

Abstract
Structural knowledge of interactions amongst the ∼ 40 proteins of the human complement system, which is central to immune surveillance and homeostasis, is expanding due primarily to X-ray diffraction of co-crystallized proteins. Orthogonal evidence, in solution, for the physiological relevance of such co-crystal structures is valuable since intermolecular affinities are generally weak-to-medium and inter-domain mobility may be important. In this current work, Förster resonance energy transfer (FRET) was used to investigate the 10 μM KD (210 kD) complex between the N-terminal region of the soluble complement regulator, factor H (FH1-4), and the key activation-specific complement fragment, C3b. Using site-directed mutagenesis, seven cysteines were introduced individually at potentially informative positions within the four CCP modules comprising FH1-4, then used for fluorophore attachment. C3b possesses a thioester domain featuring an internal cycloglutamyl cysteine thioester; upon hydrolysis this yields a free thiol (Cys988) that was also fluorescently tagged. Labeled proteins were functionally active as cofactors for cleavage of C3b to iC3b except for FH1-4(Q40C) where conjugation with the fluorophore likely abrogated interaction with the protease, factor I. Time-resolved FRET measurements were undertaken to explore interactions between FH1-4 and C3b in fluid phase and under near-physiological conditions. These experiments confirmed that, as in the cocrystal structure, FH1-4 binds to C3b with CCP module 1 furthest from, and CCP module 4 closest to, the thioester domain, placing subsequent modules of FH near to any surface to which C3b is attached. The data do not rule out flexibility of the thioester domain relative to the remainder of the complex.
Keywords: resonance energy transfer, time-resolved fluorescence, complement system, factor H, C3b, fluorescent protein labeling
Introduction
The complement system is vital to immune defense and for human health.1–3 Activated via the classical, lectin, or alternative pathways (AP), it augments cellular immunity, kills pathogens, and clears antigen–antibody complexes, and apoptotic debris. Central to complement is proteolytic cleavage, in plasma, of inactive C3 yielding active C3b. C3b can attach, via a nascent reactive thioester embedded within a thioester domain (TED), to any nearby surfaces bearing nucleophiles4 whereupon it self-propagates by forming part of a convertase (a complex of C3b plus Bb, i.e., C3b.Bb) that cleaves C3 to generate further C3b molecules.
The antibody-independent AP5 underlies the rapid responsiveness of complement. The AP is switched on permanently, albeit at a low-level, through spontaneous hydrolysis of the C3 thioester creating C3(H2O), a structural and functional analog of C3b. Thus C3(H2O) likewise forms a convertase complex (C3(H2O).Bb), which also cleaves C3 to C3b.6 The slow ubiquitous turnover of C3 to C3(H2O) consequently leads to generation of a trickle of potentially self-amplifying C3b on plasma-exposed surfaces. The regulators of complement activation (RCA) protein family7 prevent C3b self-propagation and, crucially, act preferentially on self-surfaces. The result of this selective action is protection of self-surfaces but not foreign ones. The latter become targets of massive C3b autoamplification and a rapidly escalating complement-mediated response.8
Factor H (FH) is the prominent soluble member of the RCA family. This 155 kDa glycoprotein (∼ 500 mg/L in plasma)9 regulates the AP and is of paramount importance for protection of plasma-exposed self-surfaces not protected by other membrane-bound RCAs. Indeed, inadequate FH functionality, arising from mutations or single nucleotide polymorphisms or from antiFH antibodies is linked to age-related macular degeneration10 and other diseases in which complement plays a pathogenic role.11 Factor H binds to basement membranes and cell membranes via recognition of polyanions12 as well as to C3b.13,14 It thereby suppresses C3b amplification on self-surfaces (and in fluid phase). It consists entirely of 20 units, each 60 residues in size15 called complement control protein modules (CCPs), short consensus repeats, or sushi domains. These compactly folded,16 disulfide-stabilized modules are arranged end-to-end17 in a contiguous fashion, with varying levels of inter-modular flexibility.18,19
The N-terminal four CCPs of FH (i.e., FH1-4) are necessary and sufficient for functional properties of FH.20–23 Recombinant FH1-4 accelerates irreversible decay of the AP bimolecular C3 convertase; it is also a cofactor for factor I (FI) in proteolytic C3b destruction to iC3b, which is a ligand for complement receptors 2, 3, and 4, but no longer participates in the complement cascade. These activities depend on FH1-4 binding to C3b (KD = 10 μM), and this key interaction is therefore much studied. Structural analysis of FH1-4 was undertaken using nuclear magnetic resonance (NMR)24 and small-angle X-ray scattering,25,26 while a crystal structure of the FH1-4:C3b complex was determined (PDB 2WII)27 (see Fig. 1). According to the crystal structure, FH1-4 binds with CCP 4 closest to, and contacting, the TED while CCP 1 is furthest away from the TED. In crystals, TED is packed against C3b both in isolated protein and when in complex with FH1-4 (see Fig. 1), but electron microscopy suggests partial mobility of the TED in C3b.28
Figure 1.
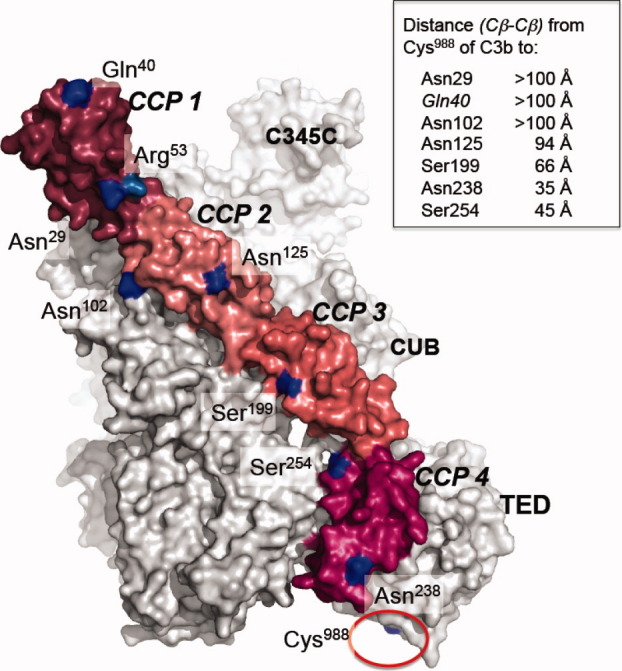
Putative locations within a C3b:FH1-4 complex of cysteine-substitution FH1-4 mutations. Positions (blue) of seven single-cysteine substitutions within crystal structure (PDB 2WII) of FH1-4 bound to C3b (gray); TED is the domain that would be tethered to a surface in the physiological setting via cleavage of the thioester bond involving Cys988 (indicated by red circle). Also shown is position of the disease-linked R53H mutation that along with Q40C (after labeling with the fluorophore) abrogated cofactor activity. Inset: Table indicates inter-atomic distances measured in the crystal structure. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
An initial crystal structure29 of another complex involving a truncated RCA fragment (of complement receptor type 2) and a C3 fragment (C3d, equivalent to the isolated TED) conflicted with mutagenesis and NMR30–32 unlike a recent structure of the same complex33 that is presumably physiologically relevant. Similarly, there are varying accounts of complexes between the C-terminal fragment of FH (FH19-20) and C3d.34,26 Generally, protein–protein complexes may be misrepresented in crystal structures because of competition between interactions driving crystallization and those driving formation of the biological complex.35 Orthogonal evidence regarding the physiological relevance of the crystallized FH1-4:C3b complex is therefore desirable, preferably via precipitant-free solution studies. One such approach is to measure Förster resonance energy transfer (FRET)36 between site-specifically conjugated fluorophores.
The thiol of nascent C3b (generated by hydrolysis of the isoglutamyl cysteine thioester of C3 to form Cys988 and Glu991) provides a unique attachment point for a maleimide-coupled fluorophore. Factor H, on the other hand, has no free thiols hence substitution by cysteines, using site-directed mutagenesis, is required. This is challenging since FH1-4 has eight disulfide bonds. We have exploited the C3b thiol, and cysteine-substitution mutagenesis, in a novel approach to using intermolecular time-resolved FRET (based on measurement of the fluorescence lifetime of the donor) for distance determination between two interacting complement proteins and we provide orthogonal support for the physiological relevance of the crystal structure of the FH1-4:C3b complex but cannot exclude motion of the TED.
Results
Protein production and characterization
To maximize coverage of FH1-4 but minimize likelihood of structural disruption, two residues per CCP module at exposed positions and well away from intermodular interfaces were selected (see Fig. 1) for mutagenesis. Native-sequence and cysteine-substitution mutant FH1-4 proteins were produced and purified to homogeneity, except for FH-1-4(N154C) that was not detected in the growth media. The C3 was purified from human plasma using standard techniques (see Methods section) and subsequently hydrolyzed to C3(H2O) before labeling and conversion to C3b.
Cofactor activity (for FI-catalyzed cleavage of C3b to iC3b) is a readily measurable biological function of the recombinant native-sequence FH-1-4 protein. This activity was assayed to confirm that the AlexaFluor555 (Ax555)-tagged adducts of the mutated versions of FH1-4 (FH1-4Ax555) retain native-like ability to bind C3b and recruit and activate FI, and hence are likely to be properly folded. Fluid-phase cofactor assays were conducted for all seven fluorescently tagged single-cysteine mutants (see Fig. 2). In the case of FH1-4(Q40C), cofactor activity is no longer detectable after fluorescent labeling, which indicates the fluorescent dye blocks a crucial binding site; this mutant was not used for FRET studies.
Figure 2.
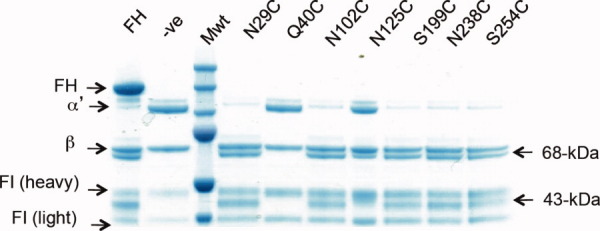
Cofactor assay of seven FH1-4 mutants. Disappearance of the α′-chain of C3b, and appearance of 43- and 68-kDa FI/FH-generated proteolytic products of the α′-chain of iC3b, were observed in Coomassie-stained gels following SDS-PAGE. Single-cysteine substitution FH1-4 mutants, as indicated, were labeled with Ax555 maleimide then samples incubated with FI and C3b (10 min, 37°C) prior to loading. Plasma-purified full-length FH or recombinant FH1-4 served as positive controls (+ve); PBS-only as a negative control (−ve). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Intermolecular FRET with donor label on C3b
In preparation for intermolecular FRET experiments, C3b was labeled with donor AlexaFluor 488 (Ax488) maleimide (yielding C3bAx488). The fluorescence decay of C3bAx488 alone (i.e., in absence of any acceptor fluorophore) required three lifetime components for a satisfactory fit (Table S1—Supporting Information). The observation of several decay components indicates that the C3b-bound Ax488 exists in a heterogeneous microenvironment, where it is subject to a variety of local quenching interactions. The longest lifetime of 4.1 ns corresponds to that of the free fluorophore in phosphate-buffered saline (Table 1.5 in Ref.37) indicating that ∼ 40% (A1) of the C3b-bound Ax488 population exists in an unquenched environment. However, the remaining 60% of the population is subject to substantial quenching giving shorter decay components (1.4 and 0.12 ns). The average lifetime, τav, of the C3bAx488 donor is thus 1.87 ns (Table I). This reduction in τav and hence in quantum yield [Eq. (2), see Methods section] of the donor is significant, because the Förster distance (R0) [Eq. (4)] depends on the donor quantum yield [Eq. (5)]. Using the respective τav's and a value of 0.92 for the quantum yield of Ax488 in PBS (Table 1.5 in Ref.37) we obtain a quantum yield for C3bAx488 of 0.42. This leads to a reduction in the value of R0, for energy transfer from Ax488 to Ax555, from 70 Å for the free donor (Table 1.6 in Ref.37) to 61 Å for the C3bAx488 donor.
Table 1.
Summary of FRET Data and Comparison of X-ray and FRET-Derived Distances
| C3bAx488:FH1-4Ax555 | C3bAx647:FH1-4Ax555 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pair | D (X-ray)a (Å) | Averageb τ (ns) | Efficiencyav | Dav (Å)c | Dmin (Å) | Dmax (Å) | Avd. τ (ns); don. vs. don. +acc. | Efficiencyav | 3Dav (Å) | Dmin (Å) | Dmax (Å) |
| C3bAx488 (alone) | – | 1.87 ± 0.04 | |||||||||
| C3bAx488: CCP 1 [N29CAx555] | 123 | 1.79 ± 0.05 | 4.4 ± 3.6 | 102 | 92 | 137 | |||||
| C3bAx488: CCP 2 [N102CAx555] | 102 | 1.73 ± 0.05 | 7.9 ± 3.6 | 92 | 86 | 102 | |||||
| C3bAx488: CCP 2 [N125CAx555] | 94 | 1.69 ± 0.06 | 9.6 ± 4.0 | 89 | 83 | 98 | |||||
| C3bAx647: CCP 3 [S199CAx555] | 66 | 1.46 ± 0.02 | 22.1 ± 2.1 | 75 | 74 | 77 | 0.52 ± 0.04 vs. 0.45 ± 0.04 | 13.2 ± 10.5 | 76 | 68 | 101 |
| C3bAx647: CCP 4 [N238CAx555] | 35 | 1.08 ± 0.06 | 42.6 ± 3.6 | 64 | 63 | 66 | 0.82 ± 0.03 vs. 0.51 ± 0.05 | 37.6 ± 6.8 | 66 | 69 | 63 |
| C3bAx647: CCP 4 [S254CAx555] | 45 | 0.49 ± 0.02 | 74.1 ± 1.4 | 51 | 50 | 52 | 0.54 ± 0.04 vs. 0.30 ± 0.03 | 43.8 ± 6.8 | 59 | 56 | 61 |
Distance (D) between Cβ of relevant side chains.
Weighted average of two or three individual lifetime (τ) components used to fit decay curve.
Average D and range, Dmin−Dmax, calculated from FRET efficiencies and associated experimental errors.
Average lifetime for donor alone (don.) versus donor plus acceptor (don. + acc.).
Having recorded data for a 400 nM sample of C3bAx488, FH1-4Ax555 was added to a final concentration of 33 μM in the sample. The fluorescence-decay curves (Fig. 3) of all donor-plus-acceptor (i.e., the C3bAx488:FH1-4Ax555) complexes were well represented as a sum of three lifetime components (Table S1—Supporting Information), except for the complex with FH1-4(S254C), which required four components. The average lifetime of C3bAx488 in each complex is given in Table I. From the average lifetime, the energy transfer efficiency [Eq. (3)], and hence the donor-acceptor distance [Eq. (4)], for each complex was determined, as also shown in Table I.
Figure 3.
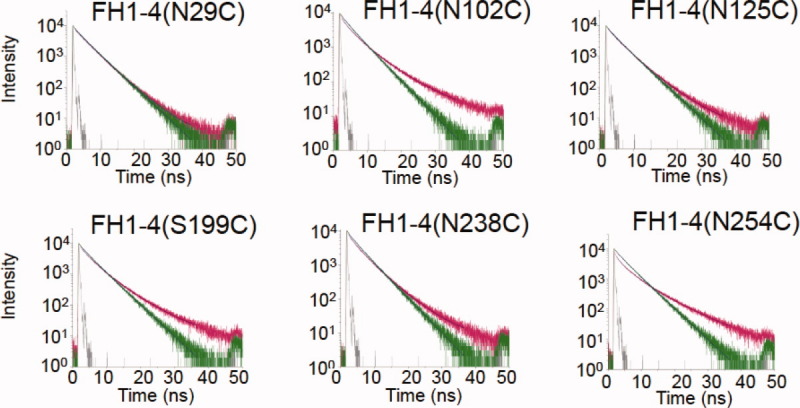
Time-resolved fluorescence-decay curves for C3bAx488:FH1-4Ax555 complexes. Fluorescence-decay curves for: C3bAx488: FH1-4(N29C)Ax555, C3bAx488: FH1-4(N102C)Ax555, C3bAx488: FH1-4(N125C)Ax555, C3bAx488: FH1-4(S199C)Ax555, C3bAx488: FH1-4(N238C)Ax555 (to exemplify quality of the fit, weighted residuals for this decay curve may be viewed in Figure S1, Supporting Information) and C3bAx488: FH1-4(N254C)Ax555. Decays of donor only (C3bAx488) are in green, of donor/acceptor complex, pink; instrumental response functions in gray. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Modules CCP 1 and CCP 2 are distant from the thioester domain in the FH1-4:C3b complex
Addition to C3bAx488 of three mutants in which fluorescently-labeled cysteine residues lie towards the N-terminus of FH1-4 [i.e., FH1-4(N29C)Ax555, FH1-4(N102C)Ax555, and FH1-4(N125C)Ax555] (see Fig. 1) each produced only marginal change in the average overall C3bAx488 fluorescence lifetime (Fig. 3, Table I). Indeed energy transfer efficiencies were calculated at <10%, generating inferred donor-acceptor distances of >90 Å. Thus while formation of C3bAx488:FH1-4Ax555 complexes modulated subtly the fluorescence decay of the donor fluorophore attached to the TED of C3b (as evidenced by differences in the lifetime components used to fit the decay curves), it is concluded that the acceptor molecules tethered to the three cysteine residues within CCPs 1 and 2 (i.e., Cys29, Cys102, and Cys125) are too far from the donor label (on C3b TED) for FRET to be reliably measured. This is consistent with distances measured (all >93 Å) in the crystal structure of FH1-4:C3b (see Fig. 1).
Modules CCP3 and CCP4 are closer to the thioester domain than CCP 1 or CCP 2
The three FH1-4Ax555:C3bAx488 complexes in which the acceptor fluorophore was attached to either CCP 3 [FH1-4(S199C)Ax555] or CCP 4 [(FH1-4(N238C)Ax555 and FH1-4(S254C)Ax555)] exhibited markedly different results to those obtained for complexes in which CCP 1 and CCP 2 were fluorescently labeled (Fig. 3). Average lifetimes of C3bAx488 (Table I) were 1.5 ns for FH1-4(S199C)Ax555, 1.1 ns for FH1-4(N238C)Ax555, and 0.5 ns for FH1-4(S254C)Ax555 (cf. the τav for the donor-only sample of 1.9 ns) yielding efficiencies of energy transfer of ∼ 22 ± 2%, 43 ± 4%, and 74 ± 1%, respectively (Table I). These values suggest that—amongst this set—the acceptor in FH1-4(S199C)Ax555 in CCP3 is the furthest away from the donor, with a distance of 74–77 Å (reasonably consistent with 66 Å in the crystal structure), followed by FH1-4(N238C)Ax555 at 63–66 Å (35 Å in the crystal structure) and FH1-4(S254C)Ax555 at 50–52 Å (45 Å in the crystal structure), in both of which the acceptor fluorophore is linked to residues in CCP 4.
Thus the general pattern of FRET-derived intermolecular distances, over the four CCPs, is consistent with the orientation of C3b-bound FH1-4 in the cocrystal structure (Fig. 1). On the other hand, the inferred interfluorophore distance between N238 of FH1-4 and Cys988 of C3b (∼ 64 Å) is longer than would be expected from the cocrystal structure given that the distance between the corresponding β-carbons is only 35 Å.
Intermolecular FRET with acceptor label on C3b
To cross-check intermolecular distances between C3b TED and the four N-terminal modules of FH in a separate experiment, the C3b component of the complex was labeled with Ax647 (instead of Ax488) and time-resolved fluorescence decay measurements were conducted with FH1-4Ax555 proteins as described earlier. Thus, in these experiments the donor fluorophore, Ax555, is attached to FH1-4 and the acceptor fluorophore is conjugated to C3b. As expected, no significant FRET was observed in the case of donors attached to CCPs 1 and 2 (not shown) and so the results described from hereon relate to the three FH1-4Ax555 proteins with donors conjugated to CCPs 3 and 4. Fluorescence decay curves recorded for the donors in the absence of acceptor (i.e., the three FH1-4Ax555 proteins alone) and in the presence of the acceptor (i.e., three FH1-4Ax555 proteins in complexes with C3bAx647) were, as before, fitted as a sum of multiple discrete lifetime components (Table S2—Supporting Information), again reflecting the heterogeneous microenvironment of the donor fluorophore. The quantum-yield-corrected R0 value for each of the three FH1-4Ax555 donors was calculated, as described earlier, using the quantum yield values given in Table S2. The fluorescence lifetime, quantum yield and R0 (for energy transfer to Ax647) of free Ax555 are 0.3 ns, 0.1 and 51 Å,, respectively. As shown in Table S2, the FH1-4Ax555R0 values are somewhat greater than those of the free donor, with values of 56 Å for both FH1-4(S199C)Ax555 and FH1-4(S254C)Ax555, and 60 Å for FH1-4(N238C)Ax555.
Average lifetimes were determined for FH1-4(S199C)Ax555 to be 0.52 ns versus 0.45 ns in absence versus presence of C3bAx647 (Table I), yielding an energy transfer efficiency of 13% (but with a large, ±10%, error) converting to a poorly defined donor-acceptor distance of between 68 and 101 Å; this may be compared to the C3bAx488 measurement (above) of 74–77 Å and the X-ray derived (inter β-carbon) distance of 66 Å. In the case of the fluorescence decay (Fig. 4) of the FH1-4(N238C)Ax555 sample, average lifetimes were determined to be 0.82 and 0.51 ns, in absence and presence of C3bAx647, respectively; this equates to a FRET efficiency of 38 ± 7% leading to a calculated donor-acceptor distance in the range of 63–69 Å (Table I), compared to 63–66 Å measured above and 35 Å (inter β-carbon) in the X-ray derived structure. Finally, in the case of the FH1-4(S254C)Ax555 sample, average lifetimes were 0.54 ns (alone) versus 0.30 ns (with C3b), the energy transfer efficiency from donor to acceptor was therefore 44 ± 7%, which corresponds to a distance of 56–61 Å, compared to 50–52 Å measured above and 45 Å (inter β-carbon) according to X-ray diffraction. As before, these results place CCP 4 closer to the thioester than CCP 3 (or CCPs 1 or 2) but the inferred distance from Asn238 to Cys988 (of C3b) is somewhat longer than that measured within the cocrystal structure.
Figure 4.
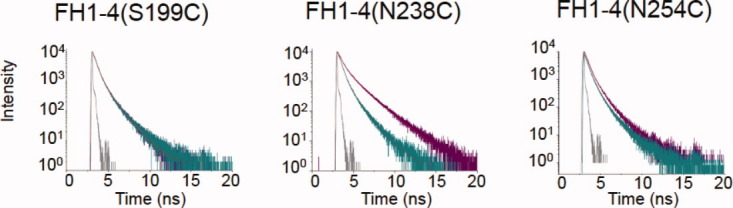
Time-resolved fluorescence-decay curves for C3bAx647: FH1-4A555 complexes. Fluorescence decay curves for C3bAx647: FH1-4(S199C)A555, C3bAx647: FH1-4(N238C)A555, and C3bAx647: FH1-4(N254C)A555. Decays of donor only (FH1-4Ax555) are purple, of the donor/acceptor complex, cyan; instrumental response functions in gray. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Discussion
Three-dimensional structures of individual protein domains (or modules) are easily determined experimentally or modeled by homology. It is less straightforward to elucidate the spatial arrangement of domains within the very large category of inherently flexible multiple-domain proteins—exemplified by FH—and their numerous, potentially transient, complexes. While FRET based on specifically positioned donor and acceptor fluorophores has been widely employed for protein-structure determination, its use to investigate interactions between the difficult-to-study multiple-domain and disulfide-rich proteins of the complement system has received only limited attention.38,39 By using FRET in the current work to validate the physiological relevance of previous crystallography and EM-based studies we have not only enhanced confidence in these results but also explored the potential of FRET for looking at further complexes, for which high-resolution structural information is lacking.
Incorporation of free cysteine residues into proteins that naturally contain multiple disulfides carries the risk of disulfide shuffling and misfolding. In this study, we showed that P. pastoris is a suitable expression host for production and secretion into the growth medium of useful quantities of a panel of single cysteine-substitution mutants of a FH fragment (FH1-4, with eight disulfides) with a success rate of seven mutants obtained out of eight. All seven successfully produced and purified mutants—that are distributed over all four CCP modules and are positioned away from intermodular interfaces—retained significant cofactor activity for FI-catalyzed cleavage of C3b (not shown); one of them—FH1-4(Q40C)—lost cofactor activity when tagged with the relatively bulky fluorescent probe, Ax555. Cofactor activity, which likely involves binding of FH1-4 to both C3b and FI, is one of the principal functions of FH and is entirely contained within, and depends upon, the structural integrity of, FH1-4. The FH1-4(Q40C) mutant was not used for further studies; in fact, the coordinates of the cocrystal structure, once they became available, showed that none of the seven replaced residues do in fact participate directly in the crystallographic C3b-FH1-4 interface (see Fig. 1). Hence, loss of function of FH1-4(Q40C) was probably not because of loss of C3b binding but more likely arose from interference between the probe and FI. Indeed Gln40 lies on the same face of CCP 1 as the R53H, aHUS-linked, mutation that was recently shown to bind normally to C3b but to have depleted cofactor activity.40 The cofactor activity of FH1-4(S254C)Ax555 was also somewhat diminished. Thus, taken together, our data support the presence of an extended composite C3b/FH 1-4 binding site for FI lying close to the proteolytic cleavage site within the CUB domain (Fig. 1).
Intermolecular FRET-based distances were measured (see Figs. 3 and 4, and Table I) in the complex of FH1-4 and C3b. Recombinant C3b was not available but the presence of the free thiol in the TED is serendipitous since it allows a fluorophore to be placed at an informative position within the C3b structure. The N-terminal (CCPs 1-4) and C-terminal (CCPs 19-20) regions of FH (155 kDa) bind cooperatively to C3b (180 kDa) producing a 1:1 (FH:C3b) complex with a KD = ∼ 1 μM (as measured by SPR). Full-length FH and its complexes have resisted all efforts at crystallization but the X-ray derived structures of FH1-4 in complex with C3b (KD = 10 μM)27 and (separately) FH19-20 in complex with the C3d (TED) derivative of C3b34,26 (KD = 4 μM), are informative. Neither of these structures have, however, been verified by orthogonal structural methods. Indeed, there is controversy over interpretation of the physiological relevance of protein–protein complexes observed in the two independently solved crystal structures of FH19-20:C3d, reminiscent of more profound questions as to the relevance of a previously solved structure of C3d and the two N-terminal modules of complement receptor type 229,33 (a close homolog of FH). The structure of FH1-4 with C3b27 is less controversial since it reveals a highly extended intermolecular interface, but the weak KD for this complex is close to what some regard as a safe limit beyond which precipitant-enhanced crystallographic interactions might outcompete weaker but biologically important ones. Indeed the orientation of FH1-4 on the C3b in this structure is counterintuitive since, for C3b tethered to a surface, it places the N-terminal part of FH furthest from the surface and the C-terminal part (from which extends an additional 16 modules in native FH) very close to the surface. Therefore the use of time-resolved FRET to measure interdomain distances in the FH1-4:C3b complex is a useful means of validating or otherwise the crystal structure.
Our strategic placement of fluorophores on FH1-4 allows its orientation as observed in the crystal structure of FH1-4:C3b to be tested since the observed orientation will place N-terminal CCP 1 furthest away from the fluorescently-tagged TED, followed by CCP 2 and then CCP 3, with CCP 4 being closest. Our results clearly showed that fluorophores on CCP 1 (attached to N29C) was beyond the upper limit of the FRET range with respect to the fluorophore on Cys988 of TED, while those on CCP 2 (N102C and N125C) were at the edge of detection; indeed the corresponding intermolecular distances (approximated as Cβ-Cβ) in the crystal complex are all >93 Å. FRET-based estimates for the distance between the fluorophores on CCPs 3 and 4 showed excellent agreement when comparing the two FRET pairs used in this study. The results are consistent with CCP 4 being closer to the TED than CCP 3. Thus even in the absence of precipitants and crystal packing forces FH1-4 bind to surface-tethered C3b with the fourth module closest to the surface. While this potentially projects subsequent CCPs counter intuitively into the glycocalyx, it is nonetheless not inconsistent with other homologous, small complement regulators in which the fourth CCP (out of four CCPs in total) immediately precedes a membrane-associating (CD55) or membrane-spanning (CD46) region binding to C3b in the same orientation.
One notable discrepancy with respect to the crystal structure is that the donor-acceptor distances measured for FH1-4(N238C)— ∼ 64 or ∼ 66 Å, depending on which FRET pair was used—were longer than those measured using S254C (∼ 51 or ∼ 59 Å) while in the cocrystal structure, CCP4 and TED are juxtaposed so that the β carbons of Asn238 and Ser254 are just 35 and 45 Å away from Cys988 Cβ, respectively. It is possible, given that free rotation has been assumed in the calculation of the Ro distances, that such discrepancies could arise from some hindrance to the free rotation of the donor or acceptor when attached to the cysteine. A more interesting possibility is that the CCP3-CCP4 junction is flexible, or more likely that the TED, which is flexibly attached to the rest of the C3b molecule according to some electron micrographs,28 retains a degree of mobility even in the presence of FH1-4 (see cartoon, Fig. 5).
Figure 5.
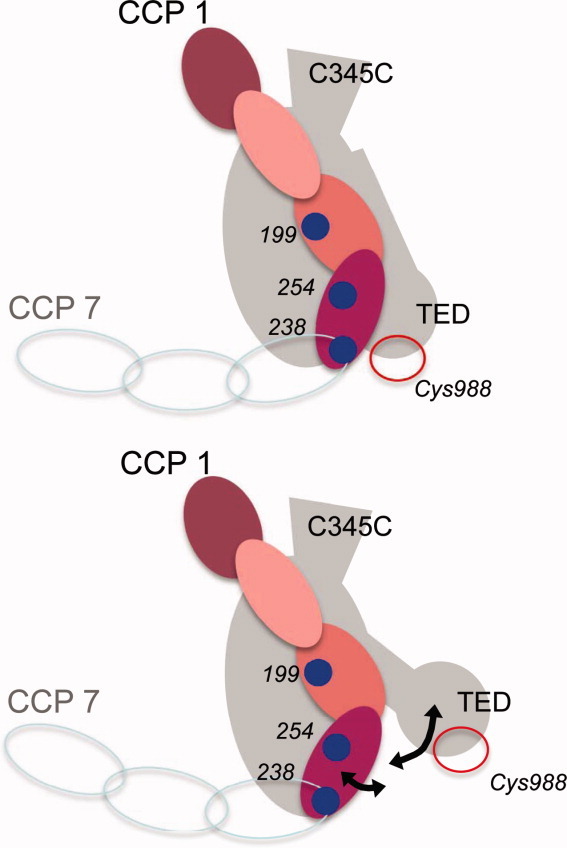
Possible flexibility within C3b/FH1-4 complex. Upper cartoon: Summarized (as in Fig. 1) are positions of CCP-3 and CCP-4 cysteine substitutions (in FH1-4) and of Cys988 (in C3b) within the crystal structure of the complex (plus possible positions of FH CCPs 5–7). Lower caroon: Alternative relative positions of CCP 4 and TED could explain the discrepancies between distance measurements in the crystal and in the fluid phase. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The currently accepted model is that full-length FH bends back on itself via a central hinge-like region41 and its C-terminus (FH19-20) binds to a distinct site on the TED immediately adjacent to the extended FH1-4 binding site (and to nearby surface-borne glycosaminoglycans). This second interaction with FH19-20 might further stabilize the conformation of C3b observed in the crystal structure with FH1-4, although it might also stabilize a different structure. The former hypothesis—in which the N-terminal part of module 19 lies close to the C-terminal region of CCP 426—could readily be tested in an extension of our approach by using fluorophore-labeled P. pastoris-expressed single cysteine-substitution mutants of FH-1-4 and FH19-20 fragments in ternary complex with C3b. Time-resolved FRET measurements of this kind could also be performed in physiological settings such as on a C3b-opsonized cell surface. More generally, many protein–protein interaction networks, for which the mammalian complement system is a paradigm, depend on highly coordinated sequences of domain rearrangements and on both intramolecular and intermolecular domain–domain recognition events. A combination of orthogonal methods is required to study such “domain choreography” and we have proved the utility of one such technique by successfully applying FRET in initial estimates of interdomain distances in proteins of the complement system.
Materials and Methods
Fluorescent probes, Alexa488 maleimide, Alexa555 maleimide, and Alexa647 maleimide were from Invitrogen (Molecular Probes). Complement proteins C3b (for cofactor activities only), FI, and plasma-purified FH were from Complement Technology (TX). Cobra venom factor:Bb complex was a gift from Viviana Ferreira (University of Texas).
The DNA coding for FH1-4 (residues 20–262, numbering includes pre–pro sequence) was amplified from full-length FH cDNA as described in Ref.24. N-terminal c-myc (EQKLISEEDL) and C-terminal hexaHis- tags were introduced. The coding region of FH1-4 was mutated to encode variants FH1-4(N29C), FH1-4(Q40C), FH1-4(N102C), FH1-4(N125C), FH1-4(N154C), FH1-4(S199C), FH1-4(N238C), and FH1-4(S254C), using QuikChange site-directed mutagenesis (Stratagene). Genes of the confirmed sequences were expressed in the Pichia pastoris strain KM71H in 500 mL of cell culture. After an induction period of four days, cells were pelleted, supernatant was harvested, and pH adjusted to 7.5.
Purifications was achieved by Ni2+-affinity chromatography (HisTrap column, GE Healthcare). Labeling with Ax555 maleimide was performed in 20 mM sodium phosphate (pH 7.0, dye:protein ratio = 2:1, 4 h, 25°C) in the dark. Unreacted dye was removed by buffer exchange in 5 mM Tris, pH 8.0. Purification from nonlabeled protein was achieved by ion-exchange chromatography using a Tricorn MonoQ (GE Healthcare) with a linear gradient from 5 mM Tris, pH 8.0 to 5 mM Tris, pH 8.0, plus 1M NaCl (flow-rate 2 mL/min). Elution of nonlabeled FH1-4 occurred at 400 mM NaCl, and of labeled FH1-4 at 600 mM NaCl. Protein concentrations were measured using λ280.
Complement component C3 was sourced from human plasma following a method described elsewhere.42 Following an established procedure,43 plasma-purified C3 was converted into the thioester- hydrolyzed form C3(H2O) by incubating C3 in water (37°C, 2 h). Remaining, unconverted C3 was removed by cation-exchange chromatrography [MonoS 5/50 (1 mL)] (GE Healthcare); C3(H2O) eluted off at ∼ 600 mM NaCl while C3 elutes earlier in the gradient. Fractions containing C3(H2O) were combined and dialyzed into PBS overnight.
To label the free thiol in C3(H2O), and simultaneously convert C3(H2O) into C3b, C3(H2O) (350 μg/mL in 400 μL), was labeled with Ax488 or Ax647 (400 μM) in the presence of 10 mM ethylenediaminetetraacetic acid (EDTA) and 8 μg cobra venom factor Bb C3 convertase. This convertase converts C3(H2O) into C3b and is used instead of the alternative pathway convertase. The reaction was incubated (2 h, room temperature, in the dark) then dialyzed into PBS/0.05% (v/v) TWEEN-20 (BioRad) (i.e., PBS-T-0.05) overnight. Size-exclusion chromatography (Superdex 16/60 S300 (GE Healthcare) in PBS-T-0.05) was performed to remove any C3b dimers from the labeled C3b (C3bAx488 or C3bAx647).
To measure biological activity of FH1-4 constructs, an endpoint fluid-phase cofactor assay was performed. For the assay, in 30 μL, 4 μg FI, and 10 μg C3b (equimolar at ∼ 2 μM) were mixed with 5 μg FH (final concentration 1 μM) for the positive-control reaction. The negative control was prepared similarly, but substituting FH with PBS. Wild-type and mutant FH1-4 (0.9 μg in PBS, final concentration 1 μM) were prepared in the same way. The reaction volume was finally made up to 30 μL using PBS. The reaction mix was then vortexed and incubated in a water bath (37°C, 15 min). An aliquot of 2× reducing SDS buffer was added to stop the reaction prior to analysis by SDS-PAGE.
Fluorescence lifetime measurements were carried out in PBS. Time-correlated single photon counting was used. Protein concentrations were either: 33 μM for fH1-4Ax555 and 400 nM for C3bAx488; or 400 nM for fH1-4Ax555, and 33 μM for C3bAx647. The excitation source was the second harmonic of the pulse-picked output of a Ti-Sapphire femtosecond laser system (Coherent, 10 W Verdi, and Mira Ti-Sapphire), consisting of pulses of ∼ 200 fs at 4.75 MHz repetition rate, with 465 nm wavelength. Fluorescence decays were measured in an Edinburgh Instruments spectrometer equipped with TCC900 photon-counting electronics. The emission from the sample was collected orthogonal to the excitation direction through a polarizer set at the magic angle with respect to the vertically polarized excitation. The fluorescence was passed through a monochromator (bandpass, 10 nm) and detected at 520 nm (Ax488 donor emission) or 570 nm (Ax555 donor emission) by a Hamamatsu Microchannel Plate Photomultiplier (R3809U-50). The instrument response of the system, measured using a Ludox scatterer, was ∼ 70 ps, full-width, half-maximum.
Fluorescence decay curves were analyzed using an iterative reconvolution method, assuming a multiexponential decay function [Eq. (1)], employing Edinburgh Instruments FAST software. To determine the number of exponential terms required to fit the decay data, the quality of the fit was judged on the basis of the reduced chi-squared statistic, χ2, and the randomness of the residuals. For our experimental system, typically, we find that χ2 <1.2 indicates an acceptable fit.
| (1) |
where Ai and τi are the fractional amplitude and fluorescence lifetime, respectively, of the ith decay component (the value of Ai gives the fractional population of the fluorescent species with lifetime τi); and B is the background count (the dark count of the detector).
The average lifetime, τav, as defined in Eq. (2) was used to calculate the FRET efficiency, Eav, [Eq. (3)]. The average lifetime is proportional to the quantum yield, φ
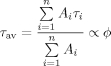 |
(2) |
| (3) |
where 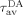 and
and 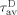 are the average fluorescence lifetimes of the donor, in the presence and absence of the acceptor, respectively.
are the average fluorescence lifetimes of the donor, in the presence and absence of the acceptor, respectively.
The energy transfer efficiency is related to the average distance, R, between donor and acceptor, according to Eq. (4).
| (4) |
where R0 is the Förster distance, at which the energy transfer efficiency = 0.5.
The Förster distance depends on the sixth root of the quantum yield of the donor, φD.
| (5) |
Acknowledgments
The authors wish to thank Dr John White (University of Edinburgh) for help with fermentation, Jochen Arlt (University of Edinburgh) for his assistance in COSMIC, and Professor Bob Sim from the University of Oxford for his help with the C3 purification. They also like to acknowledge Dr. Viviana Feirreira (University of Texas) for providing the cobra venom factor Bb complex.
Glossary
Abbreviations
- AP
alternative pathway
- Ax488 etc.
AlexaFluor 488 (etc.)
- CCP
complement control protein module
- EDTA
ethylenediaminetetraacetic acid
- FH
factor H
- FH1-4 (etc.)
CCP modules 1–4 of FH
- FI
factor I
- FRET
Förster resonance energy transfer
- NMR
nuclear magnetic resonance
- PBS
phosphate buffered saline
- RCA
regulators of complement activation
- TED
thioester domain
Supplementary material
References
- 1.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 2.Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344:1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 3.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Law SK, Dodds AW. The internal thioester and the covalent binding properties of the complement proteins C3 and C4. Protein Sci. 1997;6:263–274. doi: 10.1002/pro.5560060201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lachmann PJ. The amplification loop of the complement pathways. Adv Immunol. 2009;104:115–149. doi: 10.1016/S0065-2776(08)04004-2. [DOI] [PubMed] [Google Scholar]
- 6.Fishelson Z, Pangburn MK, Muller-Eberhard HJ. Characterization of the initial C3 convertase of the alternative pathway of human complement. J Immunol. 1984;132:1430–1434. [PubMed] [Google Scholar]
- 7.Kirkitadze MD, Barlow PN. Structure and flexibility of the multiple domain proteins that regulate complement activation. Immunol Rev. 2001;180:146–161. doi: 10.1034/j.1600-065x.2001.1800113.x. [DOI] [PubMed] [Google Scholar]
- 8.Lachmann PJ, Hughes-Jones NC. Initiation of complement activation. Springer Semin Immunopathol. 1984;7:143–162. doi: 10.1007/BF01893018. [DOI] [PubMed] [Google Scholar]
- 9.Sim RB, DiScipio RG. Purification and structural studies on the complement-system control protein beta 1H (Factor H) Biochem J. 1982;205:285–293. doi: 10.1042/bj2050285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kavanagh D, Goodship T. Genetics and complement in atypical HUS. Pediatr Nephrol. 2010;25:2431–2442. doi: 10.1007/s00467-010-1555-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meri S, Pangburn MK. Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc Natl Acad Sci U S A. 1990;87:3982–3986. doi: 10.1073/pnas.87.10.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma AK, Pangburn MK. Identification of three physically and functionally distinct binding sites for C3b in human complement factor H by deletion mutagenesis. Proc Natl Acad Sci U S A. 1996;93:10996–11001. doi: 10.1073/pnas.93.20.10996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pangburn MK. Host recognition and target differentiation by factor H, a regulator of the alternative pathway of complement. Immunopharmacology. 2000;49:149–157. doi: 10.1016/s0162-3109(00)80300-8. [DOI] [PubMed] [Google Scholar]
- 15.Kristensen T, Tack BF. Murine protein H is comprised of 20 repeating units, 61 amino acids in length. Proc Natl Acad Sci U S A. 1986;83:3963–3967. doi: 10.1073/pnas.83.11.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Norman DG, Barlow PN, Baron M, Day AJ, Sim RB, Campbell ID. Three-dimensional structure of a complement control protein module in solution. J Mol Biol. 1991;219:717–725. doi: 10.1016/0022-2836(91)90666-t. [DOI] [PubMed] [Google Scholar]
- 17.Barlow PN, Steinkasserer A, Norman DG, Kieffer B, Wiles AP, Sim RB, Campbell ID. Solution structure of a pair of complement modules by nuclear magnetic resonance. J Mol Biol. 1993;232:268–284. doi: 10.1006/jmbi.1993.1381. [DOI] [PubMed] [Google Scholar]
- 18.Perkins SJ, Haris PI, Sim RB, Chapman D. A study of the structure of human complement component factor H by Fourier transform infrared spectroscopy and secondary structure averaging methods. Biochemistry. 1988;27:4004–4012. doi: 10.1021/bi00411a017. [DOI] [PubMed] [Google Scholar]
- 19.Kirkitadze MD, Krych M, Uhrin D, Dryden DT, Smith BO, Cooper A, Wang X, Hauhart R, Atkinson JP, Barlow PN. Independently melting modules and highly structured intermodular junctions within complement receptor type 1. Biochemistry. 1999;38:7019–7031. doi: 10.1021/bi982453a. [DOI] [PubMed] [Google Scholar]
- 20.Gigli I, Fujita T, Nussenzweig V. Modulation of the classical pathway C3 convertase by plasma proteins C4 binding protein and C3b inactivator. Proc Natl Acad Sci U S A. 1979;76:6596–6600. doi: 10.1073/pnas.76.12.6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alsenz J, Lambris JD, Schulz TF, Dierich MP. Localization of the complement-component-C3b-binding site and the cofactor activity for factor I in the 38 kDa tryptic fragment of factor H. Biochem J. 1984;224:389–398. doi: 10.1042/bj2240389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulz TF, Alsenz J, Dierich MP. Localization of functionally important areas of the regulator protein factor H using monoclonal antibodies. Wien Klin Wochenschr. 1985;97:248–252. [PubMed] [Google Scholar]
- 23.Schmidt CQ, Herbert AP, Kavanagh D, Gandy C, Fenton CJ, Blaum BS, Lyon M, Uhrin D, Barlow PN. A new map of glycosaminoglycan and C3b binding sites on factor H. J Immunol. 2008;181:2610–2619. doi: 10.4049/jimmunol.181.4.2610. [DOI] [PubMed] [Google Scholar]
- 24.Hocking HG, Herbert AP, Kavanagh D, Soares DC, Ferreira VP, Pangburn MK, Uhrin D, Barlow PN. Structure of the N-terminal region of complement factor H and conformational implications of disease-linked sequence variations. J Biol Chem. 2008;283:9475–9487. doi: 10.1074/jbc.M709587200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okemefuna AI, Gilbert HE, Griggs KM, Ormsby RJ, Gordon DL, Perkins SJ. The regulatory SCR-1/5 and cell surface-binding SCR-16/20 fragments of factor H reveal partially folded-back solution structures and different self-associative properties. J Mol Biol. 2008;375:80–101. doi: 10.1016/j.jmb.2007.09.026. [DOI] [PubMed] [Google Scholar]
- 26.Morgan HP, Schmidt CQ, Guariento M, Blaum BS, Gillespie D, Herbert AP, Kavanagh D, Mertens HD, Svergun DI, Johansson CM, Uhrin D, Barlow PN, Hannan JP. Structural basis for engagement by complement factor H of C3b on a self surface. Nat Struct Mol Biol. 2011;18:463–470. doi: 10.1038/nsmb.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu J, Wu YQ, Ricklin D, Janssen BJ, Lambris JD, Gros P. Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat Immunol. 2009;10:728–733. doi: 10.1038/ni.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishida N, Walz T, Springer TA. Structural transitions of complement component C3 and its activation products. Proc Natl Acad Sci U S A. 2006;103:19737–19742. doi: 10.1073/pnas.0609791104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szakonyi G, Guthridge JM, Li D, Young K, Holers VM, Chen XS. Structure of complement receptor 2 in complex with its C3d ligand. Science. 2001;292:1725–1728. doi: 10.1126/science.1059118. [DOI] [PubMed] [Google Scholar]
- 30.Gilbert HE, Eaton JT, Hannan JP, Holers VM, Perkins SJ. Solution structure of the complex between CR2 SCR 1-2 and C3d of human complement: an X-ray scattering and sedimentation modelling study. J Mol Biol. 2005;346:859–873. doi: 10.1016/j.jmb.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 31.Hannan JP, Young KA, Guthridge JM, Asokan R, Szakonyi G, Chen XS, Holers VM. Mutational analysis of the complement receptor type 2 (CR2/CD21)-C3d interaction reveals a putative charged SCR1 binding site for C3d. J Mol Biol. 2005;346:845–858. doi: 10.1016/j.jmb.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 32.Kovacs JM, Hannan JP, Eisenmesser EZ, Holers VM. Mapping of the C3d ligand binding site on complement receptor 2 (CR2/CD21) using nuclear magnetic resonance and chemical shift analysis. J Biol Chem. 2009;284:9513–9520. doi: 10.1074/jbc.M808404200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van den Elsen JM, Isenman DE. A crystal structure of the complex between human complement receptor 2 and its ligand C3d. Science. 2011;332:608–611. doi: 10.1126/science.1201954. [DOI] [PubMed] [Google Scholar]
- 34.Kajander T, Lehtinen MJ, Hyvarinen S, Bhattacharjee A, Leung E, Isenman DE, Meri S, Goldman A, Jokiranta TS. Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc Natl Acad Sci U S A. 2011;108:2897–2902. doi: 10.1073/pnas.1017087108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krissinel E. Crystal contacts as nature's docking solutions. J Comput Chem. 2010;31:133–143. doi: 10.1002/jcc.21303. [DOI] [PubMed] [Google Scholar]
- 36.Clegg RM. Fluorescence resonance energy transfer. Curr Opin Biotechnol. 1995;6:103–110. doi: 10.1016/0958-1669(95)80016-6. [DOI] [PubMed] [Google Scholar]
- 37.Johnson I, Spence MTZ, editors. Molecular probes handbook, a guide to fluorescenet probes and labeling technologies. Invitrogren.
- 38.Cheng KH, Wiedmer T, Sims PJ. Fluorescence resonance energy transfer study of the associative state of membrane-bound complexes of complement proteins C5b-8. J Immunol. 1985;135:459–464. [PubMed] [Google Scholar]
- 39.Bexborn F, Andersson PO, Chen H, Nilsson B, Ekdahl KN. The tick-over theory revisited: formation and regulation of the soluble alternative complement C3 convertase (C3(H2O)Bb) Mol Immunol. 2008;45:2370–2379. doi: 10.1016/j.molimm.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pechtl IC, Kavanagh D, McIntosh N, Harris CL, Barlow PN. Disease-associated N-terminal complement factor H mutations perturb cofactor and decay-accelerating activities. J Biol Chem. 2011;286:11082–11090. doi: 10.1074/jbc.M110.211839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt CQ, Herbert AP, Mertens HD, Guariento M, Soares DC, Uhrin D, Rowe AJ, Svergun DI, Barlow PN. The central portion of factor H (modules 10–15) is compact and contains a structurally deviant CCP module. J Mol Biol. 2010;395:105–122. doi: 10.1016/j.jmb.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dodds AW. Small-scale preparation of complement components C3 and C4. Methods Enzymol. 1993;223:46–61. doi: 10.1016/0076-6879(93)23037-n. [DOI] [PubMed] [Google Scholar]
- 43.Usami M, Ohno Y. Preparation of complement fragments C3b and C3a from purified rat complement component C3 by activated cobra venom factor. J Pharmacol Toxicol Methods. 2005;52:260–263. doi: 10.1016/j.vascn.2004.12.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.