

Abstract
Post-translational modifications of histone H3 tails have crucial roles in regulation of cellular processes. There is cross-regulation between the modifications of K4, K9, and K14 residues. The modifications on these residues drastically promote or inhibit each other. In this work, we studied the structural changes of the histone H3 tail originating from the three most important modifications; tri-methylation of K4 and K9, and acetylation of K14. We performed extensive molecular dynamics simulations of four types of H3 tails: (i) the unmodified H3 tail having no chemical modification on the residues, (ii) the tri-methylated lysine 4 and lysine 9 H3 tail (K4me3K9me3), (iii) the tri-methylated lysine 4 and acetylated lysine 14 H3 tail (K4me3K14ace), and (iv) tri-methylated lysine 9 and acetylated lysine 14 H3 tail (K9me3K14ace). Here, we report the effects of K4, K9, and K14 modifications on the backbone torsion angles and relate these changes to the recognition and binding of histone modifying enzymes. According to the Ramachandran plot analysis; (i) the dihedral angles of K4 residue are significantly affected by the addition of three methyl groups on this residue regardless of the second modification, (ii) the dihedral angle values of K9 residue are similarly altered majorly by the tri-methylation of K4 residue, (iii) different combinations of modifications (tri-methylation of K4 and K9, and acetylation of K14) have different influences on phi and psi values of K14 residue. Finally, we discuss the consequences of these results on the binding modes and specificity of the histone modifying enzymes such as DIM-5, GCN5, and JMJD2A.
Keywords: acetylation, histone H3, methylation, torsion angles, lysine, molecular dynamics
Introduction
Nucleosomes are the fundamental structures of eukaryotic chromatin. Histone proteins are the basic components of nucleosomes which compact 1.8 m of DNA into the eukaryotic nucleus. Each nucleosome core particle consists of four histone proteins, Histone H2A, H2B, H3, and H4, and has an octameric structure, formed by a central heterotetramer of histones H3 and H4, and two heterodimers of histones H2A and H2B.1–5 Additionally a linker protein, Histone H1/H5, locks the DNA onto the core domain. All together, they constitute a spool-like structure for 146 bp of DNA to tangle around.1 Having such a basic structural role, histone proteins have N- and/or C-terminal tails that protrude from the core surface and undergo several post-translational modifications, such as acetylation, methylation, phosphorylation, ubiquitination, sumoylation, biotinylation. These post-translational modifications of histone tails make them key switch points for chromatin condensation/decondensation, affecting the cellular downstream processes such as transcription and DNA repair.2 Among the unobstructed dynamic histone tails, the Histone H3 N-terminal tail is the most extensively studied and well-known tail so far. It consists of 40 amino acids projecting from the H3 core domain, and there are 12 residues proven to be chemically modified. However, among these 12 residues, K4, K9, and K14 are the ones, the modifications of which have the most significant effect on transcriptional regulation. K4 of Histone H3 can be methylated; K9 can be methylated and acetylated, whereas K14 can only be acetylated.3
Recent studies show the dynamic interplay between different types of modifications on different residues, even between intra- and inter-nucleosomes, which was firstly proposed in the histone code hypothesis by Strahl and Allis in 2000.4 A remarkable outcome of the histone code hypothesis is the significant cross-regulation patterns of K4, K9, and K14.5 The modifications of these residues can drastically promote or inhibit each other, as well as influencing the structure of the tail and causing attractive correlations between specific residues. Furthermore, addition of methyl groups on lysine 4 residue of the Histone H3 was shown to inhibit the methylation of lysine 9 residue on the same tail, whereas the acetylation of lysine 14 is promoted. On the other hand, the methylation of lysine 9 residue of H3 inhibits both methylation of lysine 4 and acetylation of lysine 14 on the same tail.5
Inspired by the dynamic cross-talk of the H3 tail modifications, we investigated the correlations among the residues of the histone H3 tail both in the unmodified state and in the presence of modifications on the three residues (K4, K9, K14) by means of torsion angles. We carried out extensive Molecular Dynamics (MD) simulations of the histone H3 tail and we studied the alterations of torsion angles of the aforementioned three residues in the modified cases in reference to the unmodified histone H3 tail. We expected that the double modifications of the aforementioned three residues would yield more significant results than the single modifications and due to the computational costs we performed MD simulations of the dually modified histone H3 tails. On the basis of these computations, we analyzed the Ramachandran map phi and psi angles of the three residues for the unmodified and modified cases. Here, we report the structural effects of the double modifications of residues on each other as well as histone modifying enzymes by means of torsional angles.
Results and Discussion
The phi and psi torsional angles of K4, K9, and K14 residues were computed from the trajectories of entire set of MD simulations, and Ramachandran plots of these three residues were obtained which are displayed in Figures 1, 2, and 3.
Figure 1.
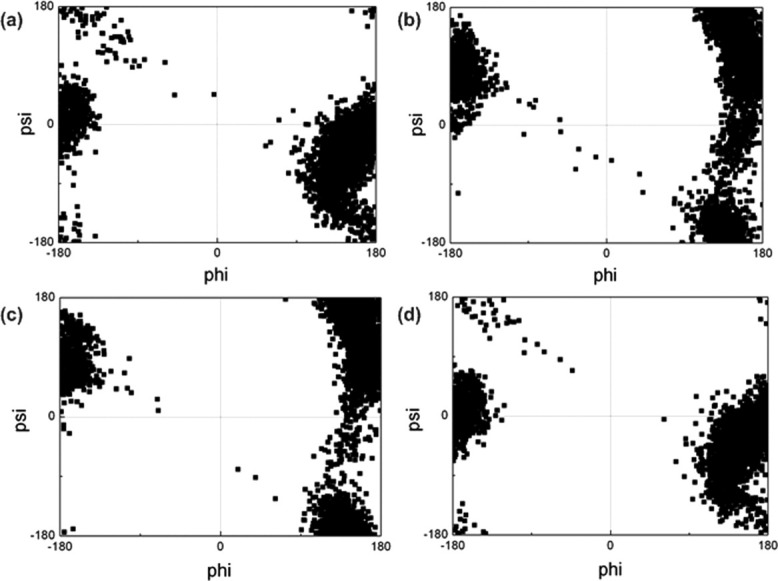
Ramachandran maps of K4 residue in (a) unmodified H3 tail, (b) K4me3K9me3 tail, (c) K4me3K14ace tail, (d) K9me3K14ace tail
Figure 2.
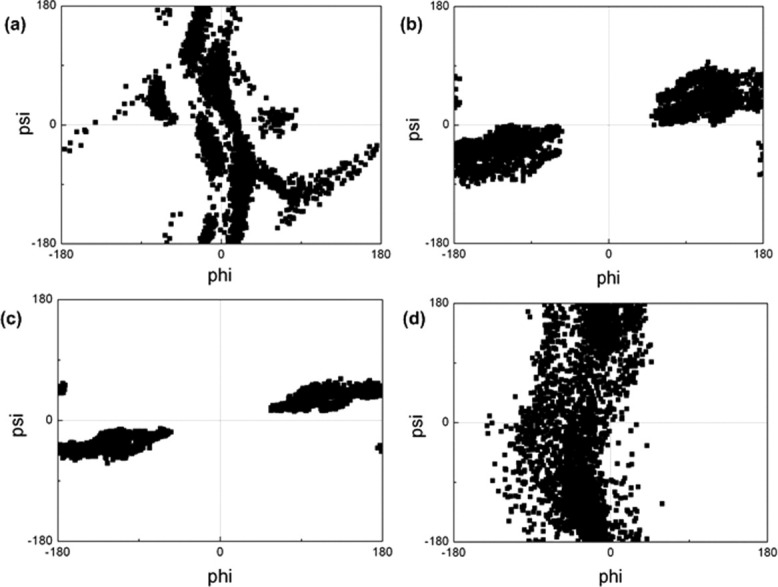
Ramachandran maps of K9 residue in (a) unmodified H3 tail, (b) K4me3K9me3 tail, (c) K4me3K14ace tail, (d) K9me3K14ace tail.
Figure 3.
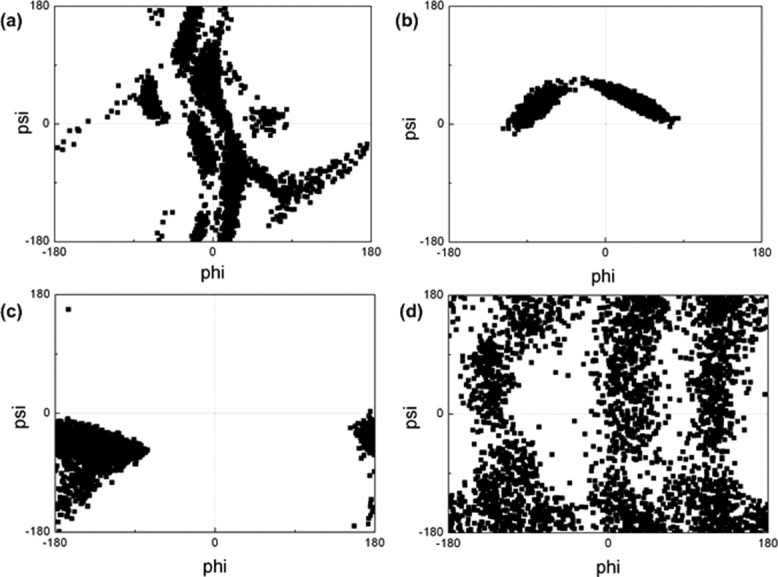
Ramachandran maps of K14 residue in (a) unmodified H3 tail, (b) K4me3K9me3 tail, (c) K4me3K14ace tail, (d) K9me3K14ace tail.
From Figure 1, one can observe that phi and psi angles of K4 residue were not affected by the modifications significantly. As it can be concluded from the K4me3K9me3 [Fig. 1(b)] and K4me3K14ace [Fig. 1(c)] Ramachandran plots compared to the unmodified Ramachandran plot of K4 residue [Fig. 1(a)], the tri-methylation of K4 residue is the major modification that brings about a minor alteration to the phi and psi angles of K4 residue. On the other hand, the simultaneous tri-methylation of K9 and acetylation of K14 [Fig. 1(d)] didn't alter the phi and psi angles of K4 compared to unmodified tail.
Additionally, the effect of tri-methylation of K4 on torsions of K9 residue can be observed from Figure 2. It is obvious that the simultaneous tri-methylation of K9 and acetylation of K14 [Fig. 2(d)] has a slight change on the torsions of K9 residue compared with the unmodified tail [Fig. 2(a)], whereas the tri-methylation of K4—irrespective of the type of the second modification (tri-methylation of K9 or acetylation of K14) [Fig. 2(b,c)]—has a drastic alteration on the phi and psi angles. A compact form of Ramachandran plots of K4me3K9me3 [Fig. 2(b)] and K4me3K14ace [Fig. 2(c)] tails for K9 residue indicates that the torsions of the K9 residue is confined to a limited set of values which designates the restricted set of accessible conformations of the K9 residue through out the trajectories.
In the case of K14 residue, it can be concluded from the Figure 3 that all of the modifications have diverse effects on torsional angles of K14 residue. However, tri-methylation of K4 [Fig. 3(b,c)] seems to have the most significant effect restricting the set of phi and psi angles and thus the accessible conformations of K14 residue, compared with the unmodified [Fig. 3(a)] and K9me3K14ace [Fig. 3(d)] tails. However, unlike K4 torsions, the type of second modification induces the area that the phi and psi angles are restricted to. On the other hand, the dual modification of K9 and K14 has a reverse effect allowing for a wide range of conformations which can be deduced from the distributed Ramachandran map of K9me3K14ace tail [Fig. 3(d)].
We further screened for the enzymes that can perform the abovementioned modifications to the three histone H3 residues. We identified four different enzymes that have PDB structures complexed with the histone H3 tails of our interest. Table I lists these enzymes together with their PDB ID, function, the residues on histone H3 tail that they recognize and the histone H3 peptides that they are complexed with.
Table I.
Histone Modifying Enzymes
| Enzyme (PDB ID) | Function | Recognition | Complexed with | Specification |
|---|---|---|---|---|
| DIM-5 (1PEG) | K9 methyltransferase | Unmodified K9 | H3 A7-G13 | Unmodified K9 |
| GCN5 (1QSN) | K14 acetyltransferase | Unmodified K14 | H3 K9-Q19 | Unmodified K9 and K14 |
| JMJD2A (2OQ6) | K9me3 demethylase | Tri-methylated K9 | H3 A7-K14 | K9me3, K14ace |
| DNMT3L (2PVC) | DNA methyltransferase | Unmodified K4 | H3 A1-A7 | Unmodified K4 |
We computed the phi and psi angles of K4, K9, and K14 residues of the histone H3 tails in the enzyme complexes listed above in order to make a comparison with the Ramachandran plots obtained from MD trajectories. The computed values of torsional angles from the enzyme complexes are given in Table II. We furthermore obtained average structures from six simulations of each tail, and investigated the conformations in a comparative manner with the enzyme complexes. The structures of the catalytic cavity of abovementioned enzymes complexed with histone H3 peptides are given in Supporting Information Figures S2–S5.
Table II.
Phi and Psi Angles of K4, K9 and K14 Residues from the Enzyme Complexes
| PDB ID | Histone H3 Residue | Phi | Psi |
|---|---|---|---|
| 1PEG | K9 | −75.52 | 140.63 |
| 1QSN | K14 | −57.78 | 56.89 |
| 2OQ6 | K9 | −62.73 | 142.02 |
| 2PVC | K4 | 179.41 | 147.79 |
The first enzyme we investigated is DIM-5 (1PEG) which is a SUV39-type histone H3 K9 methyltransferase that binds to unmodified K9 residue of histone H3 tail and attaches methyl groups to ɛ-N atom. In 2003, Zhang et al. resolved the crystal structure of DIM-5 with A7-G13 portion of the H3 tail.6 It was reported that main chain N-H and side chain hydroxyl oxygen of S10 residue form hydrogen bonds with the catalytic side residues Y283 and D209.6 The main chain carbonyl of T11 residue also hydrogen bonds to Q285 in the catalytic cavity. These hydrogen bonding interactions play key roles in H3 peptide recognition which makes S10 and T11 crucial residues for DIM-5 recognition and functionality. Additionally, A1-T6 portion and G13-A15 portion of the tail residues are also known to be important in binging of DIM-5 to histone H3.6 We computed the phi and psi angles of K9 residue of DIM-5-H3 tail complex which signifies the necessary binding configuration. Based on these values given in Table II, the torsions of the complex structure are at an accessible range considering the K9 torsions of unmodified H3 tail given in Figure 2(a), which confirms the recognition and binding of enzyme to unmodified K9 residue. However, when we consider the torsions of K9 in K4me3K14ace tail [Fig. 2(c)] which is the other H3 tail having unmodified K9, the phi and psi angles of K9 residue are confined in a limited region which is not allowable for the enzyme binding. This signifies that the tri-methylation of K4 together with the acetylation of K14 brings about a limited set of configurations for K9 residue which are not appropriate for DIM-5 binding and subsequent methylation. This result is consistent with the experimental results of Wang et. al. who demonstrate that the methylation of K4 on H3 inhibits the methylation of K9 on H3 by SUV39 methyltransferase.7 This configurational obstacle can also be observed from Figure 4 which displays the A7-G13 portion of H3 tail of DIM-5 complex and with the altered torsions of K4me3K14ace tail.
Figure 4.
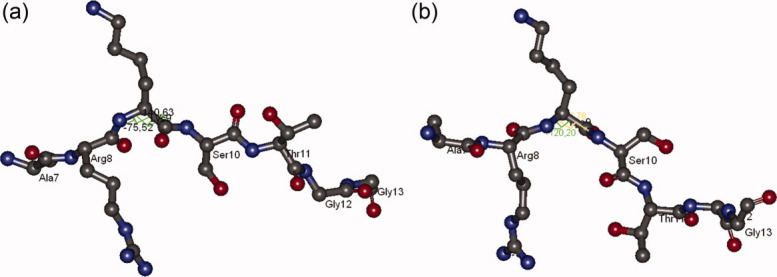
Configurations of A7-G13 portion of H3 tail in (a) DIM-5 complex and (b) in K4me3K14ace. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Table III.
Types of Histone H3 Tails with the Modifications of Interest
| K4 | K9 | K14 | Histone H3 tails |
|---|---|---|---|
| Unmodified | Unmodified | Unmodified | Unmodified tail |
| Tri-methylated | Unmodified | Unmodified | |
| Unmodified | Tri-methylated | Unmodified | |
| Unmodified | Unmodified | Acetylated | |
| Tri-methylated | Tri-methylated | Unmodified | K4me3K9me3 |
| Tri-methylated | Unmodified | Acetylated | K4me3K14ace |
| Unmodified | Tri-methylated | Acetylated | K9me3K14ace |
| Tri-methylated | Tri-methylated | Acetylated |
Figure 4 explains that the A7-G13 portion of histone H3 tail should have an extended configuration in order to fit into the catalytic cavity of DIM-5 enzyme. However, in the case of K4me3K14ace tail, there occurs a kink at K9 residue which perturbs the appropriate configuration of the tail and hinders the binding of methyltransferase. The catalytic cavity of DIM-5 complexed with histone H3 is given in Supporting Information Figure S2.
The second enzyme we examined is GCN5 (1QSN) which is another histone H3 tail lysine specific enzyme that recognizes unmodified K14 and modifies it by adding acetyl groups. It was reported by Rojas et. al. that G13, K14 and P16 are the key residues in recognition and binding of GCN5 to histone H3 tail owing to the extensive peptide–protein interactions formed through these residues.8 The computed phi and psi angles of K14 residue in enzyme complex is given in Table II as −57.78 and 56.89, respectively. When we consider the torsions of K14 residue in the unmodified H3 tail, it can be understood from the Ramachandran map of unmodified H3 tail in Figure 3(a) that the phi and psi values in the complex form are at an accessible range. This accessibility approves that the K14 residue of unmodified H3 tail adopts such conformations that can be recognize by GCN5 and allows the enzyme binding. Furthermore, when we consider the torsions of the K14 residue in K4me3K9me3 tail [Fig. 3(b)], which is the other H3 tail having unmodified K14 residue, the phi and psi angles of the K14 residue in the enzyme complex are similarly at an allowable range. This signifies that tri-methylating K4 and K9 residues of H3 tail do not disturb the recognition and binding of GCN5 to H3 tail. On the other hand, the phi and psi distributions of K14 residue in K4me3K14ace [Fig. 3(c)] and K9me3K14ace [Fig. 3(d)] tails do not comprise the values required for the binding of GCN5 which is reasonable as the enzyme can only bind to the unmodified K14 residue not the acetylated form. According to the experimental results of Martin et al. and Taverna et al., methyl-histone binding module Yng1 that recognizes tri-methylated K4 on H3 facilitates acetylation of K14 on H3 by another histone acetyltransferase (HAT) NuA3.9,10 This study points out the enhanced acetylation of K14 residue in the case of tri-methylated K4 residue on H3. Further experimental investigations are required to elucidate whether a similar enhancement exists for GCN5 activity, as well.
We also explored DNMT3L (2PVC) which is a regulatory factor that recognizes unmodified K4 of H3 and induces de novo DNA methylation by recruiting DNMT3A and DNMT3B DNA methyltransferases. In the structural study of DNMT3L-H3 tail complex, Ooi et al. was able to solve the structure of only the first seven residues of the N-terminal H3 tail, which have interactions with the binding site residues of DNMT3L.11 This finding indicates that the A1-A7 portion of H3 tail is the crucial region for the binding of the regulatory factor to H3 peptide. We obtained phi and psi angles of the unmodified K4 residue in the complex structure to be 179.41 and 147.89 as displayed in Table II. These values are at the allowable range for the unmodified K4 residue Ramachandran map of the unmodified H3 tail [Fig. 1(a)], which verifies the recognition and binding of DNMT3L to unmodified histone H3 tail. According to Figure 1, the Ramachandran maps of K4 residue are almost the same for the unmodified and K9me3K14ace tails. Hence, the torsions of the complex form are at the accessible range for K9me3K14ace tail, as well, which denotes that the recognition and binding of DNMT3L to unmodified K4 on H3 is not affected by the tri-methylation of K9 together with the acetylation of K14. This result is in accordance with the study of Lehnertz et al. who revealed an evolutionary conserved pathway between tri-methylation of K9 on H3 and DNA methylation. In their study, they showed that the tri-methylation of K9 by SUV39H is prerequisite mark for DNA methylation by DNMT3B.12 There have been other studies indicating that methylation of K9 on H3 is required for DNA methylation.13,14
Last, we studied JMJD2A (Jumonji domain containing 2A) (2OQ6) which is a histone demethylase that is selective towards di- or tri-methylated K9 and K36 of histone H3. It has been known that Jumonji domain of JMJD2A recognizes the backbone of H3 tail and removes methyl groups from K9 and K36 residues.15–17 Ng et al. solved the structure of the A7-K14 portion of the H3 tail complexed with the catalytic domain of JMJD2A and reported that the G12-G13 positioning has essential role in enzyme binding as well as S10 residue, which makes intrasubstrate interactions.15 H3 peptide in the crystal structure had tri-methylated K9 (K9me3) and acetylated K14 (K14ace) residues. We computed the phi and psi angles of K9me3 residue of the complex as −62.73 and 142.02, respectively. When compared with the Ramachandran map of K9me3 residue of K9me3K14ace tail in Figure 2(d), these values seem to be at the accessible range, which confirms the recognition of binding of JMJD2A to K9me3 in the K9me3K14ace form. On the other hand, simultaneous tri-methylation of K4 and K9 restricted the conformations of K9me3 residue in a confined range so that the abovementioned torsions are not allowable [Fig. 2(b)]. This result indicates that the double tri-methylation of K4 and K9 residues interrupts the binding of demethylase enzyme to K9 on H3 tail. The configurations of the A7-K14 portion of H3 tail of JMJD2A complex and in the case of K4me3K9me3 tail can be observed from Figure 5. The catalytic cavity of JMJD2A complexed with histone H3 is given in Supporting Information Figure S5.
Figure 5.
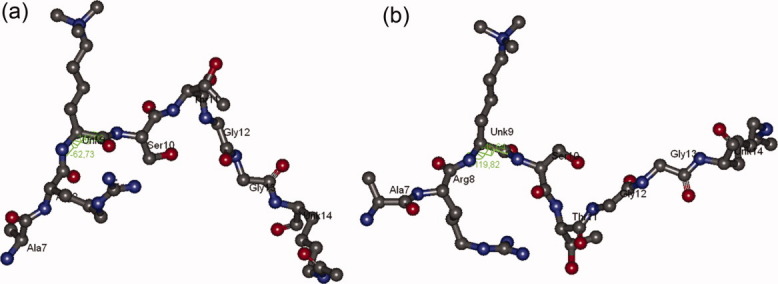
Configurations of A7-K14 portion of H3 tail in (a) JMJD2A complex and (b) in K4me3K9me3. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 5 indicates that the A7-K14 portion of histone H3 tail should have a V-shaped configuration with a kink at T11, in order to fit into the catalytic cavity of JMJD2A enzyme. In the case of K4me3K9me3 tail, there also occurs a kink at T11. However, this kink orients the conformation of G12-K14 portion in an opposite direction so that the overall tail configuration adopts an N-shaped structure. This configurational alteration seems to be an obstacle for the enzyme binding owing to the important roles of G12-G13 residue pair in JMJD2A function. Besides, JMJD2A is also known to have a Tudor domain which can recognize and bind to tri-methylated K4 of H3.18 However, the functional interplay between the Jumonji and Tudor domains remains unclear.19,20 Thus, further experimental studies are required to clarify the functional relationship between these two domains by means of K4 methylation.
Post-translational modifications of histone H3 residues have been a complex and unclear process which is mostly dominated by cell-signaling and recruitment of the modifying enzymes. The structural and experimental studies are inadequate for complete revelation of the underlying modification mechanisms. Moreover, to our knowledge, the downstream outcomes of the simultaneous modifications of more than one residue have not been clarified by experimental studies. In that respect, we believe that this computational analysis of dihedral angles of the H3 tail constitutes guidance for future experimental studies that would aim to elucidate the structural effects of double modifications on H3 tail. This study also shows that, although the post-translational modification of H3 tail residues is majorly a cell-signaling process, there is also a structural control mechanism by means of residue conformations.
Conclusion
The N-terminal tail of the histone H3 has a crucial role in epigenetics due to the post-translational modifications to which the residues are subjected. Especially, the modifications of K4, K9, and K14 residues have proven to be the switch points in cross-regulation patterns which have diverse downstream outcomes. MD simulations of the unmodified H3 tail, the K4me3K9me3 tail, the K4me3K14ace tail, and the K9me3K14ace tail were carried out to investigate the structural influence of the double modifications on the abovementioned three residues of the histone H3 tail. The choice for the simulation tails was originated from the cooperativity knowledge between the K4, K9, and K14 modifications, as well as the thought of the remarkable consequences of the double modifications.
We analyzed the torsional angles of K4, K9, and K14 residues at each double modification of H3 tail. Moreover, we investigated the structures of the tail residues in the histone modifying enzyme complexes together with the tail structures obtained from MD simulations. According to the Ramachandran maps of the aforementioned residues, the most effective modification seems to be the tri-methylation of K4 regardless of the type of second modification. Furthermore, based on the structural investigations of enzyme complexes, we conclude that the tri-methylation of K4 residue together with the acetylation of K14 residue disrupts the binding of DIM-5 methyltransferase to the unmodified K9 residue, whereas tri-methylation of K9 on H3 does not interrupt the recognition and binding of DNMT3L to unmodified K4 on H3. Additionally, the tri-methylation of K4 residue was found to disturb the H3 tail conformation in such a way that binding of JMJD2A demethylase enzyme to tri-methylated K9 residue is unavailable.
Materials and Methods
As mentioned previously, K4 and K9 residues of histone H3 tail can be tri-methylated whereas K14 residue can undergo acetylation. Based on these three types of modifications, there occurs to be eight probable modification states of histone H3 tail which are given in Table IV. From these eight probable states, we selected the histone H3 tails that have double modification together with the unmodified histone H3 tail to carry out MD simulations. We considered that the double modifications on the histone H3 tail would have remarkable consequences and lead to more significant structural alterations. Our further aim was to reduce the computational costs as well as time.
Four types of 40-residue Histone H3 tails were selected for molecular dynamics simulations, as can be observed from Table IV. These were (i) the unmodified H3 tail having no chemical modification on the residues, (ii) the tri-methylated lysine 4 and lysine 9 tail (K4me3K9me3) having 3 methyl groups added to both 4th and 9th lysine residues, (iii) the tri-methylated lysine 4 and acetylated lysine 14 tail (K4me3K14ace) having 3 methyl groups on the 4th lysine and an acetyl group on the 14th lysine residues and (iv) tri-methylated lysine 9 and acetylated lysine 14 tail (K9me3K14ace) having 3 methyl groups on the 9th lysine and an acetyl group on the 14th lysine residues. The structures of four H3 tails are given in Figure S1 in Supporting Information. The 40-residue part of the N-terminal of Histone H3 that protrudes from the nucleosome surface was obtained from the crystal structure of the nucleosome with the PDB ID 1KX5.21 Addition of methyl groups to the lysine side-chains was performed by Accelrys Discovery Studio Visualizer program.22 Since these chemical modifications affect the partial charges and chemical parameters of the lysine residues, a parameterization procedure was carried out for the modified lysine residues (see Appendix).
For each of four H3 tails, 6 MD simulations were carried out starting from different initial configurations to account for thorough sampling. These different configurations were obtained by taking random snapshots from a 2 ns simulated annealing (data not shown). The simulations were performed in explicit solvent with NAMD 2.5 package by using Duan et al. force field (2003).23,24 In order to be consistent with cellular conditions, isothermal-isobaric (NTP) ensemble was used for simulations with periodic boundary conditions. The temperature and pressure was set to 310 K and 1 bar, respectively. Computations of nonbonded interactions were carried out with a cut-off distance of 12 Å. The Particle Ewald sum was used for calculation of long-range forces, thus minimizing the error due to cut-off distance in periodic systems. For each simulation 1000 step energy minimization was performed. Simulations were recorded for 7 ns with an integration time step of 2 fs. The parameterization procedure of the modified lysine resides are described in detail in Appendix.
Appendix: Parametrization of Methylated and Acetylated Lysine Residues
The parametrization procedure was carried out in order to obtain the simulation parameters of the methylated and acetylated lysine residues. Chemical parameters for the tri-methylated and acetylated lysine residues such as bond lengths, bond angles, dihedral angles were obtained from the Duan et al. force field (2003).23 For the partial charges, Ante RED, RED III and Gaussian03 programs were used to derive RESP charges.25,26 As the initial step, the Ante RED program was executed to obtain input files for geometry optimization and MEP computation.25 Geometry optimization was carried out via Gaussian03 with the input file obtained from Ante RED. Subsequently, the RED-III program was used for MEP computation of modified lysine residues with optimized geometries.25 The MEP computation was performed by taking the dielectric constant (ɛ) 4 under continuum solvent conditions and with B3LYP/cc-pVTZ basis set that was used in Duan et al. force field (2003).23 After MEP computation RESP charge fitting was carried out and the partial charges for each atom in the modified lysine residues was obtained for MD simulations.
Supplementary material
References
- 1.Peterson CL, Laniel MA. Histones and histone modifications. Curr Biol. 2004;14:R546–R551. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 2.Loizou JI, Murr R, Finkbeiner MG, Sawan C, Wang ZQ, Herceg Z. Epigenetic information in chromatthe code of entry for DNA repair. Cell Cycle. 2006;5:696–701. doi: 10.4161/cc.5.7.2616. [DOI] [PubMed] [Google Scholar]
- 3.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 4.Biel M, Wascholowski V, Giannis A. Epigenetics—an epicenter of gene regulation: histones and histone-modifying enzymes. Angew Chem Int Ed Engl. 2005;44:3186–3216. doi: 10.1002/anie.200461346. [DOI] [PubMed] [Google Scholar]
- 5.Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Curr Opin Cell BIol. 2003;15:172–183. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 6.Zhang X, Yang Z, Khan SI, Horton JR, Tamaru H, Selker EU, Cheng XD. Structural basis for the product specificity of histone lysine methyltransferases. Mol Cell. 2003;12:177–185. doi: 10.1016/s1097-2765(03)00224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang HB, Cao R, Xia L, Erdjument-Bromage H, Borchers C, Tempst P, Zhang Y. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol Cell. 2001;8:1207–1217. doi: 10.1016/s1097-2765(01)00405-1. [DOI] [PubMed] [Google Scholar]
- 8.Rojas JR, Trievel RC, Zhou JX, Mo Y, Li XM, Berger SL, Allis CD, Marmorstein R. Structure of Tetrahymena GCN5 bound to coenzyme A and a histone H3 peptide. Nature. 1999;401:93–98. doi: 10.1038/43487. [DOI] [PubMed] [Google Scholar]
- 9.Howe L, Martin DGE, Baetz K, Shi XB, Walter KL, MacDonald VE, Wlodarski MJ, Gozani O, Hieter P. The Yng1p plant homeodomain finger is a methyl-histone binding module that recognizes lysine 4-methylated histone H3. Mol Cell Biol. 2006;26:7871–7879. doi: 10.1128/MCB.00573-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tackett AJ, Taverna SD, Ilin S, Rogers RS, Tanny JC, Lavender H, Li HT, Baker L, Boyle J, Blair LP, Chait BT, Patel DJ, Aitchison JD, Allis CD. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol Cell. 2006;24:785–796. doi: 10.1016/j.molcel.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ooi SKT, Qiu C, Bernstein E, Li KQ, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, Cheng XD, Bestor TH. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–U713. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehnertz B, Ueda Y, Derijck AAHA, Braunschweig U, Perez-Burgos L, Kubicek S, Chen TP, Li E, Jenuwein T, Peters AHFM. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol. 2003;13:1192–1200. doi: 10.1016/s0960-9822(03)00432-9. [DOI] [PubMed] [Google Scholar]
- 13.Tamaru H, Zhang X, McMillen D, Singh PB, Nakayama J, Grewal SI, Allis CD, Cheng XD, Selker EU. Trimethylated lysine 9 of histone H3 is a mark for DNA methylation in Neurospora crassa. Nature Genet. 2003;34:75–79. doi: 10.1038/ng1143. [DOI] [PubMed] [Google Scholar]
- 14.Jackson JP, Lindroth AM, Cao XF, Jacobsen SE. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature. 2002;416:556–560. doi: 10.1038/nature731. [DOI] [PubMed] [Google Scholar]
- 15.Schofield CJ, Ng SS, Kavanagh KL, McDonough MA, Butler D, Pilka ES, Lienard BMR, Bray JE, Savitsky P, Gileadi O, von Delft F, Rose NR, Offer J, Scheinost JC, Borowski T, Sundstrom M, Oppermann U. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature. 2007;448:87–91. doi: 10.1038/nature05971. [DOI] [PubMed] [Google Scholar]
- 16.Zhang G, Chen ZZ, Zang JY, Kappler J, Hong X, Crawford F, Wang Q, Lan F, Jiang CY, Whetstine J, Dai S, Hansen K, Shi Y. Structural basis of the recognition of a methylated histone tail by JMJD2A. Proc Natl Acad Sci USA. 2007;104:10818–10823. doi: 10.1073/pnas.0704525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trievel RC, Couture JF, Collazo E, Ortiz-Tello PA, Brunzelle JS. Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat Struct Mol Biol. 2007;14:689–695. doi: 10.1038/nsmb1273. [DOI] [PubMed] [Google Scholar]
- 18.Ozboyaci M, Gursoy A, Erman B, Keskin O. Molecular recognition of H3/H4 histone tails by the Tudor domains of JMJD2A: a comparative molecular dynamics simulations study. Plos One. 2011;6 doi: 10.1371/journal.pone.0014765. [PAGE #s] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. 2006;312:748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 20.Mer G, Lee J, Thompson JR, Botuyan MV. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat Struct Mol Biol. 2008;15:109–111. doi: 10.1038/nsmb1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J Mol Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 22.Inc., A. S. Discovery Studio Visualizer. Release 2.0 ed. Accelrys Software Inc; 2007. [Google Scholar]
- 23.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 24.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dupradeau FY, Cieplak P. 1995. Available at: http://q4md-forcefieldtools.org/Tutorial/Tutorial-1.php.
- 26.Inc., G. Revision C.02 ed. 2004. Gaussian 03, (CT, W., Ed.)
- 27.Ng SS, Kavanagh KL, McDonough MA, Butler D, Pilka ES, Lienard BMR, Bray JE, Savitsky P, Gileadi O, von Delft F, Rose NR, Offer J, Scheinost JC, Borowski T, Sundstrom M, Schofield CJ, Oppermann U. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature. 2007;448:87–91. doi: 10.1038/nature05971. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.