

Abstract
Hypothetically, since native HIV-1 Env trimers are exclusively recognized by neutralizing antibodies, they might induce the neutralizing antibodies in a vaccine setting. This idea has not been evaluated due to the difficulty of separating trimers from nonfunctional Env (uncleaved gp160 and gp41 stumps). The latter are immunodominant and induce nonneutralizing antibodies. We previously showed that nonfunctional Env can be selectively cleared from virus-like particle (VLP) surfaces by enzyme digests (E. T. Crooks, T. Tong, K. Osawa, and J. M. Binley, J.Virol. 85:5825, 2011). Here, we investigated the effects of these digests on the antigenicity of VLPs and their sensitivity to neutralization. Before digestion, WT VLPs (bearing wild-type Env) and UNC VLPs (bearing uncleaved gp160) were recognized by various Env-specific monoclonal antibodies (MAbs), irrespective of their neutralizing activity, a result which is consistent with the presence of nonfunctional Env. After digestion, only neutralizing MAbs recognized WT VLPs, consistent with selective removal of nonfunctional Env (i.e., “trimer VLPs”). Digests eliminated the binding of all MAbs to UNC VLPs, again consistent with removal of nonfunctional Env. An exception was MAb 2F5, which weakly bound to digested UNC VLPs and bald VLPs (bearing no Env), perhaps due to lipid cross-reactivity. Trimer VLPs were infectious, and their neutralization sensitivity was largely comparable to that of undigested WT VLPs. However, they were ∼100-fold more sensitive to the MAbs 4E10 and Z13e1, suggesting increased exposure of the gp41 base. Importantly, a scatterplot analysis revealed a strong correlation between MAb binding and neutralization of trimer VLPs. This suggests that trimer VLPs bear essentially pure native trimer that should allow its unfettered evaluation in a vaccine setting.
INTRODUCTION
Broadly neutralizing antibodies (bnAbs) are widely expected to be a crucial component of the immunity imparted by an effective HIV-1 vaccine (32, 42). These bnAbs neutralize the virus by being able to bind to native trimeric Envelope glycoprotein (Env) spikes on HIV-1 particle surfaces, thereby interfering with receptor engagement and infection (16, 26, 30, 52). These Env spikes consist of trimers of gp120/gp41 heterodimers, in which gp120 is the surface subunit and gp41 is the transmembrane-anchoring subunit. By virtue of their compact and highly glycosylated nature, Env spikes effectively resist binding by all but a few rare neutralizing monoclonal antibodies (MAbs).
To date, most Env-based vaccine candidates induce antibody responses against determinants that are inaccessible on native Env spikes and have therefore failed to induce meaningful neutralizing Ab responses (65). As the natural target of neutralizing Abs, the authentic Env spike in a native membrane context might fare better as an immunogen: logically, any antibodies induced by native Env trimers in a vaccine setting might be expected to neutralize. Most work in this area has centered on generating soluble Env trimers. However, the production of soluble Env trimers that resemble the authentic Env trimers has been hampered by their instability (12, 61). Although various modifications or mutations can resolve this instability, these alterations invariably affect trimer conformation, leading to the exposure of nonneutralizing epitopes. This is an important caveat, because it adversely affects their ability to induce antibodies that accurately target the native Env trimer. Instead, these modified trimers elicit overwhelmingly nonneutralizing responses.
Other groups have attempted to present native Env trimers in situ on membranes. A native context may naturally promote trimer stability without a need for modifications. However, progress in this area has been limited by the presence of nonfunctional Env in vaccine preparations (13, 17, 24, 27, 28, 31, 35, 37, 44, 53, 54, 64). The antigenic “promiscuity” of nonfunctional Env appears to make it immunodominant in a vaccine setting, promoting overwhelmingly nonneutralizing responses at the expense of any neutralizing responses to native trimers (19). Although the proportion of nonfunctional Env can vary, it consistently contaminates HIV-1 particles produced from a variety of cell types, including full-length molecular clones and live virus (1, 8, 33, 37, 44, 45, 47). The observation that nonneutralizing MAbs can capture live virus indicates that the nonfunctional Env is present on trimer-bearing infectious particles and is therefore not an artifact of contaminating vesicles (37, 44, 47, 53). The problem of nonfunctional Env appears to be as relevant in natural infection as it is in vaccine design: following infection, nonneutralizing binding responses are generated much earlier and at far higher titers than neutralizing responses, perhaps indicating a fitness advantage associated with incorporating nonfunctional Env (22, 57).
Considering the above observations, separating native Env trimers from nonfunctional Env may be an important step toward the discovery of an effective neutralizing antibody immunogen. Devising an appropriate purification strategy may require a better understanding of the nature of nonfunctional Env. Our recent observations indicate that, contrary to popular perceptions, nonfunctional Env is not derived from trimer dissociation (19, 20, 36, 69) but instead is a mostly static species that is coexpressed on particles alongside the trimer during synthesis. Nonfunctional Env appears to arise from the cellular stress associated with the slow Env maturation process that leads to apoptosis and contamination of nascent virus with immature forms of Env that would normally be expected to be found only in the endoplasmic reticulum. This new understanding of nonfunctional Env led us to abandon attempts to stabilize trimers (since they are in fact already stable in membranes) in favor of enzyme digests based on the perception that nonfunctional Env might be relatively enzyme sensitive just as it is more sensitive to antibody binding. This idea was successful: endo H and chymotrypsin digests removed nonfunctional Env, but native trimers remained intact (3, 19). However, there is room for improvement, as nonfunctional Env was not completely eliminated. Furthermore, in light of increasing evidence of the involvement of glycans in broadly neutralizing epitopes, such as the recently isolated N332-dependent PGT MAbs (67), endo H digests might be problematic if they remove any trimer glycans. Clearly, an ideal procedure should retain all neutralizing epitopes and eliminate all nonneutralizing epitopes. Here, we optimized our enzyme digest procedure and characterized the antigenicity and neutralization sensitivity of the resulting digested “trimer virus-like particles (trimer VLPs).”
MATERIALS AND METHODS
MAbs, soluble CD4, and monomeric gp120.
A panel of MAbs included the following: b12, VRC01, and 15e, directed to epitopes overlapping the CD4 binding site (CD4bs) of gp120 (10, 70); 2G12, directed to a unique glycan-dependent epitope of gp120 (56, 58); E51, directed to a CD4-inducible (CD4i) epitope of gp120 (71); LA21, CO11, F2A3, and 39F, reactive with clade B gp120 V3 loop sequences (18, 49, 61); PG9, PG16, PGT141, PGT142, and CH01 to CH04, directed to a conserved peptidoglycan region of gp120 variable loops 1 and 2 (V1V2) (9, 21, 67, 68); 2F5, Z13e1, and 4E10, directed to the gp41 membrane-proximal ectodomain region (MPER) (46, 72); 7B2 and 2.2B, directed to the gp41 cluster I and II epitopes, respectively (44); and PGT121, PGT125, PGT130, PGT135, and PGT136, directed to 3 distinct conserved epitope clusters involving the base of the third variable loop of gp120 (V3) and the glycan N332 at its base (group 1 includes PGT121, group 2 includes PGT125 and PGT130, and group 3 includes PGT135 and PGT136). These MAbs are either glycan sensitive (PGT121, PGT135, and PGT136) or glycan dependent (PGT125 and PGT130) (67). Recombinant monomeric JR-FL gp120 produced in CHO cells and soluble CD4 (sCD4) consisting of all 4 outer domains were gifts from Progenics Pharmaceuticals (Tarrytown, NY).
Virus-like particles.
VLPs were produced by cotransfection of 293T cells with a pCAGGS-based, Env-expressing plasmid and the Env-deficient HIV-1 genomic backbone plasmid pNL-LucR-E− (5, 38, 39). Env plasmids included pCAGGS expressing SOS, SOS uncleaved (SOS UNC) (K510S, R511S), wild-type (WT), WT UNC (K500S, R503S, K510S, R511S), E168K WT, E168K and N189A (E168K+N189A) WT, and E168K SOS mutants of JR-FL gp160ΔCT, some of which have been described previously (5, 38, 39). Mutations are numbered according to the HXB2 reference strain. A panel of Env point mutants was generated by Quikchange mutagenesis (Stratagene) and used to generate mutant VLPs (6). VLPs were purified as described previously (44).
Native PAGE Western blots.
Blue native PAGE (BN-PAGE) band shifts were used to analyze MAb binding to VLP Env (16–18, 44). VLPs were incubated with MAbs for 3 h at 37°C, washed with phosphate-buffered saline (PBS), and solubilized in 0.12% Triton X-100 in 1 mM EDTA/1.5 M aminocaproic acid and 1 μl of a protease inhibitor cocktail (Sigma). An equal volume of 2× sample buffer containing 100 mM morpholinepropanesulfonic acid (MOPS), 100 mM Tris-HCl (pH 7.7), 40% glycerol, and 0.1% Coomassie blue was then added. Samples were then loaded onto a 4 to 12% Bis-Tris NuPAGE gel (Invitrogen) and separated at 4°C for 3 h at 100 V with 50 mM MOPS/50 mM Tris (pH 7.7). Gels were then blotted onto polyvinylidene difluoride, destained, transferred to blocking buffer (4% nonfat milk in PBS), and probed with an anti-gp120 cocktail consisting of MAbs 2G12, b12, E51, and 39F and/or a gp41 cocktail consisting of MAbs 2F5, 4E10, 7B2, and 2.2B (at 1 μg/ml each), followed by an anti-human Fc alkaline phosphatase conjugate (Jackson). Band density was assessed as necessary using UN-SCAN-IT software (Silk Scientific).
SDS-PAGE Western blots.
Reducing SDS-PAGE Western blots were probed with an anti-gp120 cocktail consisting of MAbs CO11, F2A3, LA21, and 39F or a gp41 cocktail consisting of MAbs 2F5 and 4E10. Each MAb was used at 1 μg/ml. An alkaline phosphatase-labeled anti-human Fc conjugate was used to detect MAb binding (Jackson).
Enzyme digests.
VLPs (typically 5 μl of 1,000× VLP transfection supernatants) were incubated with enzymes at 37°C. Enzymes included 500 U endoglycosidase H (endo H) (New England BioLabs, Ipswich, MA), 1 μg trypsin, 1 μg chymotrypsin, 1 μg subtilisin, and/or 1 μg proteinase K (Sigma). Various incubation times and conditions were used. Specifically, we compared the effects of incubations in microtubes or PCR tubes, different reaction volumes, and a mineral overlay. In some cases, a protease inhibitor cocktail, consisting of 4-(2-aminoethyl)-benzenesulfonyl fluoride, E-64, bestatin, leupeptin, aprotinin, and sodium EDTA (P-2714; Sigma), was added to samples after digestion and washing to prevent further digestion.
gp120 and VLP ELISA.
Monomeric gp120 was coated at 5 μg/ml on enzyme-linked immunosorbent assay (ELISA) wells. VLPs (digested or undigested) were coated on ELISA wells at 20× the concentration in transfection supernatants. MAb binding was then assessed by conducting an ELISA, omitting detergent from PBS wash buffers, and probing with an anti-human Fc alkaline phosphatase conjugate (Accurate, Westbury, NY) and SigmaFAST p-nitrophenyl phosphate tablets (Sigma). Plates were read at 405 nm.
Neutralization assays.
Neutralization assays using canine CF2 cell lines were described previously (5, 18). Briefly, virus was incubated with graded dilutions of MAbs for 1 h at 37°C and then added to CF2 cells, spinoculated, and incubated for 2 h at 37°C, after which the medium was replaced. After 3 days of culture, luciferase activity in cell lysates was measured.
RESULTS
Here we sought to compare the antigenic properties of undigested VLPs and digested WT “trimer” VLPs. As before, we selected JR-FL gp160ΔCT as a prototype Env for VLP expression. This provides several significant advantages over other Envs that should facilitate the production of digested WT trimer VLPs. Specifically, the JR-FL isolate expresses well and exhibits unusually efficient gp120/gp41 processing, leading to efficient expression of native gp120/gp41 trimers. The ΔCT truncation also leads to improved Env expression compared to that of its full-length counterpart (44). For some Env isolates, gp41 tail truncation can markedly affect the Env ectodomain conformation (25, 33). However, the effect of this truncation on JR-FL neutralization sensitivity is marginal (∼2-fold) (18). Since clarifying enzyme digests are more effective against WT VLPs than SOS VLPs (19), WT VLPs were used in the present study.
To investigate the antigenicity and neutralization sensitivity of VLPs, we determined the reactivity of MAbs with VLPs using three methods: ELISA, neutralization, and BN-PAGE.
ELISA. (i) Comparative antigenicity of monomeric gp120 and undigested VLPs.
We first compared the epitope exposure of monomeric gp120 and various undigested VLPs by ELISA (Fig. 1). Env-bearing VLPs (Env-VLPs) included WT VLPs (red boxes), E168K+N189A mutant WT VLPs (black triangles) that “knock in” the neutralizing PG9, PG16, and PGT141 MAb epitopes (21), and WT UNC VLPs that bear only uncleaved gp160 (orange diamonds) (19). As a control, we also examined bald VLPs that bear no Env (green triangles). As expected, gp120 was recognized by gp120-specific MAbs but not by gp41-specific MAbs (Fig. 1, blue circles). Env-VLPs were recognized by all MAbs, regardless of their neutralizing activity (Fig. 1). The specificity of MAb binding to Env-VLPs was confirmed by their general lack of reactivity with bald VLPs. However, MAb 2F5 bound weakly to bald VLPs (>100-fold weaker binding than that to WT VLPs), suggesting a possible cross-reactivity with membrane lipids (2, 29, 60). Perhaps surprisingly, however, 4E10 binding to bald VLPs was minimal. The recently reported MAbs PGT121, PGT125, and PGT130 (67) all recognized WT VLPs, but PGT135 and PGT136 binding was minimal (Fig. 1). The binding of nonneutralizing MAbs to WT VLPs is consistent with the recognition of nonfunctional Env (44, 47).
Fig 1.

Antigenicity of monomeric gp120 and various VLPs analyzed by ELISA. Monomeric gp120 and various VLPs, as indicated, were coated on ELISA wells and probed by MAbs directed to various Env epitopes. Data are representative of at least 3 titrations of each MAb against each VLP. mann., mannose.
Although 2G12 bound gp120 and Env-VLPs equivalently, other MAbs showed preferences for particular antigens. The patterns in Fig. 1 are perhaps best appreciated by comparing MAb binding to each antigen in reference to their binding to WT VLPs. In general, MAb binding to E168K+N189A WT VLPs was stronger than MAb binding to the parent WT VLPs (Fig. 1, compare black triangles and red squares). This effect was in many cases dramatic, as for CD4-lgG2, PG9, PG16, PGT135, and PGT141, but was in other cases modest, as for b12, VRC01, PGT121, PGT125, and Z13e1. In other cases, there was merely a trend of better MAb binding to the mutant. PGT136 binding remained negligible to both Env VLPs. As we show later, the generally increased MAb binding to the E168K+N189A Env mutant reflects its somewhat higher expression (we estimate it as ∼3-fold higher). Based on these findings, this Env mutant became our new prototype.
UNC WT VLPs exhibited a MAb binding profile that was largely similar to that of WT VLPs (Fig. 1, compare red squares with orange diamonds), consistent with the idea that uncleaved gp160 (present on both VLP antigens) is immunodominant. However, MAbs PGT121, PGT125, PGT130, PGT135, PGT136, 15e, CO11, and E51 (in the presence of sCD4) recognized UNC WT VLPs far more efficiently than WT VLPs (Fig. 1) (19, 55), perhaps reflecting a preference for uncleaved gp160. CD4-IgG2 also recognized UNC WT VLPs more effectively (Fig. 1). The weaker binding of CD4-IgG2 to WT VLPs could reflect gp120 shedding and concomitant CD4-IgG2 dissociation from WT VLP surfaces.
Comparing gp120 and WT VLPs, all MAbs except for 2G12, PGT125, and PGT130 bound gp120 more effectively (Fig. 1). As expected, all gp41 MAbs, PG9, PG16, and PGT141 did not bind gp120. In the case of CD4-IgG2, as mentioned above, binding to WT VLPs may be adversely affected by gp120 shedding. In other cases, the difference in binding to gp120 and WT VLPs was marginal (e.g., VRC01). Notably, although E51 weakly binds to gp120, it was unable to bind WT VLPs in the absence of sCD4.
(ii) Antigenicity of trimer VLPs.
Previously, we showed that enzyme digests of WT VLPs using endo H followed by a cocktail of proteases, including trypsin, chymotrypsin, proteinase K, and subtilisin, substantially depleted nonfunctional Env, leaving trimers intact (19). However, remaining traces of nonfunctional Env suggested that refinements were needed to obtain pure trimers. Digests were markedly improved by using PCR tubes as reaction containers instead of 1.5-ml microtubes, presumably due to better temperature control (not shown).
A further problem with the previous digest protocol (19) is that endo H digests may remove glycans from native trimers, possibly affecting binding by glycan-dependent MAbs, such as 2G12, PG9, and PG16 (11, 50, 67, 68). Previously, PG9 binding to a clade C Du422 gp120 monomer was found to be sensitive to endo H (68) and was also unable to neutralize virus produced in the presence of kifunensine (21). To further assess the role of glycans in PG MAb binding, we digested VLPs with endo H and examined MAb binding by ELISA and BN-PAGE shifts. In both formats, endo H practically eliminated PG9 and PG16 binding, while b12 and 2G12 were unaffected (Fig. 2). The lack of trimer binding in lanes 9 and 10 of Fig. 2B suggests that endo H removes high-mannose glycans from trimers that are important for PG9 and PG16. Thus, omitting endo H from VLP digests is important to preserve these neutralizing epitopes (3, 19, 43). To try to resolve this problem, we performed digests in PCR tubes using the protease cocktail with or without endo H. This revealed that omitting endo H does not sacrifice digest efficiency, in the context of the more efficient digests performed in PCR tubes (not shown). Thus, from here on, our standard digest condition was an overnight digest using a cocktail of trypsin, chymotrypsin, proteinase K, and subtilisin protease performed in a PCR tube.
Fig 2.

Effect of endo H on PG epitopes on native trimers. E168K+N189A WT VLPs were digested with endo H as indicated and then assayed for MAb recognition by VLP ELISA (A) or BN-PAGE shifts (B), using 30 μg/ml of each MAb.
Enzyme digests dramatically affected MAb binding to E168K+N189A WT VLPs in ELISA (Fig. 3). Nonneutralizing MAb binding was reduced to background levels, reflecting the removal of nonfunctional Env on which their binding depends (Fig. 3). In contrast, neutralizing MAbs recognized digested E168K+N189A WT trimer VLPs, from here on called “trimer VLPs,” with optical densities (ODs) above a cutoff of 1.0 (Fig. 3).
Fig 3.

Effects of digests on MAb binding to E168K+N189A WT VLPs by ELISA. Data shown are representative of at least 3 titrations of each MAb.
The binding of neutralizing MAbs 2F5, PGT121, PGT125, and PGT130 was completely, or near completely, retained after digests (Fig. 3). However, most other neutralizing MAbs (e.g., b12 and Z13e1) exhibited somewhat reduced binding to trimer VLPs. In some cases, this may be due to the loss of nonfunctional Env, to which most MAbs cross-react (Fig. 1, MAb binding to UNC WT VLPs). Since the V1V2-specific MAbs PG9 and PG16 do not recognize nonfunctional Env (Fig. 1), their modestly lower binding to trimer VLPs must be explained by a partial loss of native trimer. Only three neutralizing MAbs exhibited little or no binding to trimer VLPs. CD4-IgG2 binding was weak, presumably due to gp120 shedding. The MAbs PGT135 and PGT136 have been previously reported as not being able to effectively neutralize the JR-FL isolate (67), explaining their weak (or nonexistent, in the case of PGT136) binding to JR-FL trimer VLPs.
For reference purposes, we performed a similar analysis of the effects of enzyme digests on MAb binding to UNC WT VLPs (Fig. 4). Since UNC WT VLPs bear only nonfunctional Env, all forms of Env are cleared by digests (19), as should be true, in theory at least, for any Env-specific MAb binding. This proved to be the case except for 2F5, which still bound to UNC WT VLPs, albeit quite weakly, after digests (Fig. 4, compare blue and red squares). In further experiments, the weak binding of MAb 2F5 to bald VLPs (Fig. 1) was also retained after digests, consistent with its recognition of lipids that are insensitive to digests (Fig. 4, green and orange triangles). In fact, 2F5 binding to digested UNC WT VLPs was similar to its binding to digested or undigested bald VLPs, again consistent with weak lipid binding (Fig. 4). This binding was dramatically weaker (>100-fold) than 2F5 binding to trimer VLPs (compare 2F5 binding in Fig. 3 and 4), confirming that 2F5 binding to trimer VLPs (Fig. 3) depends overwhelmingly on the native trimer.
Fig 4.

MAb binding to undigested and digested UNC WT VLPs was investigated by VLP ELISA. In addition, 2F5 binding to bald VLPs before and after digestion was assessed by ELISA. Data are representative of at least 3 titrations of each MAb against each VLP.
Neutralization sensitivity of trimer VLPs.
We next compared the neutralization sensitivities of E168K+N189A WT VLPs before and after digestion. Consistent with our earlier study (19), digested WT trimer VLPs were highly infectious, retaining approximately 30% of the infectivity of undigested WT VLPs (for example, in one experiment, the infectivity of undigested virus was ∼3 × 106 relative light units [RLU] and that of the digested virus was ∼8.5 × 105 RLU). Nonneutralizing MAbs 15e, LA21, CO11, 7B2, and 2.2B remained unable to neutralize trimer VLPs (Fig. 5). Neutralizing ligands directed to gp120 epitopes (b12, 2G12, PG9, PG16, CD4-IgG2, VRC01, PGT121, PGT125, PGT130, and PGT135) neutralized trimer VLPs with IC50s that were virtually identical to those of their undigested counterparts (Fig. 5). Although PGT136 is a broadly neutralizing MAb, it did not neutralize the JR-FL isolate in Fig. 5, consistent with a previous study (67).
Fig 5.

Effects of protease digests on VLP neutralization sensitivity. The neutralization sensitivities of undigested E168K+N189A WT VLPs and digested E168K+N189A WT trimer VLPs to various MAbs were assayed using CF2.CD4.CCR5 cells as targets. Each assay was performed in duplicate. Data are representative of at least 3 repeats.
A previous report indicated that PGT135 also does not detectably neutralize the JR-FL isolate. We therefore investigated further whether the weak PGT135 neutralization of JR-FL gp160ΔCT E168K+N189A in Fig. 5 is related to either the mutations or gp160 truncation in further neutralization assays. PGT136 neutralized both the WT parent gp160ΔCT virus and the full-length gp160 virus (data not shown), suggesting that the weak PGT136 neutralization in Fig. 5 is not related to either the gp41 truncation or the mutations. The slight discrepancy between our results and the previous report may be assay related. It is worth noting that PGT135 also modestly binds trimers in the BN-PAGE data described below, consistent with its weak neutralizing activity.
Strikingly, the gp41 MPER-specific MAbs 4E10 and Z13e1 neutralized trimer VLPs ∼100-fold more potently than undigested WT VLPs (Fig. 5). In contrast, 2F5, another MPER-specific MAb, did not show any marked increase in neutralizing activity (Fig. 5). Overall, these results show that native Env trimers retain their authentic conformation after digests, with the exception that epitopes at the very base of gp41 may become better exposed.
Analysis of MAb binding to VLP Env by BN-PAGE shifts.
BN-PAGE shifts were used as a way to visualize MAb binding to VLP Env. We initially evaluated MAb binding to undigested E168K+N189A WT VLPs (Fig. 6A). Trimer-IgG complexes often fail to resolve as clear bands in BN-PAGE, perhaps because the bivalent nature of IgG can lead to various multimers (16, 19, 44). A more reliable measure of MAb binding is therefore by depletion of unliganded trimer, as it forms complexes with trimers (3, 4, 6, 16–18, 34, 44, 46, 63).
Fig 6.

Effects of enzyme digests on native PAGE shifts of E168K+N189A WT VLPs. Undigested E168K+N189A WT VLPs (A) and enzyme-digested E168K+N189A WT trimer VLPs (B) were incubated with various MAbs or sCD4 at 30 μg/ml each for 3 h at 37°C and then washed and resolved by a BN-PAGE Western blot. An ∼200-kDa band induced by the MAbs 4E10 and Z13e1 is indicated by an asterisk in panel B.
Consistent with our previous reports, MAb binding to native trimers tracked exclusively with neutralization (Fig. 6A) (16, 44). CD4 binding site-overlapping MAbs b12 and VRC01 bound Env trimers (Fig. 6A, lanes 2 and 3). Interestingly, sCD4 binding led to a buildup of trimeric gp41 stumps (Fig. 6A, lane 4), consistent with gp120 shedding. We confirmed this interpretation by probing duplicate blots of sCD4 shifts separately with anti-gp120 or anti-gp41 cocktails (Fig. 7). As expected, the ∼100-kDa species induced by sCD4 was stained only by the gp41 cocktail (Fig. 7, lanes 5 and 6), suggesting trimeric gp41 stumps. Interestingly, omitting the wash step prior to VLP lysis led to improved sCD4-trimer binding and an increased buildup of these gp41 stumps (Fig. 7, compare lanes 2 and 3 and lanes 5 and 6). Gp120 shedding may also explain the weak binding of CD4-IgG2 to WT VLPs in an ELISA (Fig. 1), as this would cause CD4-IgG2 to dissociate from particle surfaces.
Fig 7.
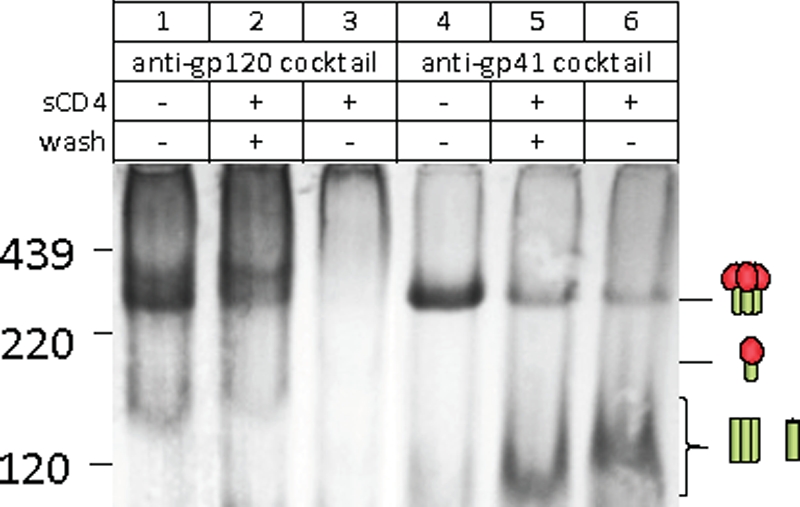
Soluble CD4 binding to E168K+N189A WT trimer VLPs causes gp120 shedding. The band induced by sCD4 binding to trimer VLPs (Fig. 6, lane 4) was investigated by probing separate blots of E168K+N189A trimer VLPs with anti-gp120 or anti-gp41 MAb cocktails as indicated. In lanes 3 and 6, the wash step that removes any unbound sCD4 before VLP lysis was omitted.
As reported previously (Fig. 6 in reference 19), MAb 2G12 induced clear trimer complexes, but trimer depletion was masked by the formation of monomer-2G12 complexes that migrate at a size similar to that of the unliganded trimer (Fig. 6A, lane 5). MAbs 2F5, 4E10, and Z13e1 also depleted trimers effectively (Fig. 6A, lanes 6 to 8). The recently isolated MAbs PG9 and PG16, targeting quaternary, glycan-dependent V1V2 epitopes, also bound to E168K+N189A WT VLP JR-FL trimers (Fig. 6A, lanes 9 and 10) but not the parent WT VLP trimers (Fig. 8A, lanes 9 and 10), consistent with the need for an E168K knock-in mutation (68). A maximum of only two copies of PG9 and PG16 appeared to bind the trimer, suggesting a steric barrier to full saturation. This contrasts with the apparent full saturation of trimers by 2G12, in which 3 copies of the MAb appear to bind (Fig. 6A, compare MAb-trimer complexes in lanes 5, 9, and 10). Other MAbs directed to similar quaternary V1V2 epitopes revealed remarkably consistent patterns of MAb-trimer binding (Fig. 6A). Specifically, the MAbs PGT141, PGT142, CH01, CH02, CH03, and CH04 induced partially saturated trimer complexes (Fig. 6A, lanes 22 to 27) that were identical to each other and to the patterns of PG9 and PG16 (Fig. 6A, lanes 9 and 10).
Fig 8.

Effects of enzyme digests on native PAGE shifts of WT VLPs. Undigested WT VLPs (A), digested WT trimer VLPs (B), and SOS UNC VLPs (C) were incubated with various MAbs or sCD4 at 30 μg/ml for 3 h at 37°C and then washed and resolved by a BN-PAGE Western blot. An ∼200-kDa band induced by the MAbs 4E10 and Z13e1 is indicated by an asterisk in panel B. A high-molecular-weight species induced by the MAb CO11 is indicated by an arrow.
The recently isolated N332 glycan-dependent MAbs PGT121, PGT125, and PGT130 all depleted the native trimer (Fig. 6A, lanes 17 to 19), consistent with their neutralizing activities. Notably, MAb PGT125 also induced an ∼200-kDa band (Fig. 6A, lane 18). PGT135 only partially depleted the trimer (Fig. 6A, lane 20), consistent with its weak neutralizing activity (Fig. 5). MAb PGT136 failed to bind trimers (Fig. 6A, lane 21), consistent with its complete lack of neutralization against JR-FL (Fig. 5).
In contrast to the neutralizing MAbs, various nonneutralizing MAbs, 15e, CO11, 39F, 7B2, and 2.2B, failed to bind to trimers (44) (Fig. 6A). Similarly, the MAb LA21, directed to the V3 loop, did not bind to native trimers (not shown).
Most MAbs partially bound to at least one form of nonfunctional Env (Fig. 6A; cartoons indicate the gp160 monomer and gp41 stumps). This was not predicted by MAb neutralizing activity, as evidenced by the depletion of the gp160 monomer by the nonneutralizing V3 MAbs CO11 and 39F (Fig. 6A, lanes 12 and 13) and the gp41 stumps by the nonneutralizing MAbs 7B2 and 2.2B (Fig. 6A, lanes 14 and 15). Paradoxically, although the MAb CO11 did not bind to the native trimer, it induced a very-high-molecular-weight complex that is more easily visible in BN-PAGE shift assays using WT VLPs (Fig. 8A and B, lane 12, indicated by an arrow) than with E168K+N189A VLPs (Fig. 6A, lane 12). Similar high-molecular-weight complexes have been previously observed in BN-PAGE shifts with other nonneutralizing MAbs (Fig. 6A in reference 16). Hypothetically, these complexes may consist of IgG complexed with two gp160 monomers (one on each Fab arm). We investigated this possibility in further BN-PAGE shift assays using UNC SOS VLPs. These VLPs bear a very prominent gp160 monomer band (Fig. 8C, lane 1) that, if our theory is correct, might accentuate the MAb-induced high-molecular-weight complexes. Revealingly, several MAbs, including 2F5, 4E10, Z13e1, 15e, CO11, and 39F, bound effectively to the UNC monomer (Fig. 8C, lanes 6, 7, 8, 11, 12, and 13, respectively), concomitant with the appearance of a prominent high-molecular-weight complex. This confirms that MAb-induced high-molecular-weight complexes in BN-PAGE shifts do not reliably track with trimer binding.
The MAbs b12 and VRC01 bound relatively poorly to the UNC SOS gp160 monomer, while other MAbs bound partially (Fig. 8C). This is consistent with reports that b12 inefficiently binds to uncleaved gp160 (48, 55) and also with our observation above that b12 binds more effectively to WT VLPs than to UNC WT VLPs by ELISA (Fig. 1). In striking contrast, the nonneutralizing CD4bs MAb 15e efficiently bound the monomer (Fig. 8C, compare lane 11 to lanes 2 and 3), consistent with its preferential binding to UNC WT VLPs by ELISA (Fig. 1). Thus, CD4bs MAbs appear to show a preference for either native trimers or UNC monomers, depending on their neutralizing activity.
BN-PAGE shifts using digested E168K+N189A WT trimer VLPs exhibited a remarkable improvement in clarity (Fig. 6, compare lane 1 in panel A and lane 1 in panel B). This was reflected in SDS-PAGE Western blots, in which digests removed uncleaved gp160 but had little effect on gp120 and gp41, consistent with the retention of the trimer (not shown). In contrast, traces of the gp160 monomer were still present in WT trimer VLPs (Fig. 8, compare lane 1 in panel A and lane 1 in panel B), as evidenced by the high-molecular-weight complex induced by the MAb CO11 (lane 12 of Fig. 8B). As described above, trimer shifts were exclusively mediated by neutralizing MAbs. Thus, in Fig. 6B, the MAbs in lanes 2 to 10, 17 to 20, 22, and 23 all depleted the trimer, but the nonneutralizing MAbs in lanes 11 to 15 did not. Similarly, in Fig. 8B, neutralizing MAbs in lanes 2 to 8 depleted WT trimers, but the MAbs in lanes 9 to 15 did not (note that PG9 and PG16 do not neutralize the WT parent). Interestingly, the MPER MAbs 4E10 and Z13e1 induced an ∼200-kDa band similar to that induced by PGT125 (Fig. 6B and 8B, marked with an asterisk in lanes 7 and 8).
Further analysis of 4E10 and Z13e1 trimer binding.
The increased 4E10/Z13e1 neutralization of trimer VLPs (Fig. 5) may be a result of improved MAb-trimer binding. Alternatively, it could relate to the ∼200-kDa Env complexes observed in BN-PAGE (Fig. 6B and 8B, lanes 7 and 8). To investigate the former possibility, we titrated 4E10 and Z13e1 in BN-PAGE shifts (Fig. 9). There was no evidence for improved 4E10 binding to digested trimer VLPs (Fig. 9A). In contrast, Z13e1 binding to the trimer was markedly stronger (Fig. 9B, compare lanes 1 to 6 to lanes 7 to 14).
Fig 9.

Effect of enzyme digestion on the trimer affinity of the MAbs 4E10 and Z13e1. 4E10 (A) and Z13e1 (B) MAbs were titrated against undigested or digested E168K+N189A WT VLPs as indicated. Below each gel, the density of the trimer band is shown as a histogram, determined using UN-SCAN-IT densitometry software (Silk Scientific). The band density in the absence of MAb is set at 100%, and any trimer binding/depletion is shown as a drop in trimer staining as the trimer becomes complexed with the MAb. Note that the trimer density of the digested E168K+N189A WT trimer VLPs is somewhat weaker than that of the undigested E168K+N189A WT VLP trimer (compare lanes 1 and 8 in panel A and lanes 1 and 7 in panel B). Therefore, the density of unliganded trimer in each of these control lanes was set at 100%.
Identifying the composition of the ∼200-kDa species induced by 4E10 and Z13e1 (Fig. 6B and 8B, lanes 7 and 8) might be informative. Two possibilities might explain the ∼200-kDa band: either it is a trimer dissociation product or a MAb complex with uncleaved gp160. To investigate, we probed duplicate blots of 4E10 or Z13e1 trimer VLP shifts separately with anti-gp120 and anti-gp41 cocktails. Both cocktails stained the 200-kDa band (Fig. 10A, marked with asterisks), suggesting that it comprises both gp120 and gp41. In further BN-PAGE shifts using biotinylated Z13e1, we found that the MAb itself formed part of the 200-kDa complex (indicated by an asterisk in lanes 2 and 5 of Fig. 10B). Overall, these data suggest that the ∼200-kDa band is either a gp160 monomer or a gp120/gp41 monomer bound to MAb. The latter explanation would require (gp120/gp41)3 dissociation in a lateral plane without gp120 shedding. Since this scenario is unlikely, the 200-kDa band more likely derives from uncleaved gp160.
Fig 10.
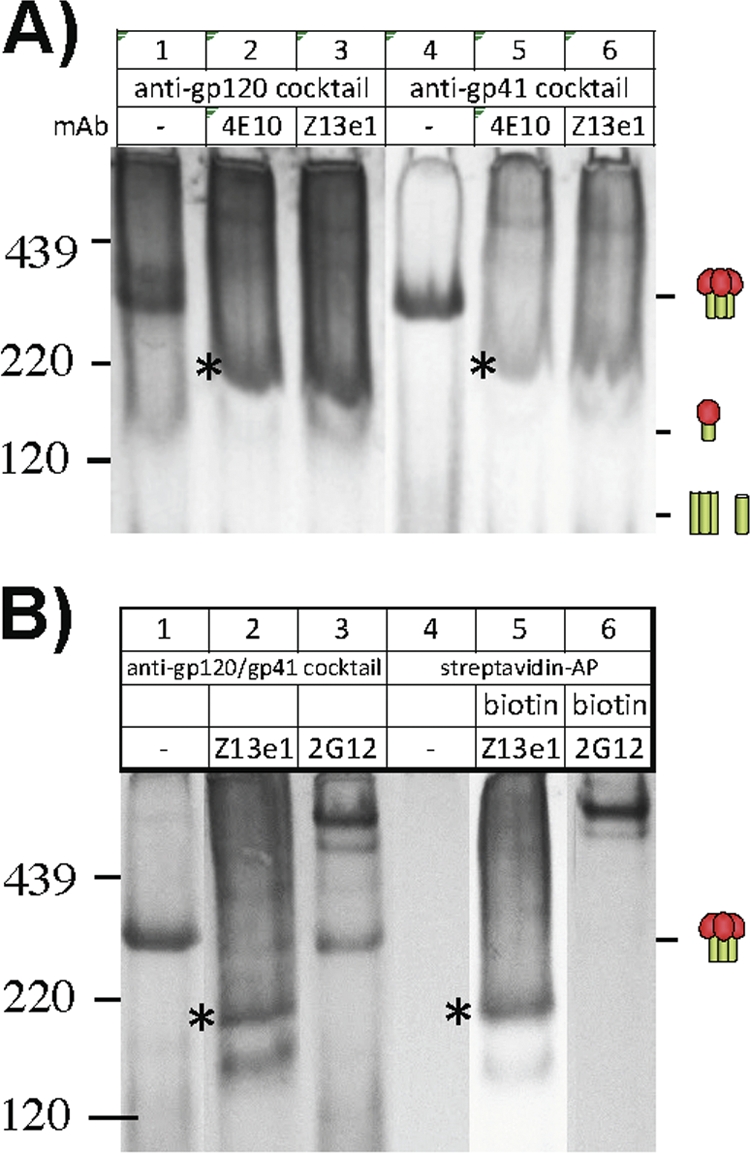
Investigation of 4E10 and Z13e1 binding to E168K+N189A WT VLPs by a BN-PAGE Western blot. The nature of the ∼200-kDa species induced by 4E10 and Z13e1 binding to E168K+N189A WT trimer VLPs (indicated by an asterisk in Fig. 6B and 8B) was investigated by probing separate BN-PAGE Western blots with anti-gp120 and anti-gp41 cocktails (A) and by using biotinylated versions of Z13e1 and 2G12 for shifts and detecting with either the gp120/gp41 cocktail or strepavidin-alkaline phosphatase (streptavidin-AP) (B).
MAb binding to trimer VLPs correlates with neutralization.
The above data, in particular Fig. 3 and 6B, suggest that only native trimers are retained after digestion of E168K+N189A WT VLPs. Therefore, we next investigated a possible correlation between MAb binding and neutralization of E168K+N189A WT trimer VLPs. For reference purposes, we first compared MAb ELISA binding titers against undigested E168K+N189A WT VLPs and their respective neutralizing IC50s in a scatter plot (Fig. 11A). MAb binding titers were taken at an optical density at 405 nm (OD405) of 1.0, as depicted by the dotted line in Fig. 3. Several nonneutralizing MAbs (Fig. 11A, red symbols) bound undigested E168K+N189A WT VLPs with titers similar to those of neutralizing MAbs (Fig. 11A, blue symbols), probably because MAb binding is dominated by nonfunctional Env (Fig. 1 and 3). As a result, there was no significant correlation between neutralization and VLP binding (r2 = 0.1768; P < 0.0730) (Fig. 11A).
Fig 11.

Correlation of VLP antigenicity and neutralization sensitivity. MAb neutralization IC50s against undigested E168K+N189A WT VLPs (A) and digested E168K+N189A WT trimer VLPs (B) were plotted against MAb binding titers from ELISAs using the same VLPs. Neutralizing MAbs are indicated by blue symbols. Nonneutralizing MAbs are indicated by red symbols. Although Z13e1 exhibits little neutralization against undigested VLPs, it is given a blue symbol because it effectively neutralizes trimer VLPs. Furthermore, despite the fact that PGT136 is a broadly neutralizing MAb, it is classified as a nonneutralizing MAb in this instance, as it fails to neutralize JR-FL. ELISA titers were taken as the concentration at which MAb binding exhibited an optical density of 1.0 at 405 nm (Fig. 3). Any titers of >30 μg/ml are plotted at 30 μg/ml. All titers are means of at least 3 repeat assays. Linear regression and P values were calculated.
In contrast, trimer VLP ELISA binding and neutralization exhibited a statistically significant correlation (r2 = 0.97; P < 0.0001) (Fig. 11B). The binding of the new PGT MAbs correlated well with their neutralizing activities, with PGT121, PGT125, and PGT130 neutralizing and binding potently, PGT135 binding and neutralizing weakly, and PGT136 neither binding nor neutralizing. However, several caveats are worth noting. Clearly, nonneutralizing MAbs favor a better correlation, as these MAbs neither neutralize nor bind to trimer VLPs appreciably (Fig. 11B and Fig. 3). Thus, when nonneutralizing MAbs were removed, the correlation became less significant (r2 = 0.3186; P < 0.0445). Although the ELISA binding titers of neutralizing MAbs to E168K+N189A WT trimer VLPs tended to be lower than their titers against undigested counterparts (compare the positions of the blue symbols on the y axis in Fig. 11A and B and the data in Fig. 3), with the exception of 4E10 and Z13e1, MAb neutralization titers were unchanged (Fig. 5). This is because the removal of nonfunctional Env affects overall MAb binding but not neutralization. Paradoxically, the markedly improved neutralization potencies of the MAbs 4E10 and Z13e1 against trimer VLPs (4E10 became the second most potently neutralizing MAb after PGT125 [Fig. 11B]) were not matched by particularly strong titers of these MAbs in a VLP ELISA. This was particularly true for Z13e1, which exhibited a low titer against trimer VLPs (compare Fig. 11A and B).
CD4-IgG2 is shown as a green symbol in Fig. 11. This is because although it neutralizes potently, it also induces gp120 shedding (Fig. 6) and is expected to fall off, along with gp120. Therefore, we reevaluated correlations that excluded CD4-IgG2 and found an even more significant linear relationship between ELISA binding and neutralization (Fig. 11B) (r2 = 0.9989, P < 0.0001). Similarly, given the unusually potent neutralizing activities of 4E10 and Z13e1 against trimer VLPs, we also assessed correlations in which they were excluded along with CD4-IgG2. This improved the linear relationship of binding and neutralization slightly further (r2 = 0.9999, P < 0.0001). Similar findings were noted when other combinations of CD4-IgG2, 4E10, and Z13e1 and nonneutralizing MAbs were excluded from correlations (Fig. 11B). In the case of undigested VLPs, only the omission of CD4-IgG2, 4E10, Z13e1, and nonneutralizing MAbs resulted in a highly significant correlation (r2 = 0.9994; P < 0.0001). This is largely because 4E10, Z13e1, and nonneutralizing MAbs bind nonfunctional Env very efficiently but fail to neutralize efficiently, thereby adversely affecting any correlation (Fig. 11A).
DISCUSSION
Our overall aim is to generate pure native Env trimers as prospective immunogens. Previous work by ourselves and others suggests that the abnormally slow cellular processing of nascent gp160 promotes apoptosis, resulting in the contamination of particle surfaces with immature gp160 (19, 36, 40). This agrees with our finding that native trimers are stable and do not readily dissociate to form nonfunctional Env (14, 19, 23). This new information led us to assess enzyme digests to selectively remove nonfunctional Env, reasoning that the compact native trimers might resist digestion in the same way they resist nonneutralizing MAbs, while the antigenically “promiscuous” nonfunctional Env might be digested (16, 18, 44). This idea proved successful (19). Here, we characterized the antigenicity of trimer VLPs.
An important part of achieving the goal of generating trimer VLPs was the selection of an appropriate prototype Env. The E168K+N189A mutant is valuable in two ways: (i) it knocks in important PG epitopes, and (ii) it expresses more trimer, as reflected by its enhanced reactivity with various neutralizing MAbs by ELISA (Fig. 1). Optimal digests led to the removal of most nonfunctional Env (Fig. 6B), consistent with the lack of nonneutralizing MAb binding in an ELISA (Fig. 3). The remarkable correlation of MAb binding and neutralization in Fig. 11B is consistent with the native trimer being the only Env antigen left on particle surfaces after digestion.
The stark differences in antigenicities of digested and undigested VLPs (Fig. 1, 3, and 4) are consistent with the major antigenic differences between cleaved and uncleaved forms of Env (7, 48). The preference of b12 for cleaved gp120/gp41 contrasts with the preference of the MAb 15e for UNC gp160 (Fig. 1, 6, and 8). This suggests that proper gp120/gp41 cleavage and maturation may be a requirement for vaccine candidates to adopt a fully authentic conformation, recognized only by neutralizing antibodies. Uncleaved forms of Env exhibit more promiscuous epitope exposure (Fig. 8C), consistent with our observation that enzyme digests clear these forms of Env from particle surfaces (Fig. 4).
Despite the efficiency of our optimized protease digests (Fig. 6B, lane 1), faint traces of partially digested material, perhaps best exemplified by the 200-kDa species formed in shifts by the MAbs 4E10, Z13e1, and PGT125, still remained (Fig. 6B, 8B, 9, and 10). Overall, however, the fact that nonneutralizing MAbs are unable to efficiently bind to trimer VLPs (Fig. 3, 6B, and 11B) strongly suggests that any antigenic interference by nonfunctional Env is all but eliminated.
Although most MAbs neutralized trimer VLPs and undigested VLPs equivalently, 4E10 and Z13e1 were ∼100-fold more potent against trimer VLPs (Fig. 5). The underlying reasons are not altogether clear. The ∼10-fold increase in Z13e1 affinity for the trimer (Fig. 9B) could at least partly explain the increase in Z13e1 neutralization potency (Fig. 5). However, the MAb 4E10 did not bind any better to the trimer after digestion (Fig. 9A). A second possible contributing factor in the increased neutralization activity of 4E10 and Z13e1 relates to the 200-kDa Env species observed in BN-PAGE shifts (Fig. 6B and 8B) which appears to be partially digested gp160. This Env species might concentrate these MPER MAbs on particle surfaces, enhancing their neutralizing activity in a manner analogous to the Fc receptor-mediated enhancement of MPER MAb neutralization reported previously (51). However, VLP ELISAs do not support this model, as Z13e1 and 4E10 binding is not especially well retained after digestion (Fig. 3). A third possible explanation for the enhanced neutralizing activity of 4E10 and Z13e1 is that the trimer is partially “triggered” by digests. However, it is not clear why such a conformational effect would be localized to the lower MPER while the rest of the gp41 and gp120 components remain unchanged (i.e., no induction of V3 or CD4i epitopes that are known to occur when the trimer is triggered by receptor binding). A fourth possibility is that the 4E10 and Z13e1 epitopes may become unusually well exposed (i.e., increased access or exposure time) in the window between receptor binding and neutralization. Unlike gp120 MAbs, MPER MAbs can neutralize virus after CD4 and coreceptor binding (5). If this is the correct explanation, the reason why 2F5 neutralization is not similarly enhanced may relate to its relative exposure on undigested virus (62). Thus, if the 2F5 epitope is already well exposed, it may not benefit from the effects that lead to increased 4E10 and Z13e1 exposure after receptor binding. Overall, whatever the mechanism, the improved activities of these MAbs targeting the base of the gp41 stump suggest that the enzyme digests probably remove protein material from the surfaces of particles that normally limit access of MPER antibodies to their epitopes on native Env. This idea is consistent with the observation that Env spikes tend to reside in protein-rich lipid rafts (41).
Pseudovirion VLPs were used as a platform in this study both for convenience and also to maximize trimer expression. Pseudoviruses are known to incorporate more nonfunctional Env than is usually detected on live virus—although nonfunctional Env is a universal particle contaminant. While this makes the challenge of removing nonfunctional Env more significant, the high yield of trimer allows a greater scope to evaluate its antigenicity. For similar reasons, we used a gp41 tail-truncated form of JR-FL Env that exhibits higher trimer expression, enhancing our ability to assay its antigenic properties. This choice does not undermine the significance of our results. Truncated JR-FL Env forms functional trimers whose neutralization sensitivity (Fig. 3) closely resembles that of full-length trimers, as we reported previously (18). For ongoing vaccine design efforts, it will be important to adapt our methods to other Envs. Given the relative resistance of native trimers to proteases, our protease digest methods should be universal. Nevertheless, modified conditions may need to be determined iteratively for each Env, perhaps using different proteases and incubation times.
One immediate application of trimer VLPs may be as baits to isolate new broadly neutralizing MAbs from the memory B cells of HIV-1-infected donors who exhibit broad serum neutralization. Despite recent successes in recovering new MAbs by several groups (15, 59, 66, 68, 70), the rarity of these MAbs and the present lack of appropriate baits has made these efforts challenging and expensive. Trimer VLP ELISAs may also be useful for small-molecule antiretroviral screening, using bald VLPs as a counter screen. The trimer VLP ELISA may also provide a rapid way to map the epitopes of new inhibitors, MAbs, and neutralizing sera, using a competitive ELISA format and a panel of biotinylated neutralizing MAbs of known epitopes.
Importantly, trimer VLPs will, for the first time, provide an opportunity to test the immunogenicity of pure native Env trimers. Previously, VLP immunogens have been marred by the presence of nonfunctional Env, which, due to its immunodominance (exemplified by WT VLPs; red boxes in Fig. 1), appears to dampen any responses to the more compact native trimer (17). In the absence of this interference, and if pure Env trimers are immunogenic, any anti-Env responses they elicit should, at least in theory, be neutralizing.
ACKNOWLEDGMENTS
This work was supported by grants AI93278, AI58763, and AI84714 (to J.M.B.) (NIH/NIAID/DAIDS), the Bill and Melinda Gates Foundation Collaboration for AIDS Vaccine Discovery (CAVD-VIMC grant no. 38619), and the Torrey Pines Institute's AIDS and Infectious Disease Science Center (to J.M.B.).
We thank C. Alving, R. W. Sanders, J. R. Mascola, and D. C. Montefiori for advice and D. R. Burton, J. Robinson, IAVI, and the NIH AIDS Research and Reference Reagent Program for providing MAbs.
Footnotes
Published ahead of print 1 February 2012
REFERENCES
- 1. Agrawal N, et al. 2011. Functional stability of unliganded envelope glycoprotein spikes among isolates of human immunodeficiency virus type 1 (HIV-1). PLoS One 6:e21339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alam SM, et al. 2007. The role of antibody polyspecificity and lipid reactivity in binding of broadly neutralizing anti-HIV-1 envelope human monoclonal antibodies 2F5 and 4E10 to glycoprotein 41 membrane proximal envelope epitopes. J. Immunol. 178:4424–4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Binley J. June 2010. Enzymatic methods to produce pure native authentic HIV-1 Env trimer immunogens. US patent 61/360,067
- 4. Binley JM. 2009. Specificities of broadly neutralizing anti-HIV-1 sera. Curr. Opin. HIV AIDS 4:364–372 [DOI] [PubMed] [Google Scholar]
- 5. Binley JM, et al. 2003. Redox-triggered infection by disulfide-shackled human immunodeficiency virus type 1 pseudovirions. J. Virol. 77:5678–5684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Binley JM, et al. 2008. Profiling the specificity of neutralizing antibodies in a large panel of plasmas from patients chronically infected with human immunodeficiency virus type 1 subtypes B and C. J. Virol. 82:11651–11668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Binley JM, et al. 2002. Enhancing the proteolytic maturation of human immunodeficiency virus type 1 envelope glycoproteins. J. Virol. 76:2606–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bonomelli C, et al. 2011. The glycan shield of HIV is predominantly oligomannose independently of production system or viral clade. PLoS One 6:e23521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bonsignori M, et al. 2011. Analysis of a clonal lineage of HIV-1 envelope V2/V3 conformational epitope-specific broadly neutralizing antibodies and their inferred unmutated common ancestors. J. Virol. 85:9998–10009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Burton DR, et al. 1994. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 266:1024–1027 [DOI] [PubMed] [Google Scholar]
- 11. Calarese DA, et al. 2003. Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science 300:2065–2071 [DOI] [PubMed] [Google Scholar]
- 12. Center RJ, et al. 2002. Oligomeric structure of the human immunodeficiency virus type 1 envelope protein on the virion surface. J. Virol. 76:7863–7867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen X, et al. 2005. Pseudovirion particle production by live poxvirus human immunodeficiency virus vaccine vector enhances humoral and cellular immune responses. J. Virol. 79:5537–5547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chertova E, et al. 2002. Envelope glycoprotein incorporation, not shedding of surface envelope glycoprotein (gp120/SU), is the primary determinant of SU content of purified human immunodeficiency virus type 1 and simian immunodeficiency virus. J. Virol. 76:5315–5325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Corti D, et al. 2010. Analysis of memory B cell responses and isolation of novel monoclonal antibodies with neutralizing breadth from HIV-1-infected individuals. PLoS One 5:e8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Crooks ET, et al. 2008. Relationship of HIV-1 and SIV envelope glycoprotein trimer occupation and neutralization. Virology 377:364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crooks ET, et al. 2007. A comparative immunogenicity study of HIV-1 virus-like particles bearing various forms of envelope proteins, particles bearing no envelope and soluble monomeric gp120. Virology 366:245–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crooks ET, et al. 2005. Characterizing anti-HIV monoclonal antibodies and immune sera by defining the mechanism of neutralization. Hum. Antibodies 14:101–113 [PMC free article] [PubMed] [Google Scholar]
- 19. Crooks ET, Tong T, Osawa K, Binley JM. 2011. Enzyme digests eliminate nonfunctional env from HIV-1 particle surfaces, leaving native Env trimers intact and viral infectivity unaffected. J. Virol. 85:5825–5839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dewar RL, Vasudevachari MB, Natarajan V, Salzman NP. 1989. Biosynthesis and processing of human immunodeficiency virus type 1 envelope glycoproteins: effects of monensin on glycosylation and transport. J. Virol. 63:2452–2456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Doores KJ, Burton DR. 2010. Variable loop glycan dependency of the broad and potent HIV-1 neutralizing antibodies PG9 and PG16. J. Virol. 84:10510–10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Doria-Rose NA, et al. 2010. Breadth of human immunodeficiency virus-specific neutralizing activity in sera: clustering analysis and association with clinical variables. J. Virol. 84:1631–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Earl PL, Moss B, Doms RW. 1991. Folding, interaction with GRP78-BiP, assembly, and transport of the human immunodeficiency virus type 1 envelope protein. J. Virol. 65:2047–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Edinger AL, et al. 2000. Characterization and epitope mapping of neutralizing monoclonal antibodies produced by immunization with oligomeric simian immunodeficiency virus envelope protein. J. Virol. 74:7922–7935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Edwards TG, et al. 2002. Truncation of the cytoplasmic domain induces exposure of conserved regions in the ectodomain of human immunodeficiency virus type 1 envelope protein. J. Virol. 76:2683–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fouts TR, Binley JM, Trkola A, Robinson JE, Moore JP. 1997. Neutralization of the human immunodeficiency virus type 1 primary isolate JR-FL by human monoclonal antibodies correlates with antibody binding to the oligomeric form of the envelope glycoprotein complex. J. Virol. 71:2779–2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hammonds J, et al. 2005. Induction of neutralizing antibodies against human immunodeficiency virus type 1 primary isolates by Gag-Env pseudovirion immunization. J. Virol. 79:14804–14814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hammonds J, Chen X, Zhang X, Lee F, Spearman P. 2007. Advances in methods for the production, purification, and characterization of HIV-1 Gag-Env pseudovirion vaccines. Vaccine 25:8036–8048 [DOI] [PubMed] [Google Scholar]
- 29. Haynes BF, et al. 2005. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science 308:1906–1908 [DOI] [PubMed] [Google Scholar]
- 30. Haynes BF, Montefiori DC. 2006. Aiming to induce broadly reactive neutralizing antibody responses with HIV-1 vaccine candidates. Expert Rev. Vaccines 5:347–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hicar MD, et al. 2010. Pseudovirion particles bearing native HIV envelope trimers facilitate a novel method for generating human neutralizing monoclonal antibodies against HIV. J. Acquir. Immune Defic. Syndr. 54:223–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hoxie JA. 2010. Toward an antibody-based HIV-1 vaccine. Annu. Rev. Med. 61:135–152 [DOI] [PubMed] [Google Scholar]
- 33. Joyner AS, Willis JR, Crowe JE, Jr, Aiken C. 2011. Maturation-induced cloaking of neutralization epitopes on HIV-1 particles. PLoS pathogens 7:e1002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kang YK, et al. 2009. Structural and immunogenicity studies of a cleaved, stabilized envelope trimer derived from subtype A HIV-1. Vaccine 27:5120–5132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. LaCasse RA, et al. 1999. Fusion-competent vaccines: broad neutralization of primary isolates of HIV. Science 283:357–362 [DOI] [PubMed] [Google Scholar]
- 36. Land A, Zonneveld D, Braakman I. 2003. Folding of HIV-1 envelope glycoprotein involves extensive isomerization of disulfide bonds and conformation-dependent leader peptide cleavage. FASEB J. 17:1058–1067 [DOI] [PubMed] [Google Scholar]
- 37. Leaman DP, Kinkead H, Zwick MB. 2010. In-solution virus capture assay helps deconstruct heterogeneous antibody recognition of human immunodeficiency virus type 1. J. Virol. 84:3382–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li M, et al. 2005. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 79:10108–10125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li M, et al. 2006. Genetic and neutralization properties of subtype C human immunodeficiency virus type 1 molecular env clones from acute and early heterosexually acquired infections in Southern Africa. J. Virol. 80:11776–11790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li Y, et al. 1996. Effects of inefficient cleavage of the signal sequence of HIV-1 gp 120 on its association with calnexin, folding, and intracellular transport. Proc. Natl. Acad. Sci. U. S. A. 93:9606–9611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liao Z, Graham DR, Hildreth JE. 2003. Lipid rafts and HIV pathogenesis: virion-associated cholesterol is required for fusion and infection of susceptible cells. AIDS Res. Hum. Retroviruses 19:675–687 [DOI] [PubMed] [Google Scholar]
- 42. Mascola JR, Montefiori DC. 2010. The role of antibodies in HIV vaccines. Annu. Rev. Immunol. 28:413–444 [DOI] [PubMed] [Google Scholar]
- 43. Means RE, Desrosiers RC. 2000. Resistance of native, oligomeric envelope on simian immunodeficiency virus to digestion by glycosidases. J. Virol. 74:11181–11190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moore PL, et al. 2006. Nature of nonfunctional envelope proteins on the surface of human immunodeficiency virus type 1. J. Virol. 80:2515–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murakami T, Freed EO. 2000. The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc. Natl. Acad. Sci. U. S. A. 97:343–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nelson JD, et al. 2007. An affinity-enhanced neutralizing antibody against the membrane-proximal external region of human immunodeficiency virus type 1 gp41 recognizes an epitope between those of 2F5 and 4E10. J. Virol. 81:4033–4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nyambi PN, et al. 1998. Mapping of epitopes exposed on intact human immunodeficiency virus type 1 (HIV-1) virions: a new strategy for studying the immunologic relatedness of HIV-1. J. Virol. 72:9384–9391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pancera M, Wyatt R. 2005. Selective recognition of oligomeric HIV-1 primary isolate envelope glycoproteins by potently neutralizing ligands requires efficient precursor cleavage. Virology 332:145–156 [DOI] [PubMed] [Google Scholar]
- 49. Pantophlet R, Aguilar-Sino RO, Wrin T, Cavacini LA, Burton DR. 2007. Analysis of the neutralization breadth of the anti-V3 antibody F425-B4e8 and re-assessment of its epitope fine specificity by scanning mutagenesis. Virology 364:441–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pejchal R, et al. 2011. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science 334:1097–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perez LG, Costa MR, Todd CA, Haynes BF, Montefiori DC. 2009. Utilization of immunoglobulin G Fc receptors by human immunodeficiency virus type 1: a specific role for antibodies against the membrane-proximal external region of gp41. J. Virol. 83:7397–7410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Phogat S, Wyatt RT, Karlsson Hedestam GB. 2007. Inhibition of HIV-1 entry by antibodies: potential viral and cellular targets. J. Intern. Med. 262:26–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Poignard P, et al. 2003. Heterogeneity of envelope molecules expressed on primary human immunodeficiency virus type 1 particles as probed by the binding of neutralizing and nonneutralizing antibodies. J. Virol. 77:353–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Poon B, et al. 2005. Induction of humoral immune responses following vaccination with envelope-containing, formaldehyde-treated, thermally inactivated human immunodeficiency virus type 1. J. Virol. 79:4927–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Roben P, et al. 1994. Recognition properties of a panel of human recombinant Fab fragments to the CD4 binding site of gp120 that show differing abilities to neutralize human immunodeficiency virus type 1. J. Virol. 68:4821–4828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sanders RW, et al. 2002. The mannose-dependent epitope for neutralizing antibody 2G12 on human immunodeficiency virus type 1 glycoprotein gp120. J. Virol. 76:7293–7305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sather DN, et al. 2009. Factors associated with the development of cross-reactive neutralizing antibodies during HIV-1 infection. J. Virol. 83:757–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Scanlan CN, et al. 2002. The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2G12 recognizes a cluster of α1→2 mannose residues on the outer face of gp120. J. Virol. 76:7306–7321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Scheid JF, et al. 2011. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 333:1633–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Scherer EM, Leaman DP, Zwick MB, McMichael AJ, Burton DR. 2010. Aromatic residues at the edge of the antibody combining site facilitate viral glycoprotein recognition through membrane interactions. Proc. Natl. Acad. Sci. U. S. A. 107:1529–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schulke N, et al. 2002. Oligomeric and conformational properties of a proteolytically mature, disulfide-stabilized human immunodeficiency virus type 1 gp140 envelope glycoprotein. J. Virol. 76:7760–7776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sun ZY, et al. 2008. HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity 28:52–63 [DOI] [PubMed] [Google Scholar]
- 63. Vaine M, et al. 2008. Improved induction of antibodies against key neutralizing epitopes by HIV-1 gp120 DNA prime-protein boost vaccination compared to gp120 protein-only vaccination. J. Virol. 82:7369–7378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vzorov AN, Lea-Fox D, Compans RW. 1999. Immunogenicity of full length and truncated SIV envelope proteins. Viral Immunol. 12:205–215 [DOI] [PubMed] [Google Scholar]
- 65. Walker LM, Burton DR. 2010. Rational antibody-based HIV-1 vaccine design: current approaches and future directions. Curr. Opin. Immunol. 22:358–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Walker LM, et al. 2010. High throughput functional screening of activated B cells from 4 African elite neutralizers yields a panel of novel broadly neutralizing antibodies, abstr OA10.06 LB. AIDS Vaccine 2010 Conf. [Google Scholar]
- 67. Walker LM, et al. 2011. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477:466–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Walker LM, et al. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Willey RL, Bonifacino JS, Potts BJ, Martin MA, Klausner RD. 1988. Biosynthesis, cleavage, and degradation of the human immunodeficiency virus 1 envelope glycoprotein gp160. Proc. Natl. Acad. Sci. U. S. A. 85:9580–9584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wu X, et al. 2010. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329:856–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Xiang SH, Doka N, Choudhary RK, Sodroski J, Robinson JE. 2002. Characterization of CD4-induced epitopes on the HIV type 1 gp120 envelope glycoprotein recognized by neutralizing human monoclonal antibodies. AIDS Res. Hum. Retroviruses 18:1207–1217 [DOI] [PubMed] [Google Scholar]
- 72. Zwick MB, Saphire EO, Burton DR. 2004. gp41: HIV's shy protein. Nat. Med. 10:133–134 [DOI] [PMC free article] [PubMed] [Google Scholar]