

Abstract
The peptide backbones of disordered proteins are routinely characterized by NMR with respect to transient structure and dynamics. Little experimental information is, however, available about the side chain conformations and how structure in the backbone affects the side chains. Methyl chemical shifts can in principle report the conformations of aliphatic side chains in disordered proteins and in order to examine this two model systems were chosen: the acid denatured state of acyl-CoA binding protein (ACBP) and the intrinsically disordered activation domain of the activator for thyroid hormone and retinoid receptors (ACTR). We find that small differences in the methyl carbon chemical shifts due to the γ-gauche effect may provide information about the side chain rotamer distributions. However, the effects of neighboring residues on the methyl group chemical shifts obscure the direct observation of γ-gauche effect. To overcome this, we reference the chemical shifts to those in a more disordered state resulting in residue specific random coil chemical shifts. The 13C secondary chemical shifts of the methyl groups of valine, leucine, and isoleucine show sequence specific effects, which allow a quantitative analysis of the ensemble of χ2-angles of especially leucine residues in disordered proteins. The changes in the rotamer distributions upon denaturation correlate to the changes upon helix induction by the co-solvent trifluoroethanol, suggesting that the side chain conformers are directly or indirectly related to formation of transient α-helices.
Keywords: aliphatic side chains, natively unfolded proteins, intrinsically disordered proteins, conformational ensemble, denatured state, hydrophobic packing
Introduction
The conformational properties of disordered proteins are studied for two reasons: first, intrinsically disordered proteins (IDPs) are abundant and perform many important functions in nature.1 Second, the denatured state of a folded protein may provide insight into the initial steps in protein folding.2 Due to their heterogeneity, these two kinds of disordered proteins pose similar challenges to the traditional methods of structural biology. NMR spectroscopy is a powerful technique for characterization of transient structure in disordered proteins. By NMR spectroscopy the conformational ensembles can be probed in detail by chemical shifts,3 paramagnetic relaxation enhancement,4,5 and residual dipolar couplings.6,7 Even though these methods provide a detailed picture of the peptide backbone,7–11 comparatively little experimental information is available about the side chains of disordered proteins.12
In disordered proteins, the NMR signals of the side chains are less dispersed than those from the backbone.13,14 The lack of chemical shift dispersion shows that the side chains of a given residue type experience a similar time-averaged environment. The side chain conformations can be determined experimentally using coupling constants that allow determination of the populations of each of the three staggered χ1-angles.15 Discrepancies between experimentally determined coupling constants from disordered proteins and those predicted from coil libraries compiled from loop regions of structured proteins suggest the formation of transient hydrophobic clusters.12,14,16 The χ1-angle distributions of disordered proteins can also be probed using residual dipolar couplings.12 This method shows only small differences in the side chain conformations within a side chain type and these variations are thus difficult to interpret. The interpretation of small differences in the rotamer distributions is complicated by the effects of neighboring residues, which may be larger than the effects of transient structures.
The most precisely measured NMR parameter is the chemical shift. In side chains, the 13C chemical shifts depend on χ-angles of the side chains17 at least partly due to the γ-gauche effect. The γ-gauche effect is an upfield shift of the 13C resonances caused by heavy atom γ-substituents in a gauche conformation.18,19 Accordingly, a 13C atom that is gauche to a single carbon γ-substituent will have a chemical shift that is approximately 5 ppm lower than an equivalent atom in a trans conformation. The γ-substituents vary between the different kinds of methyl groups in proteins, and their conformational dependence thus needs to be considered separately for each kind of methyl group. The 13C chemical shifts of the two methyl groups of leucine have a particularly simple conformational dependence as the two Cδ atoms have a single γ-substituent: the Cα. The contributions from the γ-gauche effect can be isolated by subtracting the chemical shifts of the two geminal methyl groups, and this chemical shift difference thus measures the conformation of the side chain more precisely.17,19 Due to steric repulsion, leucine side chains primarily populate the two staggered χ2-angles that position one methyl group gauche and one methyl group trans relative to Cα. As only two rotamers are significantly populated, their populations can be estimated from a linear dependence of the 13C chemical shifts.19 The chemical shift of the isoleucine Cδ obeys a similar conformational dependence as that of leucine and the χ2 distribution can similarly be estimated as a linear combination of the two most populated χ2-angles.20 The 13C chemical shifts of γ-methyl groups depend on the χ1-angle, however, since all three rotamers are usually populated it is not possible to extract the populations from the chemical shifts alone. The methyl groups of alanine, threonine, and methionine will not be considered in the following as the 13Cβ of alanine is primarily sensitive to the secondary structure of the backbone, the 13Cγ of threonine is perturbed by co-solvents12 and the 13Cɛ of methionine is extremely flexible.
For proton chemical shifts, the conformational dependence of the methyl signals is less clear than that of the 13C chemical shifts.21 In folded proteins, the dominant source of proton chemical shift dispersion for methyl groups is the ring current effect caused by aromatic rings,22 as the ring current effect of a nearby aromatic group can cause an upfield shift of more than 1 ppm.23 In protein folding studies, the disappearance of upfield shifted methyl proton signals have been used to demonstrate dynamics in the hydrophobic core of partially folded states such as molten globules.24 When the source of the ring current can be identified, the upfield shift can thus be used to probe hydrophobic interactions. Recently, a specific ring current effect on methyl groups was used to demonstrate specific hydrophobic packing in a native molten globule.25
In the following, the methyl chemical shifts of two disordered systems with transient structure have been analyzed: The intrinsically disordered activation domain of the activator for thyroid hormone and retinoid receptors (ACTR) and the acid denatured state of bovine acyl-CoA binding protein (ACBP). The activation domain of ACTR binds to the nuclear coactivator domain of CREB binding protein and forms three α-helices in the complex.26 In the free state, the activation domain of ACTR is disordered,27 but it has transient α-helices especially in the region that corresponds to the first helix formed in the complex.28,29 ACBP is a four helix bundle30 that has been investigated as a model system for protein folding.31,32 At low pH, ACBP forms a disordered state that has transient, native-like α-helices that correspond to the four helices in the folded structure.33,34 The acid denatured state of ACBP forms transient long-range native-like interactions35,36 that can be perturbed by single-site mutations suggesting a set of specific hydrophobic and transient interactions between the helices in the denatured ensemble.37
Transient secondary and tertiary structure is common in disordered proteins as is amply demonstrated by numerous NMR studies.5,35,38–40 Transient α-helices are often amphipatic, suggesting that an important factor in secondary structure formation in disordered proteins is burial of hydrophobic side chains. Similarly, long range tertiary interactions in disordered proteins can be disrupted by removal of hydrophobic side chains,37 implying that these interactions are in part driven by hydrophobic interactions as well. Even though hydrophobic side chains are important determinants of residual structure in disordered proteins, little is known about whether formation of secondary and tertiary interactions affect the conformations of the side chains. In the following, we explore the usefulness of methyl chemical shifts for probing the conformations of aliphatic side chains in disordered proteins. We use methyl chemical shifts to show that formation of transient helical structure leads to changes in the rotamer ensembles of hydrophobic side chains an order of magnitude smaller than the populations of the transient α-helices.
Results
Assignment of the methyl signals
The methyl signals in the 1H-13C HSQC are found in groups according to their amino acid types in both ACBP and ACTR (Fig. 1). As expected, the clustering shows that the hydrophobic side chains have ensemble distributions close to those of a statistical coil. The methyl group resonances can be unambiguously assigned by correlation to the backbone amide resonances of the following residue using H(CCO)NH and (H)C(CO)NH spectra41 (Supporting Information Fig. 1). A combination of these two experiments allowed the unambiguous assignment of all methyl groups from leucine, isoleucine, and valine residues in ACBP and ACTR in the presence and absence of 6M urea (Supporting Information Tables 1 and 2). The chemical shifts are site-specific suggesting that the individual methyl groups experience slightly different average environments. When urea is added to the sample, the site-specific chemical shift dispersion remains approximately the same, even though the chemical shifts are perturbed for several resonances. The difference in urea sensitivity suggests that the site-specific dispersion originates from the combined effects from nearest neighbors and residual structure.
Figure 1.
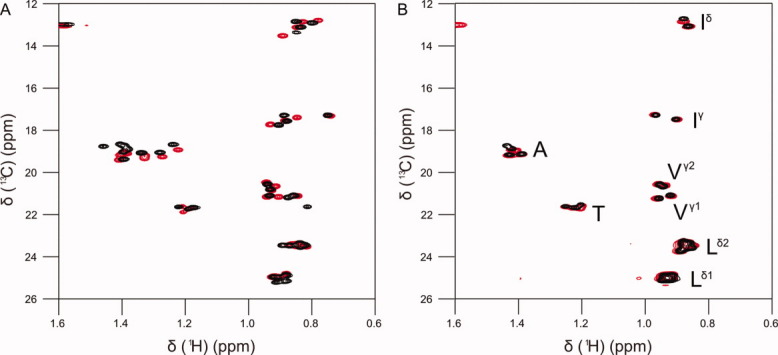
Constant time 1H-13C HSQC spectra of the methyl regions of acid denatured ACBP (A) and ACTR (B). The experiment was run without urea (black) and in the presence of 6M urea (red).
Precision of chemical shift measurements
As the chemical shift dispersion in disordered proteins is small, it is worthwhile to consider the precision of the measurements beforehand. The precision of chemical shift measurements depend on the signal-to-noise ratio and the line width42 and can be estimated to lw/2SN.43 For 13C chemical shifts, the precision of the chemical shift measurements depend on whether the peaks are resolved in the constant time HSQC spectrum as is indicated in the Supporting Information Tables 1 and 2 for each resonance. The line widths in the constant time HSQC are on average 15 Hz (12–18 Hz), while the line widths in C(CO)NH experiment are approximately 72 Hz (64–79 Hz). With a signal-to-noise ratio of approximately 1:50, this translates into measurement uncertainties of 0.8 ppb and 3.6 ppb. For the 1H chemical shifts, the line widths are similar in the H(CCO)NH experiment and the HSQC and have measurement uncertainties of approximately 0.5 ppb. In practice, however, the error associated with the measurements may be larger due to slight phase distortions or overlap, so we will not consider secondary chemical shifts below 10 ppb for 13C and 1 ppb for 1H to be significant. For leucines residues, this translates into an uncertainty of the χ2-distribution of 0.2% from the chemical shift measurement alone.19
13C methyl chemical shifts probe the individual side chain conformations in disordered proteins
In the two model proteins, the chemical shifts of the geminal methyl groups in the leucine residues correlate as shown in Figure 2. For the protons, this correlation suggests that the chemical environment affects the protons of the two methyl groups similarly. Correlated chemical shift changes could, for example, be caused by electrostatics or ring currents from nearby aromatic groups. In contrast, the 13C chemical shifts of the geminal methyl groups are weakly anticorrelated [Fig. 2(B)] more for ACTR than for ACBP (R = −0.74 vs. R = −0.54). This difference most likely reflect the absence of aromatic residues in ACTR and hence the absence of correlated chemical shift perturbations from ring currents. The anticorrelation is not significantly perturbed by denaturation with urea (both proteins with urea: R = −0.7, without urea R = −0.66), suggesting that it is a result of factors intrinsic to the amino acid sequence rather than transient secondary or tertiary interactions. Anticorrelated changes in the 13C chemical shifts is most likely due to the γ-gauche effect and changes in the rotamer distribution, as the two dominant χ2-rotamers of leucine side chains have one methyl group gauche and one methyl group trans relative to Cα.19 Redistribution between these two rotamers will thus lead to an increase in the γ-gauche effect for one methyl group and a decrease for the neighboring methyl group. The anticorrelation of the 13C chemical shifts of geminal methyl groups suggests that small differences between the side chain rotamer ensembles contribute to the dispersion of methyl chemical shifts in disordered proteins. This implies that the 13C methyl chemical shifts may be used to probe the individual side chain conformations in disordered proteins.
Figure 2.
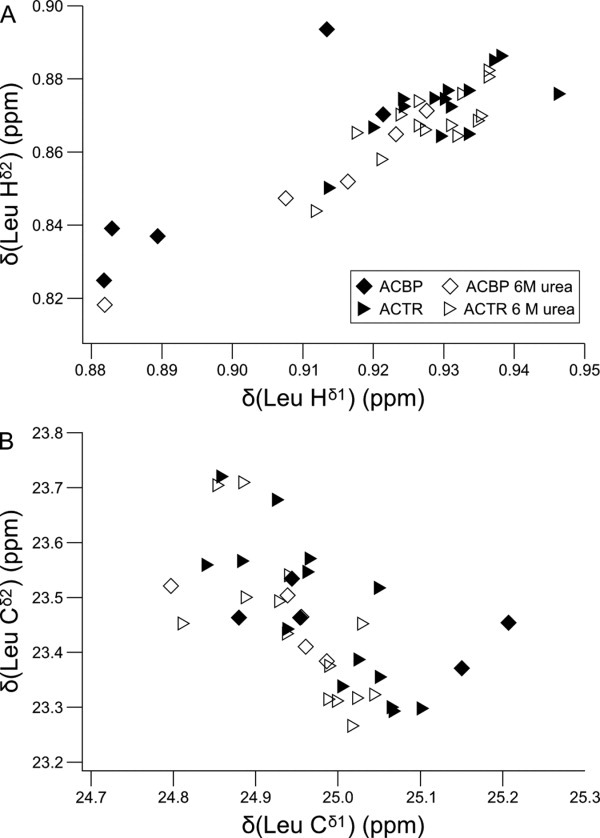
Correlation between chemical shifts from the geminal methyl groups of Leu side chains of ACBP and ACTR with and without urea. The 1Hδ chemical shifts are correlated (R = 0.85) (A) and the 13Cδ chemical shifts (B) are anticorrelated (R = −0.62). The anticorrelation of the 13C chemical shifts is better for ACTR alone (R = −0.74). The anticorrelation of the 13C chemical shifts suggests that variations in the χ2-distributions affect the methyl chemical shifts.
Trifluoroethanol titration of ACTR
To test whether methyl chemical shifts are affected by helical structure stabilization in the backbone, ACTR was titrated with trifluoroethanol (TFE). TFE stabilizes helices in disordered proteins through a mechanism that is not fully understood,44 and is accordingly expected to increase the populations of the transient helices in ACTR. The helical populations were monitored using the backbone chemical shifts, Cα and C′, that are most sensitive to the transient secondary structure. Upon addition of TFE, the 13C backbone chemical shifts only increase in the regions that form transient helices in the disordered state [Fig. 3(A,B)].28,29 TFE thus induces helicity specifically in regions with an intrinsic propensity for helix formation. Simultaneously, the side chain populations of the individual leucines were monitored using 13C methyl chemical shifts, which allow determination of the population of trans rotamers based on the difference in 13C chemical shifts between the geminal methyl groups.19 The methyl chemical shifts suggest that the side chains ensembles change with increasing TFE concentration [Fig. 3(C)]. The chemical shift changes may be both positive and negative depending on which χ2-rotamer is more stable in the helical conformation. The methyl chemical shift changes are largest in the regions where helical structure is induced in the backbone [Fig. 3(D)], suggesting that the side chain conformational distributions are connected to transient secondary structure in the backbone.
Figure 3.
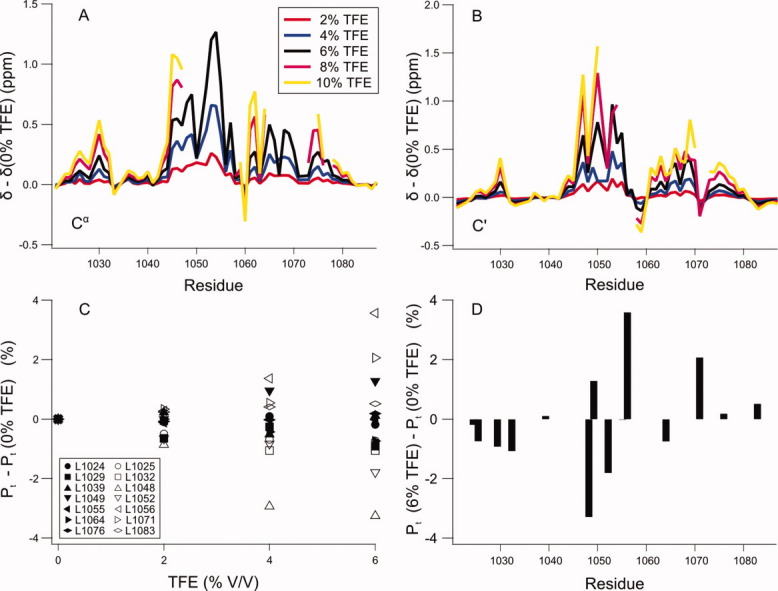
TFE titration of ACTR suggests interplay between backbone and side chain conformations. ACTR was titrated with the helix inducing co-solvent TFE, which results in increasing populations of the transient helices as demonstrated by Cα (A) and C′ (B) chemical shifts. The side chain distributions in the disordered state changes approximately linearly with increasing TFE concentration (C). The largest changes are roughly in the regions where transient helices are formed (D) suggesting that there is an interplay between the backbone and the side chain conformations. The populations of the trans rotamer Pt is calculated based on the difference in 13C chemical shifts between the two geminal methyl groups.19 At the highest TFE concentrations, the chemical shifts in the helical segments are missing due to exchange broadening, suggesting formation of relatively long-lived interactions stabilized by TFE.
Secondary chemical shifts for methyl groups
Secondary chemical shifts are ubiquitously used to probe the backbone conformations of disordered proteins. Here, we examine how side chain secondary chemical shifts may be useful for defining the side chain conformations. The secondary chemical shift is calculated by subtracting a statistical coil chemical shift from the experimentally determined chemical shift. The statistical coil values are typically determined in small unstructured peptides. In this study, we have measured the methyl chemical shifts of two peptide series with the sequences Ac-GGXGG-NH2 and Ac-QQXQQ-NH2 (Table I), which have both recently been used to determine the backbone statistical coil chemical shifts.45,46 The Ac-GGXGG-NH2 peptide series is similar to most of the random coil peptide series in the literature and is commonly used for backbone chemical shift analysis.47,48 It has recently been proposed that the Ac-QQXQQ-NH2 peptides have more representative conformational distributions than the glycine containing peptides.46 The methyl chemical shifts of these two peptide series are similar except for the 13C chemical shifts of Val and Ile where differences of 0.3–0.4 ppm are seen. These differences suggest neighbor dependent changes in the conformational distributions of the side chains.
Table I.
Random Coil Chemical Shift of Methyl Groups in Two Peptide Series
| Ac-GGXGG-NH2 | Ac-QQXQQ-NH2 | ||||
|---|---|---|---|---|---|
| Atom | 1H | 13C | 1H | 13C | |
| Ala | β | 1.421 | 19.16 | 1.406 | 19.05 |
| Ile | γ | 0.939 | 17.46 | 0.908 | 17.38 |
| δ | 0.888 | 13.04 | 0.873 | 12.71 | |
| Leu | δ1 | 0.944 | 24.95 | 0.945 | 24.86 |
| δ2 | 0.892 | 23.30 | 0.884 | 23.35 | |
| Met | ɛ | 2.118 | 16.92 | 2.106 | 16.91 |
| Thr | γ | 1.226 | 21.52 | 1.222 | 21.66 |
| Val | γ 1 | 0.963 | 21.06 | 0.938 | 21.07 |
| γ 2 | 0.960 | 20.28 | 0.962 | 20.69 | |
The statistical coil chemical shifts in Table I were used to calculate secondary chemical shifts of the methyl groups for all leucine, isoleucine, and valine residues of ACBP and ACTR (Supporting Information Figs. 2 and 3). These secondary chemical shifts have large variations, but neither of the peptide random coil datasets result in secondary chemical shifts that correlate to regions of known structure in the backbone. This could be due to the effects of sequential neighbors that are known to have a large effect on the statistical coil chemical shifts of backbone nuclei.48 If the neighboring residues affect the side chain chemical shifts as well, this would obscure the detection of transient secondary or tertiary structure. To examine this effect a model system study was performed.
The effects of neighboring residues on the statistical coil chemical shifts
To investigate the effect of neighboring residues on the chemical shifts, we examined a series of peptides containing an isoleucine preceded or succeeded by either a small hydrophilic residue (S), a negatively charged residue (D), a positively charged residue (R), an aliphatic residues (L), or one of the three aromatic residues (F, Y, W). Isoleucine was chosen as the invariant residue as it contains both a γ- and a δ-methyl group. For proton chemical shifts, the neighbor effect is small for all non-aromatic residues [Fig. 4(A)]. The aromatic residues, however, have a significant impact on the proton chemical shifts of the methyl groups in the neighboring residues. This is in accordance with the neighbor effects observed for backbone nuclei48 and is caused by a ring current from the aromatic side chain. The methyl group neighbor effects of the aromatic side chains are of the same magnitude as the upfield shifts seen for the methyl groups in acid denatured ACBP (Fig. 1). For the 13C chemical shift, the neighbor effect is more complicated. The individual neighbor effects are of approximately the same magnitude as the chemical shift dispersion observed in the two disordered proteins (Fig. 1). If this neighbor effect is not taken into account, it will dominate the side chain secondary chemical shifts.
Figure 4.
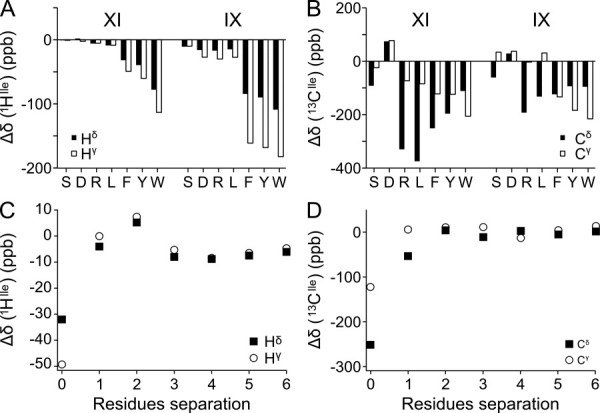
Methyl chemical shift perturbations from sequential neighbors. 1H (A) and 13C (B) methyl secondary chemical shifts of Ile were obtained for a series of Ac-GGXIGG-NH2 and Ac-GGIXGG-NH2 peptides. The X residue is indicated on the X-axis. 1H (C) and 13C (D) methyl secondary chemical shifts were determined in the peptide series Ac-GGFGnIGG-NH2 for separations up to 6 residues. The secondary chemical shifts were calculated using the chemical shifts from the peptide Ac-GGIGG-NH2 in 1M urea as random coil reference.
For protons, the neighbor effect was generally larger for the γ-methyl than the δ-methyl, whereas for 13C the effect was largest for the δ-methyl group. The two types of nuclei report on different aspects of the local structure as the protons primarily report on the magnitude of the ring current, where the carbons report on the χ1/χ2 distribution. The γ-methyl is only affected by χ1 averaging and is thus less dynamic than the δ-methyl, which is affected by both χ1 and χ2 averaging. The rotational freedom of the side chain is consistent with the buildup of a larger ring current effect on the less flexible γ-methyl, and a larger perturbation of the side chain rotamer distribution for the more flexible δ-methyl. To compare the effect of the preceding and the following residue, the isoleucine was placed both before and after the variable residue. For 1H resonances, the upfield shift was larger when the aromatics were placed after the isoleucine, suggesting that an aromatic side chain exerts a larger ring current effect on the preceding residue. For 13C, the neighbor effect varies considerably dependent on the residue type. There is not, however, an immediately apparent connection between the structure of the neighbor and the size of the neighbor effect. Determination of neighbor correction factors like those reported for the backbone nuclei48 would require a large peptide library and is not practical, which suggests that another method has to be adopted to correct for the neighbor effects.
To probe whether more distant residues affect the methyl chemical shifts, we designed a peptide series containing an isoleucine and a phenylalanine separated by a flexible linker consisting of up to six glycine residues. The ring current from the aromatic group allows detection of even a slight interaction. Figure 4 reveals that the perturbation of the chemical shifts of the methyl groups is small when the two hydrophobic residues are separated by one or more glycines. Unless medium or long range interactions bring residues into close contact, effects beyond the nearest neighbor can thus safely be ignored. A small, but significant, distance-dependent change of secondary chemical shift can be seen for protons even at larger separations. The secondary chemical shifts have a small, positive maximum at two intervening residues, suggesting that at this separation the isoleucine methyl groups are more likely to be in the plane of the aromatic ring than above or below the ring. At longer separations, the secondary chemical shift decreases as the length of the intervening linker is increased as expected for an unspecific hydrophobic interaction. The small magnitude, however, suggests that the two hydrophobic residues only interact in a small fraction of the ensemble, implying that they are unable to bury sufficient hydrophobic surface area to compensate for the loss of conformational entropy associated with the formation of a medium range hydrophobic interaction.
Urea titration of ACTR
In backbone secondary chemical shift analyses, the neighbor effects can be removed by using a highly disordered state of the same protein as a reference, which gives clean secondary chemical shifts.29,34 Many studies of chemically disordered proteins have demonstrated that even under highly denaturing conditions transient structure can still be observed.6,38,49–51 Therefore, it is necessary to study the urea dependence of the secondary structure propensity. A urea titration series was recorded for ACTR from 0 to 6M urea, where the transient helicity was monitored by C′ and Cα chemical shifts. Figure 5 shows the secondary chemical shifts of residues from the helix 1 as a function of urea concentration. The secondary chemical shifts decrease most rapidly at low urea concentrations, but do not reach zero even at 6M urea. This is consistent with the numerous studies demonstrating transient helicity in chemically denatured states. At high urea concentrations, the chemical shift change levels off but is not constant similar to what have been observed for ACBP previously.34 To be consistent with previous studies, we will use a urea concentration of 6M in the following.29,34 The protein is not fully disordered in this state, but since intrinsic random coil referencing compares relative structural propensities it only requires that the protein is significantly more disordered than the native state.
Figure 5.
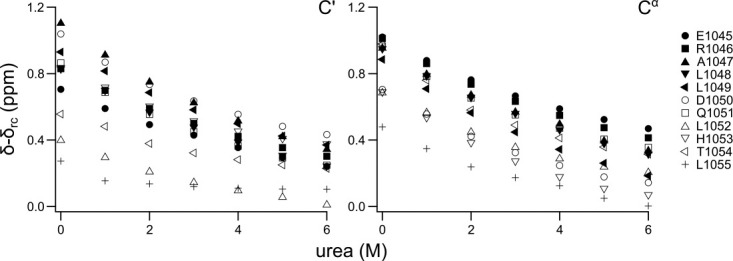
Urea titration of ACTR. The backbone chemical shifts of ACTR are observed with increasing urea concentration for the first transiently formed helix (residues 1045–1055). The helical population decreases rapidly at low concentrations of urea, but levels of at higher concentration. The secondary chemical shifts do not reach zero, which suggests a small helical population in the urea denatured state consistent with other studies of denatured proteins. Random coil chemical shifts are based on work from Ref. 46.
Secondary chemical shift analysis by intrinsic statistical coil referencing
We used the urea unfolded state as the intrinsic statistical coil chemical shifts for the methyl chemical shifts of ACBP and ACTR (Fig. 6) to obtain the sequence specific random coil chemical shifts of the residue types of valine, leucine, and isoleucine. The magnitudes of the secondary chemical shifts are significantly smaller when referenced to the urea denatured state compared to those referenced to peptide derived random coil chemical shifts (Supporting Information Figs. 2 and 3) consistent with the removal of contributions from sequential neighbors.
Figure 6.

Methyl secondary chemical shifts were determined by intrinsic statistical coil referencing for ACBP (A,C) and ACTR (B,D). The chemical shifts determined for the highly disordered state in 6M urea are used as the intrinsic statistical coil chemical shifts and are subtracted from the chemical shifts of the partially folded states to give secondary chemical shifts. V77 has a particularly large 1H secondary chemical shift, which suggests a ring current effect. The insert shows the conformations of V77 and Y73 the NMR structure of ACBP (PDB:1NTI). The cylinders show the locations of the α-helices in the folded state of ACBP30 and the complex between ACTR and CBP.26 The shading of the cylinders represents the approximate populations of the helices in the free state with fully formed helices being solid black.
For ACBP, the valine and leucine residue 13C methyl group secondary chemical shifts range from −50 to 450 ppb and the separation between the shifts of the intraresidue methyl groups between −100 and up to 250 ppb. For the isoleucine γ2 and δ methyl groups, secondary chemical shifts between −200 and 100 ppb were observed. Most of these residues are located in the segments of the protein that form the four helices in native ACBP. Having eliminated the contribution from neighboring residues by using intrinsic statistical chemical shift referencing, the contributions from the γ-gauche effect is more readily available, in particular, for leucines. These relatively large secondary chemical shifts may be due to be long- or short-range interactions favored by electrostatic or hydrophobic interactions in association with ring current shift perturbations. In the urea referenced secondary chemical shifts, a large effect is seen for all nuclei of V77 in ACBP. The magnitude of the proton secondary chemical shifts and the correlation between the two 13C chemical shifts suggest that the large secondary chemical shifts are due to a ring current effect and not just due to a change in the side chain rotamer distribution. V77 does not have any aromatic neighbors, but Y73 is one turn away in a transient helix. In the folded state of ACBP, these side chains interact and a large ring current effect would be predicted for V77 [Fig. 6(C) insert]. In this case, the secondary chemical shifts report on a medium range, native-like interaction between hydrophobic side chains that forms transiently in the disordered state. For ACTR, the intrinsic statistical chemical shift referencing of the valine, leucine, and isoleucine secondary 13C methyl group shifts result in a considerably smaller chemical shift range between −100 and 100 ppb, and the largest secondary shifts for the segment of residues between 1048 and 1056 coincide with the first helix in the folded form of the protein. Most of the secondary shifts from residues in other parts are very small.
Extracting rotamer population from 13C methyl secondary chemical shifts
For isoleucine and leucine, the populations of χ2-angles can be estimated from the 13C chemical shifts.19,20 Figure 7 shows the difference in the population of the trans rotamer between the partially folded state and the highly disordered state in urea. For ACTR, the population differences are close to zero. A notable exception is the region from residue 1048 to 1056 where perturbations of up to ±1% are seen. This region corresponds to the first helix in the complex between ACTR and CBP,26 which forms transiently in the unbound state.28,29 When the effects of sequence are removed, the 13C secondary chemical shifts show that transient helix formation is accompanied by a small but significant change in the side chain rotamer distribution. Figure 6 suggests that I1073 of ACTR experience a larger change of rotamer population than the helical region. While leucine side chain populations can be estimated by the difference between two chemical shifts, the isoleucine rotamer population is based on just one chemical shift. Apart from the γ-gauche effect, factors affecting the chemical shifts are likely to be similar for the two geminal methyl groups as illustrated by the correlation between the proton chemical shifts of geminal methyl groups. By subtracting the two methyl chemical shifts in the leucine side chain, other contributions to the chemical shifts are removed. This is not possible for isoleucine and therefore the estimation of the χ2-distribution is likely to be less precise for isoleucine than for leucine. It is thus not certain whether the large secondary chemical shift of I1073 represents changes in the χ2-distribution or small chemical shift perturbations from other factors. The same consideration applies to the isoleucine side chains of ACBP. The only completely unfolded region in acid denatured ACBP is the loop between helices 2 and 3. Consistent with the low content of secondary structure, the loop region has χ2 population perturbations that are close to zero. The residues in the helix segments have a shift in their χ2 populations up to 3% compared to the urea unfolded form, which is significantly larger than the population shifts observed in ACTR.
Figure 7.
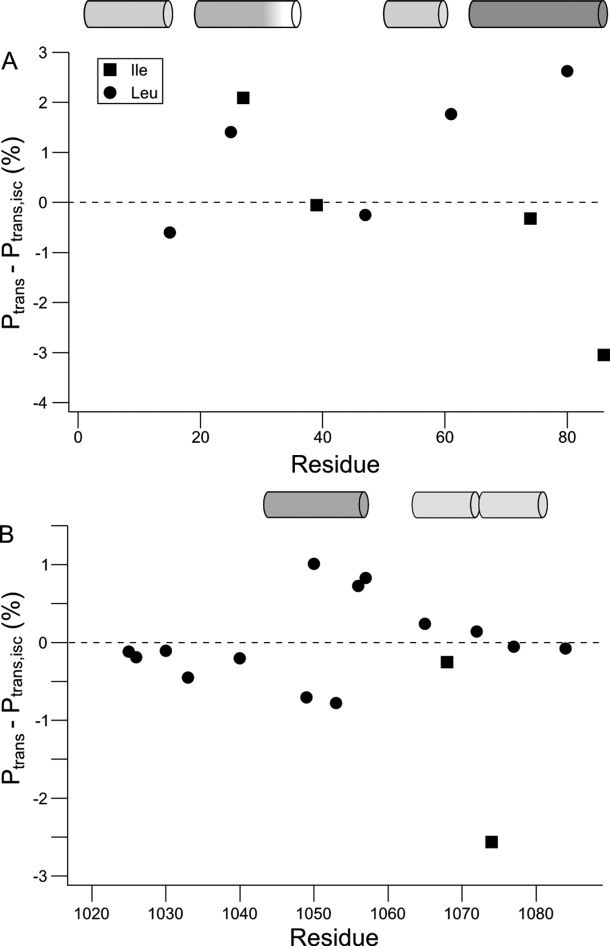
Perturbations of the χ2-rotamer distributions between the partially folded states and the highly disordered state in urea for ACBP (A) and ACTR (B). The population of the trans rotamer of the χ2-angle was estimated from the difference between δ(Cδ1) and δ(Cδ2) for leucine or from δ(Cδ) for isoleucine using the linear relationships reported previously.19,20
If the deviations from the intrinsic statistical coil rotamer distribution observed under native conditions are due to the transient α-helices, then we would expect to see opposite changes under helix stabilizing conditions and denaturing conditions, that is, TFE and urea, respectively. In Figure 8, the change in the population of trans rotamers between TFE and buffer is correlated to the change between buffer and urea. The changes in the rotamer distributions are correlated with a correlation coefficient of 0.76, which suggest that it is the same kind of structural change occurring.
Figure 8.
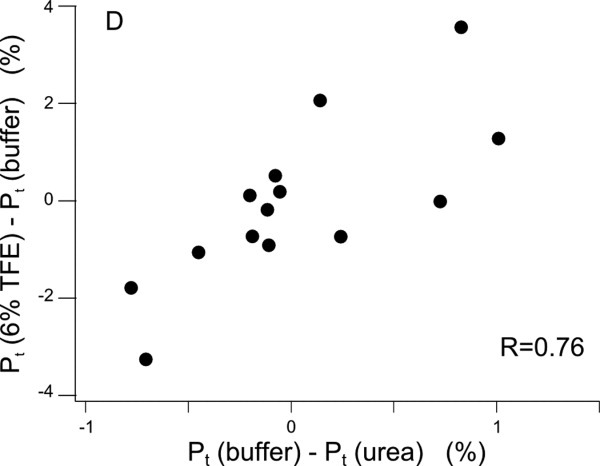
Correlation between changes in the rotamer distribution in solvent conditions stabilizing (TFE) and destabilizing α-helices (urea) suggests that the subtle rearrangements in the side chain rotamer distributions are mainly caused by helix formation
Discussion
We have shown that when the effect of neighbors is removed, small secondary chemical shifts can be measured for methyl groups in disordered proteins. The 13C chemical shifts report primarily on the rotamer ensemble distribution, while the 1H chemical shifts report on a mixture of other structural factors. This prompts the question whether these secondary chemical shifts probe local structural preferences or long-range interactions. In both of the two model proteins used in this study, the changes in the rotamer distribution where largest in the regions containing transient helices, and furthermore stabilization of the helices in ACTR leads to changes in the rotamer distributions. These observations suggest that formation of an α-helix leads to changes in the rotamer distribution of the side chains. These changes are, however, relatively subtle as the population changes are an order of magnitude smaller than the populations of the helices. In ACBP, the secondary chemical shifts are much larger than in ACTR, and accordingly the perturbations of the rotamer ensembles are up to threefold larger. This difference is larger than what would have been expected from the populations in the transient helices in these two proteins. The individual helix segments in acid unfolded ACBP have pairwise interactions that may contribute to the secondary chemical shifts observed35,37 explaining the larger magnitude of the secondary chemical shifts relative to ACTR. Furthermore, ACBP has several aromatic residues that causes ring current effects. In the fourth helix segment in ACBP, the interaction between the two residues Y73 and V77 results in an up-field shift of the methyl resonances of V77. The methyl chemical shifts of this residue thus report on a medium-range side chain interaction in the transient helix that according to the random peptides in Figure 4 would not be detectable in a coil structure. This suggests that methyl secondary chemical shifts can be a useful probe of medium- to long-range structure in disordered proteins if the source of ring current effects can be traced.
The transient α-helices found in both ACTR and acid denatured ACBP are amphipathic, which means their formation is largely driven by the burial of the side chains on the hydrophobic side. Bunching all the hydrophobic side chains together on one side of a helix thus allows the side chains to interact and are thus less exposed to the solvent. Furthermore, hydrophobic clusters in disordered proteins are often involved in long-range interactions suggesting that they are also driven by burial of hydrophobic groups. Due to these hydrophobic interactions, the rotamer distributions of the side chains change relative to the random coil, but the population changes are an order smaller than populations of the structure in the backbone. This suggests that even though the side chains interact to become partially buried, their rotamer distributions are only perturbed slightly, suggesting that the interactions do not require specific hydrophobic packing. This is consistent with a model of protein folding, where the proteins collapses initially due to non-specific hydrophobic interactions, and rigid side chain packing occurs as the last step in the protein folding pathway.
Materials and Methods
Protein preparation
15N and 13C labeled ACBP was expressed and purified as described previously.29,52 ACTR was coexpressed with the nuclear coactivator binding domain of CBP using a biscistronic expression vector.26 The bacteria pellet resuspended in 10 mM phosphate buffer, pH 7, and heat treated at 55°C for 5 min and sonicated. Solid urea was added to the supernatant and loaded onto a HiTrap Q FF column. ACTR was eluted with a gradient from 0 to 0.5M NaCl and purified by RP-HPLC as described previously.29 The NMR sample of ACTR contained 2 mM labeled protein, 20 mM phosphate buffer pH 6.5, 10% D2O, 0.02% NaN3, and 1 mM DSS. The NMR sample of ACBP contained 0.75 mM labeled protein, 50 mM phosphate buffer pH 2.4, 50 mM NaCl, 0.02% NaN3, and 50 μM DSS. Similar samples were prepared containing 6M urea and concentrations of 1 mM and 0.75 mM, respectively, for ACTR and ACBP.
NMR spectroscopy
For all four NMR samples, a constant time 13C HSQC,53 a (H)C(CO)NH,41 and a H(CCO)NH41 experiment were acquired at 25°C on a Varian Unity 800 MHz spectrometer equipped with either a cold probe or a room temperature probe. A FLOPSY854 mixing sequence was used to ensure maximal sensitivity on the methyl signals. Chemical shifts were referenced to internal DSS as described previously.55 NMR data were processed using NMRPipe56 and analyzed in CCPNMR Analysis 2.1.5.57 Assignment of methyl resonances were performed using (H)C(CO)NH and H(CCO)NH experiments using previously reported backbone resonance assignments in the absence28,33 and presence of urea.29,34 For non-overlapped peaks, the chemical shifts were extracted from the constant time 13C HSQC experiment, whereas for overlapping peaks the chemical shifts were determined from the (H)C(CO)NH and H(CCO)NH experiments. Furthermore, TFE was titrated into a sample containing 1 mM double labeled ACTR under similar conditions to that described above. A 15N HSQC, a 13C constant time HSQC of the methyl region, a (H)C(CO)NH, a 2D HNCO, and a 2D HNCOCA spectrum were acquired at 0, 2, 4, 6, 8, and 10 %(V/V) TFE. A urea titration series was acquired by recording seven datasets consisting of a 15N HSQC, a 2D HNCO, and a 2D HNCOCA spectrum from 0 to 6M urea by addition of small quantities from a protein solution containing 10.4M urea.
All peptides were purchased from KJ Ross-Petersen ApS (Klampenborg, Denmark) and were purified to more than 95% purity by reversed phase HPLC and their identities were confirmed by mass spectroscopy. The effect of the immediate neighbor was investigated using two peptide series with the sequences Ac-GGXIGG-NH2 and Ac-GGIXGG-NH2, where X was either of (S, D, R, L, F, Y, W). The distance dependence of the neighbor effect was investigated using a peptide series with the sequence Ac-GGFGnIGG-NH2, where the number of separating residues, n, was varied from 0 to 6. In these peptide series, NMR samples were prepared by dissolving 2–3 mg peptide into 500 μL 20 mM phosphate buffer pH 6.5 containing 5% D2O, 3 mM NaN3, and 1 mM DSS. pH was adjusted to the desired pH by addition of small quantities of HCl. Two series of random coil peptides with the sequence Ac-GGXGG-NH2 and Ac-QQXQQ-NH2 (X = A, I, L, M, T, V) were used to determine the statistical coil chemical shift values of the methyl chemical shifts. The solvent conditions were identical to previously used for determination of random coil backbone chemical shifts on the same peptides,45,46 that is, pH 6.5 20 mM phosphate buffer, 5% D2O, 3 mM NaN3, and 1 mM DSS. The Ac-GGXGG-NH2 peptides additionally contained 1M urea and was recorded at 25°C, while the Ac-QQXQQ-NH2 peptide samples did not contain urea and were recorded at 5°C. For all peptides, the chemical shifts were determined from a 1H-13C HSQC spectrum recorded at 25°C and processed as described above.
Acknowledgments
Authors thank Kaare Teilum and Søren Brander for constructive comments to the manuscript and the Swedish NMR Centre for spectrometer time.
Supplementary material
References
- 1.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Bio. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 2.Shortle D. The denatured state (the other half of the folding equation) and its role in protein stability. FASEB J. 1996;10:27–34. doi: 10.1096/fasebj.10.1.8566543. [DOI] [PubMed] [Google Scholar]
- 3.Jensen MR, Salmon L, Nodet G, Blackledge M. Defining conformational ensembles of intrinsically disordered and partially folded proteins directly from chemical shifts. J Am Chem Soc. 2010;132:1270–1272. doi: 10.1021/ja909973n. [DOI] [PubMed] [Google Scholar]
- 4.Gillespie JR, Shortle D. Characterization of long-range structure in the denatured state of staphylococcal nuclease. I. Paramagnetic relaxation enhancement by nitroxide spin labels. J Mol Biol. 1997;268:158–169. doi: 10.1006/jmbi.1997.0954. [DOI] [PubMed] [Google Scholar]
- 5.Lietzow MA, Jamin M, Dyson HJ, Wright PE. Mapping long-range contacts in a highly unfolded protein. J Mol Biol. 2002;322:655–662. doi: 10.1016/s0022-2836(02)00847-1. [DOI] [PubMed] [Google Scholar]
- 6.Shortle D, Ackerman MS. Persistence of native-like topology in a denatured protein in 8 M urea. Science. 2001;293:487–489. doi: 10.1126/science.1060438. [DOI] [PubMed] [Google Scholar]
- 7.Bernado P, Blanchard L, Timmins P, Marion D, Ruigrok RWH, Blackledge M. A structural model for unfolded proteins from residual dipolar couplings and small-angle X-ray scattering. Proc Natl Acad Sci USA. 2005;102:17002–17007. doi: 10.1073/pnas.0506202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jensen MR, Markwick PR, Meier S, Griesinger C, Zweckstetter M, Grzesiek S, Bernado P, Blackledge M. Quantitative determination of the conformational properties of partially folded and intrinsically disordered proteins using NMR dipolar couplings. Structure. 2009;17:1169–1185. doi: 10.1016/j.str.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 9.Marsh JA, Forman-Kay JD. Structure and disorder in an unfolded state under nondenaturing conditions from ensemble models consistent with a large number of experimental restraints. J Mol Biol. 2009;391:359–374. doi: 10.1016/j.jmb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 10.Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M. Structural polymorphism of 441-residue tau at single residue resolution. PLos Biol. 2009;7:e34. doi: 10.1371/journal.pbio.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang JR, Grzesiek S. Ensemble calculations of unstructured proteins constrained by RDC and PRE data: a case study of urea-denatured ubiquitin. J Am Chem Soc. 2010;132:694–705. doi: 10.1021/ja907974m. [DOI] [PubMed] [Google Scholar]
- 12.Vajpai N, Gentner M, Huang JR, Blackledge M, Grzesiek S. Side-chain chi(1) conformations in urea-denatured ubiquitin and protein G from (3)J coupling constants and residual dipolar couplings. J Am Chem Soc. 2010;132:3196–3203. doi: 10.1021/ja910331t. [DOI] [PubMed] [Google Scholar]
- 13.Alexandrescu AT, Abeygunawardana C, Shortle D. Structure and dynamics of a denatured 131-residue fragment of Staphylococcal nuclease—a heteronuclear NMR-study. Biochemistry. 1994;33:1063–1072. doi: 10.1021/bi00171a004. [DOI] [PubMed] [Google Scholar]
- 14.Hennig M, Bermel W, Spencer A, Dobson CM, Smith LJ, Schwalbe H. Side-chain conformations in an unfolded protechi(1) distributions in denatured hen lysozyme determined by heteronuclear C-13, N-15 NMR spectroscopy. J Mol Biol. 1999;288:705–723. doi: 10.1006/jmbi.1999.2722. [DOI] [PubMed] [Google Scholar]
- 15.West NJ, Smith LJ. Side-chains in native and random coil protein conformations. Analysis of NMR coupling constants and chi1 torsion angle preferences. J Mol Biol. 1998;280:867–877. doi: 10.1006/jmbi.1998.1911. [DOI] [PubMed] [Google Scholar]
- 16.Mathieson SI, Penkett CJ, Smith LJ. Characterisation of side-chain conformational preferences in a biologically active but unfolded protein. Pac Symp Biocomput. 1999:542–553. doi: 10.1142/9789814447300_0054. [DOI] [PubMed] [Google Scholar]
- 17.London RE, Wingad BD, Mueller GA. Dependence of amino acid side chain 13C shifts on dihedral angle: application to conformational analysis. J Am Chem Soc. 2008;130:11097–11105. doi: 10.1021/ja802729t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tonelli AE, Schilling FC. C-13 NMR chemical-shifts and the microstructure of polymers. Acc Chem Res. 1981;14:233–238. [Google Scholar]
- 19.Mulder FAA. Leucine side-chain conformation and dynamics in proteins from C-13 NMR chemical shifts. Chembiochem. 2009;10:1477–1479. doi: 10.1002/cbic.200900086. [DOI] [PubMed] [Google Scholar]
- 20.Hansen DF, Neudecker P, Kay LE. Determination of isoleucine side-chain conformations in ground and excited states of proteins from chemical shifts. J Am Chem Soc. 2010;132:7589–7591. doi: 10.1021/ja102090z. [DOI] [PubMed] [Google Scholar]
- 21.Sahakyan AB, Vranken WF, Cavalli A, Vendruscolo M. Structure-based prediction of methyl chemical shifts in proteins. J Biomol NMR. 2011;50:331–346. doi: 10.1007/s10858-011-9524-2. [DOI] [PubMed] [Google Scholar]
- 22.Perkins SJ, Wuthrich K. Ring current effects in the conformation dependent NMR chemical shifts of aliphatic protons in the basic pancreatic trypsin inhibitor. Biochim Biophys Acta. 1979;576:409–423. doi: 10.1016/0005-2795(79)90416-1. [DOI] [PubMed] [Google Scholar]
- 23.Haigh CW, Mallion RB. Ring current theories in nuclear magnetic-resonance. Prog Nucl Magn Reson Spect. 1979;13:303–344. [Google Scholar]
- 24.Redfield C. NMR studies of partially folded molten-globule states. Methods Mol Biol. 2004;278:233–254. doi: 10.1385/1-59259-809-9:233. [DOI] [PubMed] [Google Scholar]
- 25.Kjaergaard M, Teilum K, Poulsen FM. Conformational selection in the molten globule state of the nuclear coactivator binding domain of CBP. Proc Natl Acad Sci USA. 2010;107:12535–12540. doi: 10.1073/pnas.1001693107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Demarest SJ, Martinez-Yamout M, Chung J, Chen H, Xu W, Dyson HJ, Evans RM, Wright PE. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature. 2002;415:549–553. doi: 10.1038/415549a. [DOI] [PubMed] [Google Scholar]
- 27.Demarest SJ, Deechongkit S, Dyson HJ, Evans RM, Wright PE. Packing, specificity, and mutability at the binding interface between the p160 coactivator and CREB-binding protein. Protein Sci. 2004;13:203–210. doi: 10.1110/ps.03366504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ebert MO, Bae SH, Dyson HJ, Wright PE. NMR relaxation study of the complex formed between CBP and the activation domain of the nuclear hormone receptor coactivator ACTR. Biochemistry. 2008;47:1299–1308. doi: 10.1021/bi701767j. [DOI] [PubMed] [Google Scholar]
- 29.Kjaergaard M, Norholm AB, Hendus-Altenburger R, Pedersen SF, Poulsen FM, Kragelund BB. Temperature-dependent structural changes in intrinsically disordered proteins: formation of α-helices or loss of polyproline II? Protein Sci. 2010;19:1555–1564. doi: 10.1002/pro.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andersen KV, Poulsen FM. Three-dimensional structure in solution of acyl-coenzyme A binding protein from bovine liver. J Mol Biol. 1992;226:1131–1141. doi: 10.1016/0022-2836(92)91057-v. [DOI] [PubMed] [Google Scholar]
- 31.Kragelund BB, Robinson CV, Knudsen J, Dobson CM, Poulsen FM. Folding of a four-helix bundle: studies of acyl-coenzyme A binding protein. Biochemistry. 1995;34:7217–7224. doi: 10.1021/bi00021a037. [DOI] [PubMed] [Google Scholar]
- 32.Kragelund BB, Osmark P, Neergaard TB, Schiodt J, Kristiansen K, Knudsen J, Poulsen FM. The formation of a native-like structure containing eight conserved hydrophobic residues is rate limiting in two-state protein folding of ACBP. Nat Struct Biol. 1999;6:594–601. doi: 10.1038/9384. [DOI] [PubMed] [Google Scholar]
- 33.Thomsen JK, Kragelund BB, Teilum K, Knudsen J, Poulsen FM. Transient intermediary states with high and low folding probabilities in the apparent two-state folding equilibrium of ACBP at low pH. J Mol Biol. 2002;318:805–814. doi: 10.1016/S0022-2836(02)00159-6. [DOI] [PubMed] [Google Scholar]
- 34.Modig K, Jurgensen VW, Lindorff-Larsen K, Fieber W, Bohr HG, Poulsen FM. Detection of initiation sites in protein folding of the four helix bundle ACBP by chemical shift analysis. FEBS Lett. 2007;581:4965–4971. doi: 10.1016/j.febslet.2007.09.027. [DOI] [PubMed] [Google Scholar]
- 35.Teilum K, Kragelund BB, Poulsen FM. Transient structure formation in unfolded acyl-coenzyme A-binding protein observed by site-directed spin labelling. J Mol Biol. 2002;324:349–357. doi: 10.1016/s0022-2836(02)01039-2. [DOI] [PubMed] [Google Scholar]
- 36.Fieber W, Kristjansdottir S, Poulsen FM. Short-range, long-range and transition state interactions in the denatured state of ACBP from residual dipolar couplings. J Mol Biol. 2004;339:1191–1199. doi: 10.1016/j.jmb.2004.04.037. [DOI] [PubMed] [Google Scholar]
- 37.Bruun SW, Iesmantavicius V, Danielsson J, Poulsen FM. Cooperative formation of native-like tertiary contacts in the ensemble of unfolded states of a four-helix protein. Proc Natl Acad Sci USA. 2010;107:13306–13311. doi: 10.1073/pnas.1003004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neri D, Billeter M, Wider G, Wuthrich K. NMR determination of residual structure in a urea-denatured protein, the 434-repressor. Science. 1992;257:1559–1563. doi: 10.1126/science.1523410. [DOI] [PubMed] [Google Scholar]
- 39.Gillespie JR, Shortle D. Characterization of long-range structure in the denatured state of staphylococcal nuclease. II. Distance restraints from paramagnetic relaxation and calculation of an ensemble of structures. J Mol Biol. 1997;268:170–184. doi: 10.1006/jmbi.1997.0953. [DOI] [PubMed] [Google Scholar]
- 40.Mohana-Borges R, Goto NK, Kroon GJA, Dyson HJ, Wright PE. Structural characterization of unfolded states of apomyoglobin using residual dipolar couplings. J Mol Biol. 2004;340:1131–1142. doi: 10.1016/j.jmb.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 41.Grzesiek S, Anglister J, Bax A. Correlation of backbone amide and aliphatic side-chain resonances in C-13/N-15-enriched proteins by isotropic mixing of C-13 magnetization. J Magn Reson. 1993;101:114–119. [Google Scholar]
- 42.Kontaxis G, Clore GM, Bax A. Evaluation of cross-correlation effects and measurement of one-bond couplings in proteins with short transverse relaxation times. J Magn Reson. 2000;143:184–196. doi: 10.1006/jmre.1999.1979. [DOI] [PubMed] [Google Scholar]
- 43.Bax A, Kontaxis G, Tjandra N. Dipolar couplings in macromolecular structure determination. Methods Enzymol. 2001;339:127–174. doi: 10.1016/s0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- 44.Buck M. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q Rev Biophys. 1998;31:297–355. doi: 10.1017/s003358359800345x. [DOI] [PubMed] [Google Scholar]
- 45.Kjaergaard M, Brander S, Poulsen FM. Random coil chemical shift for intrinsically disordered proteins: effects of temperature and pH. J Biomol NMR. 2011;49:139–149. doi: 10.1007/s10858-011-9472-x. [DOI] [PubMed] [Google Scholar]
- 46.Kjaergaard M, Poulsen FM. Sequence correction of random coil chemical shifts: correlation between neighbor correction factors and changes in the Ramachandran distribution. J Biomol NMR. 2011;50:157–165. doi: 10.1007/s10858-011-9508-2. [DOI] [PubMed] [Google Scholar]
- 47.Schwarzinger S, Kroon GJ, Foss TR, Wright PE, Dyson HJ. Random coil chemical shifts in acidic 8 M urea: implementation of random coil shift data in NMRview. J Biomol NMR. 2000;18:43–48. doi: 10.1023/a:1008386816521. [DOI] [PubMed] [Google Scholar]
- 48.Schwarzinger S, Kroon GJ, Foss TR, Chung J, Wright PE, Dyson HJ. Sequence-dependent correction of random coil NMR chemical shifts. J Am Chem Soc. 2001;123:2970–2978. doi: 10.1021/ja003760i. [DOI] [PubMed] [Google Scholar]
- 49.Bhavesh NS, Sinha R, Mohan PM, Hosur RV. NMR elucidation of early folding hierarchy in HIV-1 protease. J Biol Chem. 2003;278:19980–19985. doi: 10.1074/jbc.M301615200. [DOI] [PubMed] [Google Scholar]
- 50.Lindorff-Larsen K, Kristjansdottir S, Teilum K, Fieber W, Dobson CM, Poulsen FM, Vendruscolo M. Determination of an ensemble of structures representing the denatured state of the bovine acyl-coenzyme a binding protein. J Am Chem Soc. 2004;126:3291–3299. doi: 10.1021/ja039250g. [DOI] [PubMed] [Google Scholar]
- 51.Meier S, Strohmeier M, Blackledge M, Grzesiek S. Direct observation of dipolar couplings and hydrogen bonds across a beta-hairpin in 8 M urea. J Am Chem Soc. 2007;129:754–755. doi: 10.1021/ja067522k. [DOI] [PubMed] [Google Scholar]
- 52.Mandrup S, Hojrup P, Kristiansen K, Knudsen J. Gene synthesis, expression in Escherichia-coli, purification and characterization of the recombinant bovine acyl-CoA-binding protein. Biochem J. 1991;276:817–823. doi: 10.1042/bj2760817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kay LE, Keifer P, Saarinen T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J Am Chem Soc. 1992;114:10663–10665. [Google Scholar]
- 54.Mohebbi A, Shaka AJ. Improvements in C-13 broad-band homonuclear cross-polarization for 2d and 3d NMR. Chem Phys Lett. 1991;178:374–378. [Google Scholar]
- 55.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 56.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Nmrpipe—a multidimensional spectral processing system based on Unix pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 57.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas P, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.