

Abstract
Pathogenic yeasts of the genus Candida produce secreted aspartic proteinases, which are known to enhance virulence. We focused on Sapp1p proteinase secreted by Candida parapsilosis and studied the final stage of its passage through the cell wall and release into the extracellular environment. We found that Sapp1p displays enzyme activity prior to secretion, and therefore, it is probably fully folded within the upper layer of the cell wall. The positioning of cell surface-associated Sapp1p was detected by cell wall protein labeling using biotinylation agents, extraction of cell wall proteins by β-mercaptoethanol, immunochemical detection, and mass spectrometry analysis. All lysine residues present in the structure of soluble, purified Sapp1p were labeled with biotin. In contrast, the accessibility of individual lysines in cell wall-associated Sapp1p varied with the exception of four lysine residues that were biotinylated in all experiments performed, suggesting that Sapp1p has a preferred orientation in the cell wall. As the molecular weight of this partially labeled Sapp1p did not differ among the experiments, we can assume that the retaining of Sapp1p in the cell wall is not a totally random process and that pathogenic yeasts might use this cell-associated proteinase activity to enhance degradation of appropriate substrates.
Keywords: Candida parapsilosis, secreted aspartic proteinases, Sapp1p, cell wall, biotin, proteolytic activity
Introduction
Candida parapsilosis is one of the most frequently occurring opportunistic fungal pathogens. In epidemiological surveys it ranks as the second or third most common Candida species, depending on the medical setting and geographical area.1,2 It is a major cause of nosocomial infections by yeasts, and high incidence of C. parapsilosis has been reported in neonatal intensive care units.3,4 C. parapsilosis infections are often exogenous. The fungus can be transmitted via contaminated invasive therapeutic or monitoring equipment. In addition, C. parapsilosis is isolated from the hands of healthy individuals and healthcare personnel more often than any other yeast species.5
Factors that enhance the virulence of pathogenic Candida spp. include efficient adhesion to host surfaces, morphological diversity, biofilm formation, and secretion of hydrolytic enzymes. Secreted aspartic proteinases, together with lipases and phospholipases, facilitate yeast adhesion in the initial stages of infection, enable penetration of the pathogens into host tissues, and provide a source of nutrients for the pathogen by breaking down host molecules.6–8 Pathogenic Candida spp. usually possess a gene family encoding secreted aspartic proteinases (SAPs). Historically, C. parapsilosis was considered to possess three such genes: SAPP1, SAPP2,9 and SAPP3 (NCBI accession number AF339513). Recently, a phylogenetic analysis of the published C. parapsilosis genome10,11 revealed 14 sequences potentially encoding secreted aspartic proteinases. However, no further information about the expression of these genes and proteinases is available. Only Sapp1p and Sapp2p proteinases have been biochemically characterized.12–14 Production of Sapp1p is induced in the presence of an exogenous protein as a sole nitrogen source, as in the case of the Sap2 enzyme from C. albicans. Sapp1p, the main secreted isoenzyme of C. parapsilosis, has a broad substrate specificity and higher catalytic activity than Sapp2p, which has been shown to be secreted constitutively under all conditions tested but in a concentration at least one order of magnitude lower.15
The SAP genes of pathogenic Candida spp. encode pre-pro-enzymes that consist of a leader peptide followed by a pro-peptide and mature protease. The signal peptide is removed in the rough endoplasmic reticulum, and the zymogen is activated after transport to the Golgi apparatus by Kex2 proteinases or alternative pathways.13,16,17 The mature proteinases are transported via the secretory pathway to the cell surface and have to pass the protective cell wall, which consists of two main layers of different electron density. The inner layer contains a network of 1,3-β-glucan, 1,6-β-glucan, and chitin molecules, and the dense outer layer is enriched with mannoproteins covalently linked to the nonreducing ends of 1,6-β-glucans via glycosyl phosphatidylinositol (GPI) anchors. Additional cell wall proteins (CWPs) are linked to 1,3-β-glucan via a linkage sensitive to alkali treatment (non-GPI-CWPs or Pir-CWPs; for review see Chaffin).18. The temporarily cell wall associated proteins and noncovalently linked proteins can be released from the yeast cell wall by ionic detergents or chaotropic agents.19,20 In addition, the extraction of cell wall fractions with a reducing agent such as β-mercaptoethanol (βME) leads to release of a distinct set of proteins, which might be bound to other cell wall proteins by disulfide bridges.21,22
In this study, we show that Sapp1p is temporarily retained in the cell wall prior to secretion into the extracellular space. While associated with the cell surface, Sapp1p can cleave substrates that occur in the surrounding environment, demonstrating the accessible positioning of the proteinase active site in the cell wall.
Results
Sapp1p is temporarily associated with the cell wall and can cleave extracellular substrates
To analyze the presence of the Sapp1p proteinase of C. parapsilosis in the cell wall and to detect its proteolytic activity, we collected C. parapsilosis cells grown in the presence of BSA as a sole nitrogen source, conditions known to induce the expression of Sapp1p. Washing the cells with water or with PTB buffer removed remnants of the medium, including soluble secreted Sapp1p. We detected Sapp1p in the first wash fractions, and further washing of the cells did not release detectable amounts of Sapp1p [Fig. 1(A)]. When we used PTB with 0.5% βME, Sapp1p was found even in the fourth wash [Fig. 1(B)], indicating that a substantial amount of Sapp1p is retained in the cell wall and that βME treatment causes its release. Similar results were obtained when the cells were treated with PTB containing 1% SDS (data not shown). Sapp1p was not present in any additional fractions of cell wall protein isolation (NaOH-fraction, Lyticase mediated fraction, data not shown).
Figure 1.
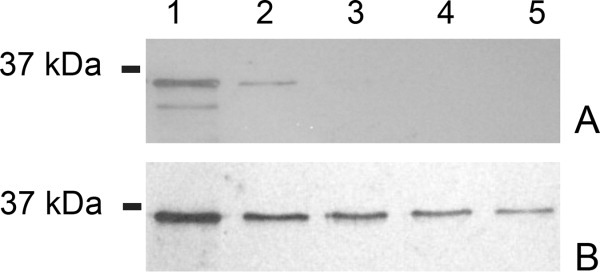
Western blot detection of Sapp1p associated with the cell surface of C. parapsilosis. Panel A: fractions obtained by cell surface washing with PTB buffer; Panel B: fractions from washing with PTB containing 0.5% βME. Lane 1, fraction after 10-min incubation; Lanes 2–5, fractions after repeated washings. Detection was performed using polyclonal antibodies raised against Sapp1p.
To obtain a sufficient amount of cell wall-associated Sapp1p for further experiments, we treated the yeast cell pellet with extraction buffer containing 1% βME. SDS-PAGE and Western blot analyses showed that the molecular weight of the Sapp1p fraction extracted from the cell wall corresponded to that of the secreted mature proteinase (Fig. 2). Furthermore, the N-terminal sequence of the cell wall-associated proteinase (DSISL-) was the same as that of fully processed, active Sapp1p. Mass spectrometry analysis of the immunochemically reactive bands presented in Figure 2 confirmed that only the band at a molecular weight of 36 kDa contains Sapp1p. The band corresponding to a molecular weight of about 34 kDa and the band at the top of Lane 2, which contains protein aggregates, are unknown proteins extracted from the cell wall that nonspecifically cross react with anti-Sapp1p polyclonal antibodies.
Figure 2.
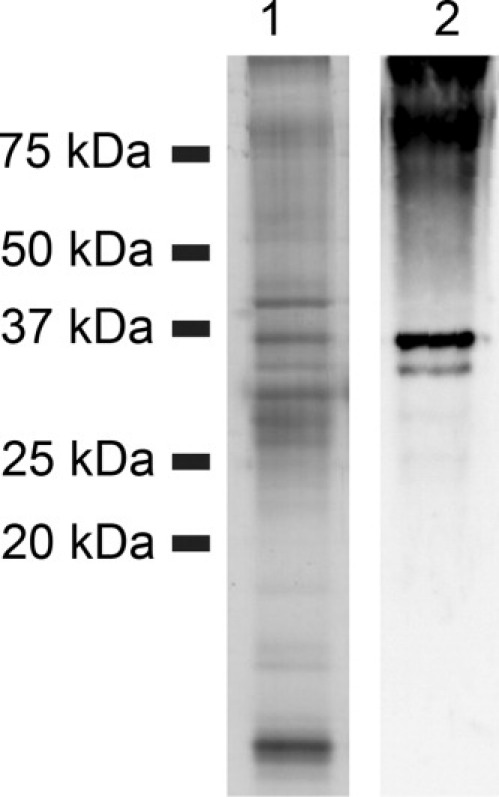
Profile of proteins isolated from the C. parapsilosis cell wall with 1% βME. Lane 1, silver-stained SDS-PAGE; Lane 2, Western blot of cell wall protein samples detected with polyclonal antibodies raised against a peptide corresponding to part of the Sapp1p sequence (anti-Sapp1p/186-199).
We also analyzed the βME extraction fractions for the presence of Sapp2p, another secreted isoenzyme of C. parapsilosis, using Western blot and specific anti-Sapp2p antibodies. We detected only traces of Sapp2p in the cell wall fractions (data not shown). This is not surprising, since the concentration of Sapp2p in medium upon induction is always at least one order of magnitude lower than that of Sapp1p.15
To examine whether cell wall-associated Sapp1p can cleave substrates present in the extracellular space, we incubated thoroughly washed C. parapsilosis cells with a fluorescent peptide substrate for 30 minutes. The substrate was readily hydrolyzed, and its cleavage was sensitive to the presence of pepstatin A, a potent inhibitor of Sapp1p23 (Fig. 3). The fluorescent substrate that we used for the activity assay can differentiate between the Sapp1p and Sapp2p izoenzymes.15 However, no Sapp2p proteolytic activity was detected in the reaction mixture, which correlates with very low level of Sapp2p in the cell wall detected by Western blot. To confirm that the substrate cleavage was mediated by the cell wall-associated enzyme and not by Sapp1p released into the reaction mixture during the incubation, we preincubated the washed cells in the proteinase activity buffer for 30 min, removed the cells, and incubated the cell-free solution with the proteinase substrate for an additional 30 min. The cell-free reaction mixture did not display any proteolytic activity. These results demonstrate that cell wall-associated Sapp1p is proteolytically active even prior to release and that its conformation enables sufficient substrate cleavage.
Figure 3.
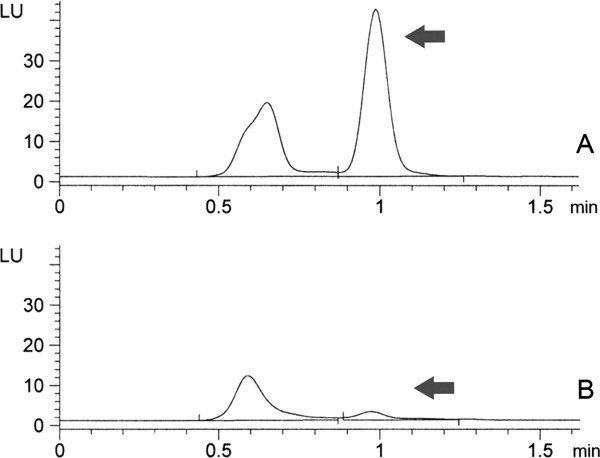
Cleavage of the fluorescent substrate Dabcyl-EHVKLVE-EDANS by cell-associated Sapp1p. Panel A: cleavage products obtained from incubation of C. parapsilosis cells with the substrate. Panel B: substrate cleavage products from incubation of C. parapsilosis cells in the presence of 1 μM pepstatin A. Arrows indicate the position of Sapp1p-specific cleavage product. The position of the cleavage product was verified by in vitro substrate cleavage with purified Sapp1p.
Biotinylation of the cell wall reveals exposed parts of the Sapp1p molecules
The finding that cell surface-associated Sapp1p can cleave an exogenous substrate indicates that the proteinase active site is accessible for substrates prior to the enzyme's release. We set out to analyze the positioning of Sapp1p in the cell wall by biotin labeling, a technique that has been successfully used for labeling of C. albicans cell wall proteins.24,25 We used both sulfo-NHS-biotin and sulfo-NHS-LC-biotin, which contains a longer spacer that may facilitate access of the reagent to more complex protein structures.
Sapp1p contains 15 lysine residues and an N-terminal aspartate that might be labeled with biotin. To examine whether all of the potential sites in Sapp1p are accessible to biotinylation, we performed labeling of the purified secreted proteinase. Incubation of Sapp1p aliquots with a dilution series of sulfo-NHS-biotin or sulfo-NHS-LC-biotin resulted in a stepwise increase in the molecular weight of the proteinase (Fig. 4). The molecular weight of biotinylated Sapp1p determined by mass spectrometry analysis confirmed that 15 molecules of biotin are bound upon incubation of Sapp1p with stock solutions of both biotinylation reagents. N-terminal sequencing of biotinylated Sapp1p showed that the N-terminal aspartate remained unbiotinylated, indicating that all the lysine residues in Sapp1p are accessible for biotinylation in vitro.
Figure 4.
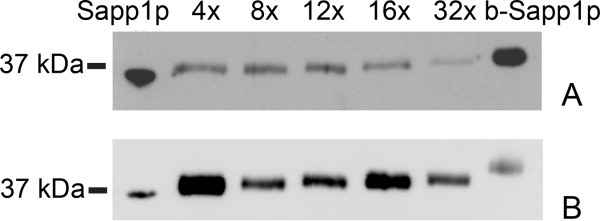
Western blot analysis of stepwise biotinylation of purified Sapp1p using a 4-, 8-, 12-, 16-, or 32-fold molar excess of biotinylation reagent. The blot was probed with anti-Sapp1p polyclonal antibodies (anti-Sapp1p/186-199). Panel A: biotinylation performed with sulfo-NHS-biotin; Panel B: biotinylation performed with sulfo-NHS-LC-biotin. Sapp1p, purified nonbiotinylated proteinase; b-Sapp1p, purified fully biotinylated proteinase.
Next, we treated C. parapsilosis cells with sulfo-NHS-biotin or sulfo-NHS-LC-biotin and extracted the cell wall proteins using βME as described in Methods. Western blot analysis indicated that surface-associated Sapp1p was only partially biotinylated, since its molecular weight was higher than that of the nonbiotinylated Sapp1p control sample and lower than that of fully biotinylated proteinase (Fig. 5). During repeated experiments, this Western blot pattern did not significantly change, even when higher amounts of the reagents were used or when the incubation time was prolonged.
Figure 5.
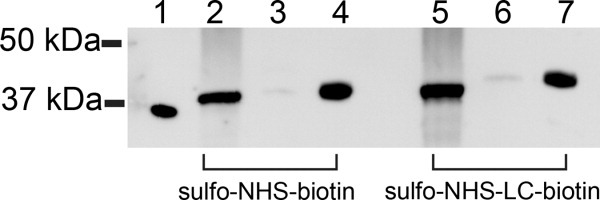
Western blot analysis of biotinylation of intact C. parapsilosis cells with sulfo-NHS-biotin (left) or sulfo-NHS-LC-biotin (right) using polyclonal anti-Sapp1p antibodies. Lane 1, unbiotinylated Sapp1p; Lanes 2, 5, βME extract after biotinylation of the intact cells; Lanes 3, 6, material released from the cell surface during incubation with biotinylation reagents; Lanes 4, 7, purified, fully biotinylated Sapp1p.
The SDS-PAGE band corresponding to cell wall-extracted biotinylated Sapp1p was analyzed using in-gel digestion with trypsin and chymotrypsin and mass spectrometry (MS). In three repeated biotinylation experiments, we obtained peptide fragments containing the N-terminal aspartate and 14 lysine residues. Only Lys24 was not detected in any of the extracted peptides. Table I shows a typical set of analyses of tryptic and chymotryptic digests of partially labeled Sapp1p obtained from the biotinylation experiment. The final Sapp1p sequence coverage was 73% (Fig. 6). Despite this high sequence coverage, Lys17, Lys90, and Lys149 were identified only in tryptic peptide fragments as C-terminal amino acids. MS analysis of proteinase fragments showed that labeling of most of the lysines was ambiguous. In separate experiments, we detected lysines 148, 181, 185, 207, 239, and 329 either with or without the biotin label. In contrast, lysines 49, 68, 152, and 275 were biotinylated in all the MS measurements performed. Interestingly, all unbiotinylated or undetected lysines were present in the N-terminal domain of the molecule (see Fig. 6). These results indicate that Sapp1p molecules have different orientations when they are retained in the cell wall. There is, however, a sufficient population of those molecules whose active sites are accessible to external substrates. Proteolytic degradation of proteins or peptides occurring in the environment surrounding C. parapsilosis cells can thus proceed prior to the release of Sapp1p into the extracellular space.
Table I.
MS Analysis of Biotinylation of Cell Surface-Associated Sapp1p
| Position of primary amine group | B/S | |
|---|---|---|
| Trypsin digestion | Chymotrypsin digestion | |
| Asp1 | 0/2 | n.d. |
| Lys17 | 0/2 | n.d. |
| Lys24 | n.d. | n.d. |
| Lys49 | n.d. | 1/1 |
| Lys54 | n.d. | 0/1 |
| Lys68 | n.d. | 1/1 |
| Lys90 | 0/2 | n.d. |
| Lys148 | 2/4 | n.d. |
| Lys149 | 0/2 | n.d. |
| Lys152 | 1/1 | n.d. |
| Lys181 | 0/2 | 1/2 |
| Lys185 | 2/2 | 1/2 |
| Lys207 | 0/2 | 1/2 |
| Lys239 | 0/2 | 2/3 |
| Lys275 | n.d. | 1/1 |
| Lys329 | n.d. | 1/2 |
The band containing Sapp1p was cut from a gel and digested with trypsin or chymotrypsin (see Materials and Methods for details). B, number of peptide fragments containing bitonylated residue; S, total number of relevant peptide fragments; n.d., peptide fragment was not detected.
Figure 6.

Positions of lysine residues in the three-dimensional (Panel A) and primary (B) structures of Sapp1p. The unbiotinylated lysine residues are indicated in red, the biotinylated lysines in green, and the undetected lysine residue (Lys24) in black. The sequence regions obtained from the combined in-gel digest of biotinylated Sapp1p are framed in gray in panel B.
Discussion
Secretion of hydrolytic enzymes, especially aspartic proteinases, is important for the virulence of pathogenic Candida species. While secreted proteinases have been thoroughly studied in C. albicans, the SAPP gene family of C. parapsilosis has been only partially described. In this study, we focused on the C. parapsilosis enzyme Sapp1p, which is induced in a similar manner as Sap2 of C. albicans but displays certain enzymological and structural differences.23,26 To study the passage of Sapp1p through the yeast cell wall during secretion into the extracellular space, we used biotin labeling of cell wall proteins followed by mass spectroscopy analysis. Due to the dynamics of protein secretion and the complexity of the cell wall, this experimental approach provides a global picture of the trafficking of a major population of Sapp1p molecules, rather than a detailed description of possible topological variants of the proteinase on the cell surface.
Labeling of cell surface proteins with a biotinylation reagent has been successfully used for identification of yeast cell wall proteins.21,24 The recently solved Sapp1p X-ray structure26 showed that the active molecule has an open structure and that lysine residues are present in both N- and C-terminal proteinase domains. Moreover, all 15 lysines are surface-exposed, which makes it possible to efficiently label Sapp1p with biotin. Indeed, we showed that Sapp1p can be fully biotinylated in vitro. Successful labeling of all 15 lysines in purified Sapp1p provided a sufficient control for further experiments performed with whole C. parapsilosis cells. Biotinylation of cell wall proteins yielded only partially labeled Sapp1p. In repeated experiments, we observed bands of similar molecular weight for extracted, modified Sapp1p; however, in-gel digests of these bands revealed different labeling of the individual lysines, likely reflecting variations in the positioning of Sapp1p in the cell wall. Nevertheless, we found four lysine residues that were biotinylated in all the experiments performed. The accessibility of the remaining lysines for biotinylation varied. The unlabeled lysines might be located in the Sapp1p region that preferably interacts with other cell wall proteins, and this interaction may hinder the accessibility of the lysine residues. This indicates that the position of Sapp1p in the cell wall is not totally random and that Sapp1p has a preferred orientation. It also raises the question of what types of interactions occur between Sapp1p and cell wall structures before the release of the proteinase into the extracellular space. Since we succeeded in extracting Sapp1p using the β-mercaptoethanol extraction method, we might infer that Sapp1p is retained in the cell wall by disulfide bridges. However, the Sapp1p crystal structure showed that the four cysteine residues present in the structure form two disulfide bridges, which contribute to the maintenance of the proper proteinase conformation and thus to the proteolytic activity. Since the cell wall-associated Sapp1p is proteolytically active, we can assume that the proteinase cysteines do not form disulfide bridges with cysteines from other cell wall proteins but rather form two intramolecular S-S bridges. Treatment of the cell wall with βME therefore likely releases S-S linkages in other proteins that contribute to proteinase capture in the cell wall. Moreover, we succeeded in extracting Sapp1p from the cell wall with 1% SDS, which can release temporarily associated, noncovalently bound cell wall proteins. Sapp1p is likely bound in the cell wall by interactions with other cell wall proteins.
In C. albicans, localization of Sap2 in the cell wall was detected during experimental infection of model reconstituted human vaginal epithelium27 and also in the vaginas of infected ovariectomized estrogen-treated rats using immunoelectron microscopy with anti-Sap2 antibodies.28 Nevertheless, it was not clear which conformation Sap2 adopts in the cell wall, whether it is temporarily retained there, and if so, whether it is proteolytically active prior to its release. These questions have not been addressed for C. albicans or for other pathogenic Candida species. It has been assumed that Saps hydrolyze host proteins after secretion. Our present work shows that peptides occurring in the environment surrounding C. parapsilosis cells can be cleaved by Sapp1p before the enzyme is secreted into the extracellular environment. This feature may be common to protein-induced Candida proteinases, such as Sap2 from C. albicans or Sapt1p from C. tropicalis. The ability of yeast cells to retain active Sap molecules in the cell wall before their secretion into extracellular space might provide a great benefit for pathogenic Candida spp., mostly during biofilm formation, but also during adhesion and colonization of host tissues.
Besides secreted aspartic proteinases, C. albicans possesses two aspartic proteinases that are covalently linked to the cell wall and plasma membrane via GPI-anchors (Sap9 and Sap10). These proteinases are known to perform many cell wall-related functions, ranging from maintenance of cell wall integrity to interaction with epithelial cells and the cleavage of cell surface-associated proteins.29,30 They cleave the substrate at basic or dibasic residues, in contrast to the pepsin-like secreted proteinase Sap2, which prefers the presence of hydrophobic amino acids around the cleavage site. Secreted proteinases retained in the cell wall can thus increase the proteolytic activity associated with the surface of yeast cells. For C. parapsilosis, no typical GPI-specific sequence has been identified in the available SAPP genes.11 Cell wall-associated Sapps in C. parapsilosis might therefore play an important role in processes in which cell-associated proteolytic activity of C. parapsilosis is needed.
Materials and Methods
Strains, media, and growth conditions
The Candida parapsilosis strain used for this work was clinical isolate CP69 obtained from the mycological collection of the Faculty of Medicine, Palacky University, Olomouc, Czech Republic.
Yeast was incubated either in the induction medium, which consisted of 1.2% yeast carbon base (YCB) supplemented with 0.2% bovine serum albumin (BSA), pH 3.5, or in YPD (1% yeast extract, 2% peptone, and 2% glucose), which was used as a noninduction medium. Yeast was cultivated at 30°C.
Cell surface washing
Soluble secreted proteins from the surface of C. parapsilosis cells were washed out from the harvested cells with water or with 50 mM Tris-HCl, pH 7.5, containing 10 mM NaN3, and 10 mM NaF (pretreatment buffer, PTB).31
Extraction of noncovalently bound cell wall proteins
Noncovalently bound cell wall proteins were extracted using a modification of the method described by Casanova et al.24 Briefly, the yeast cells were washed once with water and then resuspended in 1/10 of the original cultivation volume of 10 mM phosphate buffer, pH 7.4, with 1% (v/v) β-mercaptoethanol (βME). The cells were incubated with gentle shaking at 37°C for 30 min. The suspension was centrifuged (5 min, 5000 g), and the supernatant was filtered through a 0.22 μm filter, dialyzed against water for 48 h, and lyophilized.
Isolation of cell wall proteins
The isolation protocol was based on the method described by Yin et al.32 The yeast cells were harvested and washed with water. The cell pellet was then resuspended in an equal volume of water containing 0.2 mM Pefabloc, and cells were broken by French press according to the manufacturer's instructions. The suspension was centrifuged (5 min, 2000 g), and the supernatant was stored at −20°C. The sediment containing cell debris was extensively washed with 1M NaCl and resuspended in SDS/ME buffer (2% SDS, 0.1 M EDTA, 40 mM β-mercaptoethanol, 50 mM Tris-HCl, pH 7.8). To release the cell wall proteins, the suspension was heated at 100°C for 5 min and then centrifuged (5 min, 2000 g). The sediment was again resuspended in SDS/ME buffer, and the extraction was repeated. The supernatants were pooled, dialyzed against water, and lyophilized. The cell debris remaining after the extraction procedure was washed with water three times, resuspended in 30 mM NaOH, and incubated at 4°C overnight. After centrifugation, the supernatant was dialyzed against water and lyophilized. The sediment was washed with water and resuspended in spheroplasting buffer (0.8 M sorbitol, 50 mM monopotassium phosphate, 40 mM β-mercaptoethanol, pH 7.5) supplemented with Lyticase (1 mg/100 mg wet cell debris). The mixture was incubated for 2 h at 37°C, then centrifuged. The supernatant was dialyzed against water and lyophilized.
Biotinylation of the cell surface proteins
The biotinylation protocol was based on that of Casanova et al.24 Yeast was cultivated in induction medium for a minimum of 48 h. Cells were collected and washed once with water. The cells were resuspended in 1/5 of the original culture volume of 0.1 M sodium phosphate buffer, pH 8.5. Sulfo-NHS-biotin or sulfo-NHS-LC-biotin was added to the cell suspension to a final concentration of 2 mg/mL, and the mixture was incubated for 30 min with gentle shaking at ambient temperature. The incubation was stopped by addition of 0.1 M glycine. After 5 min, the cells were centrifuged (5 min, 5000g) and washed once with 10 mM phosphate buffer, pH 7.4.
Biotinylation of purified Sapp1p
Purified Sapp1p (4 mg/mL) was dialyzed against 0.1 M sodium phosphate buffer, pH 8.5. Stock solutions of sulfo-NHS-biotin (1 mg/mL) and sulfo-NHS-LC-biotin (10 mg/mL) were prepared in the same phosphate buffer. A dilution series was prepared (4×, 8×, 12×, 16×, and 32× molar excess of activated biotin to purified Sapp1p) and used for stepwise Sapp1p biotinylation. The stock solutions were used to fully biotinylate Sapp1p. Mixtures of Sapp1p and activated biotin were incubated for 30 min at ambient temperature. The reactions were stopped by addition of 0.1 M glycine. The solutions were dialyzed against water and lyophilized if needed.
Western blotting
Proteins separated by SDS-PAGE were transferred onto a nitrocellulose membrane. For Sapp1p detection, we used rabbit polyclonal antibodies raised against mature proteinase or against a unique sequence segment (residues 186–199 of Sapp1p). For Sapp2p detection, we used antibodies raised against a unique sequence (residues 389–404 of Sapp2p).15 The protein bands were visualized using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific).
N-terminal sequencing of proteins
Proteins separated by SDS-PAGE were transferred onto a poly(vinylidene difluoride) (PVDF) membrane using a semidry transfer protocol. The membrane was stained with BioSafe Coomassie (Bio-Rad). Bands of interest were cut out of the membrane, and the N-terminal protein sequence was determined by Edman degradation performed on an ABI Procise sequencer (Applied Biosystems).
Proteinase activity assay
The activity of secreted Sapp1p in cell-free culture supernatants was examined using a fluorogenic substrate (Dabcyl-Glu-His-Val-Lys-Leu-Val-Glu-EDANS) in 0.1 M sodium citrate buffer, pH 3.5, as previously described.14,15 Analysis of the cleavage products was performed using an HPLC system equipped with an Agilent C-18 column and a fluorescence detector set at an excitation wavelength of 350 nm and an emission wavelength of 480 nm. A linear methanol gradient was used for elution.
Controls containing pepstatin A, a specific inhibitor of aspartic proteinases, were prepared similarly. Pepstatin was dissolved in dimethylsulfoxide and added to reaction mixtures up to a final concentration of 1 μM.
Detection of cell wall-associated proteinase activity
The C. parapsilosis cells were harvested by centrifugation (4000 g), washed with water, and resuspended in 0.1 M sodium citrate buffer, pH 3.5, so that the final OD550 was ∼1.5. A 100 μL aliquot of this suspension was added to the reaction mixture, which contained 200 μL of 0.1 M sodium citrate buffer and 10 μL of stock solution of the fluorogenic substrate Dabcyl-Glu-His-Val-Lys-Leu-Val-Glu-EDANS. The stock solution contained 0.5 mg of the substrate per 1 mL of dimethylsulfoxide. The mixture was incubated for 30 min at 37°C. The reaction was stopped by addition of 60 μL 20% trifluoroacetic acid, and the mixture was filtered through a 0.22-μm filter. Analysis of cleavage products was performed using HPLC. To rule out the possibility that the substrate was cleaved by a proteinase that was fully secreted from the cells during incubation, the cells resuspended in sodium citrate buffer were incubated for 30 min at 37°C. Then, this suspension was filtered through a 0.22 μm filter, and the proteinase activity in the filtrate was tested as described above.
Mass spectrometry
Proteins were reduced with 25 mM DTT for 1 h at 37°C, and cysteine residues were alkylated using 25 mM iodoacetamide solution for 20 min at room temperature in the dark. Samples were digested with trypsin for 12 h at 37°C at an estimated protein : trypsin ratio of 50 : 1 or with chymotrypsin for 12 h at 25°C in 25 mM bicarbonate buffer at a protein : chymotrypsin ratio of 25 : 1. Peptides were extracted from in-gel digests using a gradient of acetonitrile in 1% TFA and 3 × 15 min of sonication on ice. Peptide mixtures were evaporated using a SpeedVac concentrator and dissolved in 0.1% formic acid. The resulting peptides were analyzed on an LTQ Orbitrap XL mass spectrometer (Thermo) coupled to a Rheos 2000 2D capillary chromatography platform (Flux Instruments). The first-dimension column was a monolithic PS-DVB (200 μm × 10 mm; Dionex), and the second dimension column was a PepMap C18 (75 μm × 150 mm × 3 μm, LC Packings). The mass data were processed with Seques and Bioworks software (Thermo). The precursor mass of peptides was determined in orbitrap operating in high-resolution mode (R = 100,000). CID in linear ion trap was used for fragmentation, and product spectra were collected in orbitrap (R = 15,000). Lysine biotinylation was searched as a standard variable modification, and five possible missed cleavages were used for data processing. For positive identification of (un)biotinylated lysine residues, we used a 3ppm mass tolerance of precursor ions and 5ppm tolerance of fragment ions. These parameters were used for all measurements of digested samples.
Acknowledgments
The authors thank Elena Dolejší for excellent technical assistance, Zdeněk Voburka for N-terminal protein sequencing, and Hillary Hoffman for language editing.
Glossary
Abbreviations
- sulfo-NHS-biotin
sulfosuccinimidyl biotin
- sulfo-NHS-LC-biotin
sulfosuccinimidyl 6-(biotinamido) hexanoate
References
- 1.Pfaller MA, Diekema DJ. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev. 2007;20:133–163. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pfaller MA, Diekema DJ, Gibbs DL, Newell VA, Ng KP, Colombo A, Finquelievich J, Barnes R, Wadula J, the Global Antifungal Surveillance Group Geographic and temporal trends in isolation and antifungal susceptibility of Candida parapsilosis: a global assessment from the ARTEMIS DISK antifungal surveillance program, 2001-2005. J Clin Microbiol. 2008;46:842–849. doi: 10.1128/JCM.02122-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levy I, Shalit I, Askenazi S, Klinger G, Sirota L, Linder N. Duration and outcome of persistent candidaemia in newborn infants. Mycoses. 2006;49:197–201. doi: 10.1111/j.1439-0507.2006.01231.x. [DOI] [PubMed] [Google Scholar]
- 4.Trofa D, Gacser A, Nosanchuk JD. Candida parapsilosis, an emerging fungal pathogen. Clin Microbiol Rev. 2008;21:606–625. doi: 10.1128/CMR.00013-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonassoli LA, Bertoli M, Svidzinski TIE. High frequency of Candida parapsilosis on the hands of healthy hosts. J Hosp Infect. 2005;59:159–162. doi: 10.1016/j.jhin.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 6.Heynes K. Virulence in Candida species. Trends Microbiol. 2001;9:591–596. doi: 10.1016/s0966-842x(01)02237-5. [DOI] [PubMed] [Google Scholar]
- 7.Naglik JR, Challacombe SJ, Hube B. Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol Mol Biol Rev. 2003;67:400–428. doi: 10.1128/MMBR.67.3.400-428.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hrušková-Heidingsfeldová Secreted proteins of Candida albicans. Front Biosci. 2008;13:7227–7242. doi: 10.2741/3224. [DOI] [PubMed] [Google Scholar]
- 9.de Viragh PA, Sanglard D, Togni G, Falchetto R, Monod M. Cloning and sequencing of two Candida parapsilosis genes encoding acid proteases. J Gen Microbiol. 1993;139:335–342. doi: 10.1099/00221287-139-2-335. [DOI] [PubMed] [Google Scholar]
- 10.Parra-Ortega B, Cruz-Torres H, Villa-Tanaca L, Hernandez-Rodriguez C. Phylogeny and evolution of the aspartyl protease family from clinically relevant Candida species. Mem Inst Oswaldo Cruz. 2009;104:505–512. doi: 10.1590/s0074-02762009000300018. [DOI] [PubMed] [Google Scholar]
- 11.Butler G, Rasmussen MD, Lin MF, Santos MAS, Sakthikumar S, Munro CA, Rheinbay E, Grabherr M, Forche A, Reedy JL, Agrafioti I, Arnaud MB, Bates S, Brown AJP, Brunke S, Costanzo MC, Fitzpatrick DA, de Groot PWJ, Harris D, Hoyer LL, Hube B, Klis FM, Kodira C, Lennard N, Logue ME, Martin R, Neiman AM, Nikolaou E, Quail MA, Quinn J, Santos MC, Schmitzberger FF, Sherlock G, Shah P, Silverstein KAT, Skrzypek MS, Soll D, Staggs R, Stansfield I, Stumpf MPH, Sudbery PE, Srikantha T, Zeng QD, Berman J, Berriman M, Heitman J, Gow NAR, Lorenz MC, Birren BW, Kellis M, Cuomo CA. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature. 2009;459:657–662. doi: 10.1038/nature08064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fusek M, Smith EA, Monod M, Foundling SI. Candida parapsilosis expresses and secretes two aspartic proteinases. FEBS Lett. 1993;327:108–112. doi: 10.1016/0014-5793(93)81050-a. [DOI] [PubMed] [Google Scholar]
- 13.Dostál J, Dlouhá H, Maloň P, Pichová I, Hrušková-Heidingsfeldová O. The precursor of secreted aspartic proteinase Sapp1p from Candida parapsilosis can be activated both autocatalytically and by a membrane-bound processing proteinase. Biol Chem. 2005;386:791–799. doi: 10.1515/BC.2005.093. [DOI] [PubMed] [Google Scholar]
- 14.Merkerová M, Dostál J, Hradilek M, Pichová I, Hrušková-Heidingsfeldová O. Cloning and characterization of Sapp2p, the second aspartic proteinase isoenzyme from Candida parapsilosis. FEMS Yeast Res. 2006;6:1018–1026. doi: 10.1111/j.1567-1364.2006.00142.x. [DOI] [PubMed] [Google Scholar]
- 15.Hrušková-Heidingsfeldová O, Dostál J, Majer F, Havliková J, Hradilek M, Pichová I. Two aspartic proteinases secreted by the pathogenic yeast Candida parapsilosis differ in expression pattern and catalytic properties. Biol Chem. 2009;390:259–268. doi: 10.1515/BC.2009.034. [DOI] [PubMed] [Google Scholar]
- 16.Togni G, Sanglard D, Quadroni M, Foundling SI, Monod M. Acid proteinase secreted by Candida tropicalis: functional analysis of preproregion cleavages in C. tropicalis and Saccharomyces cerevisiae. Microbiology. 1996;142:493–503. doi: 10.1099/13500872-142-3-493. [DOI] [PubMed] [Google Scholar]
- 17.Newport G, Agabian N. KEX2 influences Candida albicans proteinase secretion and hyphal formation. J Biol Chem. 1997;272:28954–28961. doi: 10.1074/jbc.272.46.28954. [DOI] [PubMed] [Google Scholar]
- 18.Chaffin WL. Candida albicans cell wall proteins. Microbiol Mol Biol Rev. 2008;72:495–544. doi: 10.1128/MMBR.00032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruiz-Herrera J, Elorza VM, Valentin E, Sentandreu R. Molecular organisation of the cell wall of Candida albicans and its relation to pathogenicity. FEMS Yeast Res. 2006;6:14–29. doi: 10.1111/j.1567-1364.2005.00017.x. [DOI] [PubMed] [Google Scholar]
- 20.Klis FM, Sosinska GJ, de Groot PWJ, Brul S. Covalently linked cell wall proteins of Candida albicans and their role in fitness and virulence. FEMS Yeast Res. 2009;9:1013–1028. doi: 10.1111/j.1567-1364.2009.00541.x. [DOI] [PubMed] [Google Scholar]
- 21.Mrša V, Seidl T, Gentzsch M, Tanner W. Specific labelling of cell wall proteins by biotinylation. Identification of four covalently linked O-mannosylated proteins of Saccharomyces cerevisiae. Yeast. 1997;13:1145–1154. doi: 10.1002/(SICI)1097-0061(19970930)13:12<1145::AID-YEA163>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 22.Cappellaro C, Mrsa V, Tanner W. New potential cell wall glucanases of Saccharomyces cerevisiae and their involvement in mating. J Bacteriol. 1998;180:5030–5037. doi: 10.1128/jb.180.19.5030-5037.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pichová I, Pavlíčková L, Dostál J, Dolejší E, Hrušková-Heidingsfeldová O, Weber J, Ruml T, Souček M. Secreted aspartic proteases of Candida albicans, Candida tropicalis, Candida parapsilosis, Candida lusitaniae: inhibition with peptidomimetic inhibitors. Eur J Biochem. 2001;268:2669–2677. doi: 10.1046/j.1432-1327.2001.02152.x. [DOI] [PubMed] [Google Scholar]
- 24.Casanova M, Lopez-Ribot JL, Martinez JP, Sentandreu R. Characterization of cell wall proteins from yeast and mycelilal cells of Candida albicans by labelling with biotcomparison with other techniques. Infect Immun. 1992;60:4898–4906. doi: 10.1128/iai.60.11.4898-4906.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gozalbo D, Gil-Navarro I, Azorín I, Renau-Piqueras J, Martínez JP, Gil ML. The cell wall-associated glyceraldehyde-3-phosphate dehydrogenase of Candida albicans is also a fibronectin and laminin binding protein. Infect Immun. 1998;66:2052–2059. doi: 10.1128/iai.66.5.2052-2059.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dostál J, Brynda J, Hrušková-Heidingsfeldová O, Sieglová I, Pichová I, Řezáčová P. The crystal structure of the secreted aspartic protease 1 from Candida parapsilosis in complex with pepstatin A. J Struct Biol. 2009;167:145–152. doi: 10.1016/j.jsb.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 27.Schaller M, Bein M, Korting HC, Baur S, Hamm G, Monod M, Beinhauer S, Hube B. The secreted aspartyl proteinases Sap1 and Sap2 cause tissue damage in an in vitro model of vaginal candidiasis based on reconstituted human vaginal epithelium. Infect Immun. 2003;71:3227–3234. doi: 10.1128/IAI.71.6.3227-3234.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stringaro A, Crateri P, Pellegrini G, Arancia G, Cassone A, de Bernardis F. Ultrastructural localization of the secretory aspartyl proteinase in Candida albicans cell wall in vitro and in experimentally infected rat vagina. Mycopathologia. 1997;137:95–105. doi: 10.1023/a:1006897208863. [DOI] [PubMed] [Google Scholar]
- 29.Albrecht A, Felk A, Pichova I, Naglik JR, Schaller M, de Groot P, Maccallum D, Odds FC, Schafer W, Klis F, Monod M, Hube B. Glycosylphosphatidylinositol-anchored proteases of Candida albicans target proteins necessary for both cellular processes and host-pathogen interactions. J Biol Chem. 2006;281:688–694. doi: 10.1074/jbc.M509297200. [DOI] [PubMed] [Google Scholar]
- 30.Schild L, Heyken A, de Groot PW, Hiller E, Mock M, de Koster C, Horn U, Rupp S, Hube B. Proteolytic cleavage of covalently linked cell wall proteins by Candida albicans Sap9 and Sap10. Eukaryot Cell. 2011;10:98–109. doi: 10.1128/EC.00210-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaiser CA, Chen EJ, Losko S. Subcellular fractionation of secretory organelles. Methods Enzymol. 2002;351:325–338. doi: 10.1016/s0076-6879(02)51855-3. [DOI] [PubMed] [Google Scholar]
- 32.Yin QY, de Groot PWJ, Dekker HL, de Jong L, Klis FM, de Koster CG. Comprehensive proteomic analysis of Saccharomyces cerevisiae cell walls. J Biol Chem. 2005;280:20894–20901. doi: 10.1074/jbc.M500334200. [DOI] [PubMed] [Google Scholar]