Fig. 10B). Consistently, anti-A27 antibody detected A27 protein in the 70-kDa and 90-kDa protein complexes in the supernatant (Fig. 10C). These results provide direct evidence that A26 protein, alone or in complex with A27 protein, dissociates from the MV particles in response to the low-pH condition.
DISCUSSION
In this study, we initially observed that vaccinia virus MV A26 protein interacted with four components, A16, G9, G3, and L5, of the viral EFC in virus-infected HeLa cells. Using coimmunoprecipitation in transiently transfected cells and GST pulldown assays, we demonstrated that the A26 protein bound to the A16 and G9 proteins in vivo and in vitro, suggesting that two other EFC components, G3 and L5, were indirectly associated with A26 protein. Furthermore, A26 interacted individually with the A16 and G9 proteins, suggesting different binding domains on the A26 protein. Indeed, binding of A26 to A16 and G9 appeared not to be competitive since single or double expression of the latter two proteins in the same cells did not affect the binding of A26 protein. The specificity of such interactions was corroborated by the fact that other abundant virion proteins, A4 and D8, and EFC components, G3 and L5, did not bind to A26 protein in all experiments. However, our data do not exclude the involvement of other EFC components binding to A26 protein since we did not have antibodies against every component and since some EFC component proteins were poorly expressed, making their inclusion in the study difficult.
Previously, Senkevich et al. used mass spectrometry to identify vaccinia virus EFC components associated with H2 protein (47); however, A26 protein was not detected in their coimmunoprecipitation analyses. It could be that they used membrane materials floating at the top of a 36% sucrose cushion, which was described as “enriched in viral membrane proteins and was presumably derived from immature virions or their precursors membrane materials” (47), whereas we used total cell lysates for the coimmunoprecipitation experiments reported here. Nevertheless, whether this is indeed the cause of different results is not known.
Studies on membrane fusion induced by poxviruses have utilized two cell fusion systems: fusion from without (FFWO) triggered by incoming MVs and fusion from within (FFWI) triggered by cell-produced EVs (Table 1) (17, 19, 31, 36). Both FFWO and FFWI are regulated by virus-encoded proteins; i.e., A56/K2 proteins suppress FFWI (24, 30, 55, 63), and A26 protein suppresses FFWO (8). Both fusion suppressions are overcome by acidic treatment (19, 28), and both fusions require the EFC to execute the fusion step (36, 57, 59). A56/K2 proteins were previously shown to copurify with A16 and G9 proteins, and both A16 and G9 proteins are required for binding to A56/K2 proteins, showing that A56/K2 proteins bind to a protein complex composed of A16 and G9 proteins (58, 59). In contrast, here we showed that A26 could bind to either A16 or G9 protein individually (Table 1). It is worth noting that although the A26 and A56/K2 proteins were all present in the infected cells, our GST-A26 pulldown and coimmunoprecipitation results clearly showed that they bind to different A16 and G9 proteins. This conclusion is not totally unexpected since the A56/K2 proteins are mainly located on the cell surface, whereas A26 is in the cytoplasm of the infected cells. Our results are also consistent with a previous report from Wagenaar and Moss, who identified K2, A16, G9, I1, C3, J5, and O2 proteins, but not A26 protein, associated with A56-TAP (where TAP is a tandem affinity purification tag) constructs in infected cell lysates (57). Taken together, these results indicate that vaccinia virus has evolved a common mechanism shared by MVs and EVs to suppress virus-induced membrane fusion by physically targeting the viral EFC. The data also imply that although the EFC is a complex of 12 proteins required for membrane fusion, not all components work at the same step during membrane fusion. Rather, some components such as A16 and G9 may be required for fusion “priming,” whereas others are needed for membrane fusion execution. Although 12 EFC components form a complex in MV membranes, their protein topology is not the same. A21, A28, G3, H2, L5, and O3 contain a transmembrane region at or close to the N terminus, and A16, F9, G9, I2, J5, and L1 contain a transmembrane region at the C terminus (5, 37, 44, 47). Whether these structures reflect different functions during membrane fusion activation remains to be investigated.
Table 1.
Vaccinia virus suppression of membrane fusion
| Virus suppression factor | Role/presence of factor by type of fusion |
|
|---|---|---|
| MV-mediated FFWO | EV-mediated FFWI | |
| Suppressor protein | A26 | A56/K2 |
| Sensitivity to low pH | + | + |
| Interaction with EFC components | Bind to A16 and G9 individually | Bind to A16-G9 protein complexa |
According to reference 59.
Sensitivity to acidic environments is another common feature shared by the above fusion suppressors (Table 1). Documented experimental procedures (19, 23, 49, 52) usually used acidic buffer of pH 4.7 to 5.0, mimicking intracellular vesicular pH, to trigger FFWO and FFWI. How acidic pH neutralizes these viral fusion suppressors in order to activate EFC-mediated membrane fusion remains unknown. Here, we investigated whether A26 protein structure is acid sensitive by monitoring the protein hydrophilicity/hydrophobicity shift in acidic solution containing Triton X-114, which was previously used for investigation of a low-pH-induced conformational change of the fusion proteins of alphavirus and herpes simplex virus (16, 27). Furthermore, we tested whether a low-pH treatment of purified MVs resulted in the dissociation of A26 protein from MVs. Indeed, our results described here support the idea that A26 protein is acid sensitive; however, it remains puzzling to us that not all of the A26 protein on MVs is equally sensitive to low pH and that a significant amount of A26 protein (and 70- and 90-kDa A26-A27 protein complexes) remained resistant to Triton X-114 extraction to aqueous phase or dissociation from virions. Although we previously showed that WRΔA26L, a mutant vaccinia virus with a deletion of the A26 WR ORF, caused significant FFWO in HeLa cells at neutral pH (8), these acid-treated vaccinia virus MVs did not exhibit a fusogenic phenotype at neutral pH (data not shown), suggesting that additional steps are required to fully nullify the fusion suppression function of A26 protein. This conclusion seemed to echo the study by Townsley and Moss, who proposed that two distinct low-pH steps are required to promote vaccinia virus entry (49). In the future, specific issues regarding A26 protein structure and pH relationship will be addressed in order to gain an understanding of the poxviral fusion activation mechanism.
ACKNOWLEDGMENTS
We thank Sue-Ping Lee and Shu-Ping Tsai for assistance with EM analyses. We also thank Ya-Fan Chung for purification of recombinant GST proteins.
This work was supported by grants from Academia Sinica and the National Science Council of the Republic of China (NSC-100-2320-B-001-006).
Footnotes
Published ahead of print 25 January 2012
REFERENCES
- 1. Armstrong JA, Metz DH, Young MR. 1973. The mode of entry of vaccinia virus into L cells. J. Gen. Virol. 21:533–537 [DOI] [PubMed] [Google Scholar]
- 2. Bengali Z, Townsley AC, Moss B. 2009. Vaccinia virus strain differences in cell attachment and entry. Virology 389:132–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bisht H, Weisberg AS, Moss B. 2008. Vaccinia virus l1 protein is required for cell entry and membrane fusion. J. Virol. 82:8687–8694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bordier C. 1981. Phase separation of integral membrane proteins in Triton X-114 solution. J. Biol. Chem. 256:1604–1607 [PubMed] [Google Scholar]
- 5. Brown E, Senkevich TG, Moss B. 2006. Vaccinia virus F9 virion membrane protein is required for entry but not virus assembly, in contrast to the related L1 protein. J. Virol. 80:9455–9464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carter GC, Law M, Hollinshead M, Smith GL. 2005. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J. Gen. Virol. 86:1279–1290 [DOI] [PubMed] [Google Scholar]
- 7. Chang A, Metz DH. 1976. Further investigations on the mode of entry of vaccinia virus into cells. J. Gen. Virol. 32:275–282 [DOI] [PubMed] [Google Scholar]
- 8. Chang SJ, Chang YX, Izmailyan R, Tang YL, Chang W. 2010. Vaccinia virus A25 and A26 proteins are fusion suppressors for mature virions and determine strain-specific virus entry pathways into HeLa, CHO-K1, and L cells. J. Virol. 84:8422–8432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chang SJ, et al. 2009. Poxvirus host range protein CP77 contains an F-box-like domain that is necessary to suppress NF-B activation by tumor necrosis factor alpha but is independent of its host range function. J. Virol. 83:4140–4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ching YC, et al. 2009. Disulfide bond formation at the C termini of vaccinia virus A26 and A27 proteins does not require viral redox enzymes and suppresses glycosaminoglycan-mediated cell fusion. J. Virol. 83:6464–6476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chiu WL, Lin CL, Yang MH, Tzou DL, Chang W. 2007. Vaccinia virus 4c (A26L) protein on intracellular mature virus binds to the extracellular cellular matrix laminin. J. Virol. 81:2149–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chung CS, et al. 2006. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J. Virol. 80:2127–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chung CS, Hsiao JC, Chang YS, Chang W. 1998. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 72:1577–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Condit RC, Moussatche N, Traktman P. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv. Virus Res. 66:31–124 [DOI] [PubMed] [Google Scholar]
- 15. Dales S, Kajioka R. 1964. The cycle of multiplication of vaccinia virus in Earle's strain L cells. I. Uptake and penetration. Virology 24:278–294 [DOI] [PubMed] [Google Scholar]
- 16. Dollery SJ, Delboy MG, Nicola AV. 2010. Low pH-induced conformational change in herpes simplex virus glycoprotein B. J. Virol. 84:3759–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Doms RW, Blumenthal R, Moss B. 1990. Fusion of intra- and extracellular forms of vaccinia virus with the cell membrane. J. Virol. 64:4884–4892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Engelstad M, Smith GL. 1993. The vaccinia virus 42-kDa envelope protein is required for the envelopment and egress of extracellular virus and for virus virulence. Virology 194:627–637 [DOI] [PubMed] [Google Scholar]
- 19. Gong SC, Lai CF, Esteban M. 1990. Vaccinia virus induces cell fusion at acid pH and this activity is mediated by the N-terminus of the 14-kDa virus envelope protein. Virology 178:81–91 [DOI] [PubMed] [Google Scholar]
- 20. Guan KL, Dixon JE. 1991. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal. Biochem. 192:262–267 [DOI] [PubMed] [Google Scholar]
- 21. Hsiao JC, Chung CS, Chang W. 1998. Cell surface proteoglycans are necessary for A27L protein-mediated cell fusion: identification of the N-terminal region of A27L protein as the glycosaminoglycan-binding domain. J. Virol. 72:8374–8379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hsiao JC, Chung CS, Chang W. 1999. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 73:8750–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang CY, et al. 2008. A novel cellular protein, VPEF, facilitates vaccinia virus penetration into HeLa cells through fluid phase endocytosis. J. Virol. 82:7988–7999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ichihashi Y, Dales S. 1971. Biogenesis of poxviruses: interrelationship between hemagglutinin production and polykaryocytosis. Virology 46:533–543 [DOI] [PubMed] [Google Scholar]
- 25. Ichihashi Y, Oie M. 1983. The activation of vaccinia virus infectivity by the transfer of phosphatidylserine from the plasma membrane. Virology 130:306–317 [DOI] [PubMed] [Google Scholar]
- 26. Izmailyan RA, Huang CY, Mohammad S, Isaacs SN, Chang W. 2006. The envelope G3L protein is essential for entry of vaccinia virus into host cells. J. Virol. 80:8402–8410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kielian M, Helenius A. 1985. pH-induced alterations in the fusogenic spike protein of Semliki Forest virus. J. Cell Biol. 101:2284–2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kohno K, Sambrook J, Gething M-J. 1988. Effect of lysosomotropic agents on the entry of vaccinia virus into CV-1 cells. J. Cell. Biochem. 38(Suppl 12):13220878 [Google Scholar]
- 29. Laliberte JP, Moss B. 2009. Appraising the apoptotic mimicry model and the role of phospholipids for poxvirus entry. Proc. Natl. Acad. Sci. U. S. A. 106:17517–17521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Law KM, Smith GL. 1992. A vaccinia serine protease inhibitor which prevents virus-induced cell fusion. J. Gen. Virol. 73:549–557 [DOI] [PubMed] [Google Scholar]
- 31. Law M, Carter GC, Roberts KL, Hollinshead M, Smith GL. 2006. Ligand-induced and nonfusogenic dissolution of a viral membrane. Proc. Natl. Acad. Sci. U. S. A. 103:5989–5994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lin CL, Chung CS, Heine HG, Chang W. 2000. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J. Virol. 74:3353–3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Locker JK, et al. 2000. Entry of the two infectious forms of vaccinia virus at the plasma membrane is signaling-dependent for the IMV but not the EEV. Mol. Biol. Cell 11:2497–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mercer J, Helenius A. 2008. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320:531–535 [DOI] [PubMed] [Google Scholar]
- 35. Moss B. 2007. Poxviridae: the viruses and their replication, p 2905–2945 In Knipe DM, et al. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 36. Moss B. 2006. Poxvirus entry and membrane fusion. Virology 344:48–54 [DOI] [PubMed] [Google Scholar]
- 37. Nichols RJ, Stanitsa E, Unger B, Traktman P. 2008. The vaccinia virus gene I2L encodes a membrane protein with an essential role in virion entry. J. Virol. 82:10247–10261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oie M, Shida H, Ichihashi Y. 1990. The function of the vaccinia hemagglutinin in the proteolytic activation of infectivity. Virology 176:494–504 [DOI] [PubMed] [Google Scholar]
- 39. Ojeda S, Domi A, Moss B. 2006. Vaccinia virus G9 protein is an essential component of the poxvirus entry-fusion complex. J. Virol. 80:9822–9830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ojeda S, Senkevich TG, Moss B. 2006. Entry of vaccinia virus and cell-cell fusion require a highly conserved cysteine-rich membrane protein encoded by the A16L gene. J. Virol. 80:51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Payne L. 1978. Polypeptide composition of extracellular enveloped vaccinia virus. J. Virol. 27:28–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Payne LG, Norrby E. 1978. Adsorption and penetration of enveloped and naked vaccinia virus particles. J. Virol. 27:19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Resch W, Hixson KK, Moore RJ, Lipton MS, Moss B. 2007. Protein composition of the vaccinia virus mature virion. Virology 358:233–247 [DOI] [PubMed] [Google Scholar]
- 44. Satheshkumar PS, Moss B. 2009. Characterization of a newly identified 35-amino-acid component of the vaccinia virus entry/fusion complex conserved in all chordopoxviruses. J. Virol. 83:12822–12832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Seki M, Oie M, Ichihashi Y, Shida H. 1990. Hemadsorption and fusion inhibition activities of hemagglutinin analyzed by vaccinia virus mutants. Virology 175:372–384 [DOI] [PubMed] [Google Scholar]
- 46. Senkevich TG, Moss B. 2005. Vaccinia virus H2 protein is an essential component of a complex involved in virus entry and cell-cell fusion. J. Virol. 79:4744–4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Senkevich TG, Ojeda S, Townsley A, Nelson GE, Moss B. 2005. Poxvirus multiprotein entry-fusion complex. Proc. Natl. Acad. Sci. U. S. A. 102:18572–18577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Senkevich TG, Ward BM, Moss B. 2004. Vaccinia virus entry into cells is dependent on a virion surface protein encoded by the A28L gene. J. Virol. 78:2357–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Townsley AC, Moss B. 2007. Two distinct low-pH steps promote entry of vaccinia virus. J. Virol. 81:8613–8620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Townsley AC, Senkevich TG, Moss B. 2005. The product of the vaccinia virus L5R gene is a fourth membrane protein encoded by all poxviruses that is required for cell entry and cell-cell fusion. J. Virol. 79:10988–10998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Townsley AC, Senkevich TG, Moss B. 2005. Vaccinia virus A21 virion membrane protein is required for cell entry and fusion. J. Virol. 79:9458–9469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Townsley AC, Weisberg AS, Wagenaar TR, Moss B. 2006. Vaccinia virus entry into cells via a low-pH-dependent endosomal pathway. J. Virol. 80:8899–8908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Turner PC, et al. 2007. Vaccinia virus temperature-sensitive mutants in the A28 gene produce non-infectious virions that bind to cells but are defective in entry. Virology 366:62–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Turner PC, Moyer RW. 1995. Orthopoxvirus fusion inhibitor glycoprotein SPI-3 (open reading frame K2L) contains motifs characteristic of serine proteinase inhibitors that are not required for control of cell fusion. J. Virol. 69:5978–5987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Turner PC, Moyer RW. 1992. An orthopoxvirus serpinlike gene controls the ability of infected cells to fuse. J. Virol. 66:2076–2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Turner PC, Moyer RW. 2008. The vaccinia virus fusion inhibitor proteins SPI-3 (K2) and HA (A56) expressed by infected cells reduce the entry of superinfecting virus. Virology 380:226–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wagenaar TR, Moss B. 2007. Association of vaccinia virus fusion regulatory proteins with the multicomponent entry/fusion complex. J. Virol. 81:6286–6293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wagenaar TR, Moss B. 2009. Expression of the A56 and K2 proteins is sufficient to inhibit vaccinia virus entry and cell fusion. J. Virol. 83:1546–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wagenaar TR, Ojeda S, Moss B. 2008. Vaccinia virus A56/K2 fusion regulatory protein interacts with the A16 and G9 subunits of the entry fusion complex. J. Virol. 82:5153–5160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Whitbeck JC, Foo CH, Ponce de Leon M, Eisenberg RJ, Cohen GH. 2009. Vaccinia virus exhibits cell-type-dependent entry characteristics. Virology 385:383–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wolfe CL, Moss B. 2011. Interaction between the G3 and L5 proteins of the vaccinia virus entry-fusion complex. Virology 412:278–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wolfe CL, Ojeda S, Moss B. 2012. Transcriptional repression and RNA silencing act synergistically to demonstrate the function of the eleventh component of the vaccinia virus entry-fusion complex. J. Virol. 86:293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhou J, Sun XY, Fernando GJ, Frazer IH. 1992. The vaccinia virus K2L gene encodes a serine protease inhibitor which inhibits cell-cell fusion. Virology 189:678–686 [DOI] [PubMed] [Google Scholar]