

Abstract
In severe acute respiratory syndrome coronavirus, the envelope heptad repeat 2 (HR2) plays a critical role in viral entry. Moreover, HR2 is both the target for novel antiviral therapies and, as an isolated peptide, presents a potential antiviral therapeutic. The structure of HR2, as determined by NMR spectroscopy in the presence of the co-solvent trifluoroethanol (TFE), is a trimer of parallel helices, whereas the structure of HR2, as determined by X-ray crystallography, is a tetramer of anti-parallel helices. In this work, we added a nitroxide spin label to the N-terminal region of HR2 and used paramagnetic relaxation enhancement to assess the orientation of the HR2 helices under different solution conditions. We find that the relaxation effects are consistent with an orientation corresponding to a trimer of parallel helices in both the presence and absence of TFE. This work suggests that the different orientation and oligomerization states observed by NMR and X-ray are due to the 11 additional residues present at the N-terminus of the NMR construct.
Keywords: NMR, spin label, SARS, heptad repeat, virus entry
Introduction
In severe acute respiratory syndrome coronavirus (SARS-CoV), the envelope protein, termed S or spike, plays a critical role in viral entry.1 In the first step of entry, S binds to the target cell receptor, human angiotensin-converting enzyme 2 (hACE2), and the virus enters the cell via receptor-mediated endocytosis.1,2 In the second step of entry, proteolysis by proteases of the cathespin family trigger conformational changes within S that bring heptad repeat domains, termed HR1 and HR2, into contact, thereby providing the driving force for fusion of the viral and endosomal membranes.3–5 Interestingly, the HR1 and HR2 domains of other enveloped viruses including Ebola, HIV, and influenza are thought to function in a similar manner and are targets of antiviral therapies.6 Moreover, the HR2 of the HIV envelope is a potent inhibitor of HIV entry, presumably by disrupting the intramolecular HR1–HR2 interaction during the entry process.7 On the other hand, the HR2 of S is a relatively modest inhibitor of SARS-CoV entry.8,9
Structural studies of the HR1 and HR2 of SARS-CoV demonstrate that they form a “six helix bundle” that is analogous to that formed by other viral envelope proteins such as HIV/SIV gp41.3,4,8,10–13 More recently, the SARS-CoV HR2 has been studied in isolation to lend insight into prefusion conformations of envelope.13–16 Interestingly, the previous studies of SARS-CoV HR2 have resulted in two different structures for the prefusion state.13,14 As shown in Figure 1, the NMR structure, in which the co-solvent trifluoroethanol (TFE) was present, indicated that the prefusion state of HR2 is a trimer of parallel helices, while the X-ray structure of HR2 is a tetramer of anti-parallel helices. In both cases, analytical ultracentrifugation confirmed the oligomeric state of the construct used. For example, the NMR construct was a trimer in the presence of 30% TFE and the X-ray construct was a tetramer under aqueous conditions.13,14
Figure 1.

(a) NMR structure of SARS-CoV HR2 (residues 1141–1193 of S). (b) X-ray structure of SARS-CoV HR2 (residues 1150–1193 of S). The buffer conditions for the NMR structure were 10 mM NaPO4, pH 7.0, 30% TFE.14 The buffer conditions for the X-ray crystallization were 100 mM Tris-HCl, pH 8.5, 10 mM NiCl2 with 22% PEG MME 2000.13 (c) Amino acid sequence of SARS-CoV HR2. The additional residues present in the NMR construct are boxed. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
There could be several explanations for the disparity in the NMR and X-ray structures. First, the NMR structure was determined in the presence of TFE, a co-solvent used to stabilize helical structures17 and thus the oligomerization state and relative orientation of the helices could be induced by the co-solvent. However, analytical ultracentrifugation studies of the NMR construct under aqueous conditions (i.e., in the absence of TFE) suggested that HR2 was in equilibrium between monomeric and trimeric forms.15 Second, the determination of a protein complex, the HR2 trimer, by NMR is relatively demanding because it is based on a small number of intermolecular NOEs that could possibly be misinterpreted. Finally, the NMR construct that we used contained an 11 residue extension at the N-terminus (9 native residues and 2 residues from a cloning artifact), which may affect the orientation and oligomerization state of HR2. Clearly, it is important to resolve the orientation and oligomerization states of HR2 for a better understanding of SARS-CoV envelope-mediated viral entry and the mechanism of HR2-based inhibitory peptides. In this work, we added a nitroxide spin label to the N-terminal region of HR2 and used paramagnetic relaxation enhancement to assess the orientation of the HR2 helices under different solution conditions.
Results and Discussion
As discussed in the introduction, there are significant differences in the structure of SARS-CoV HR2 determined by NMR and X-ray crystallography (Fig. 1). To determine whether HR2 is organized in a parallel or anti-parallel manner, we mutated residue S14, which is found in the N-terminal region of HR2, to cysteine. Subsequently, we attached the nitroxide spin label MTSL to the newly introduced cysteine to form S14C-MTSL. This has the effect of introducing a paramagnetic center to this region of the protein, which has previously been shown to result in increased line-widths of resonances within 25 Å of the spin label.18,19 If the HR2 complex is in a parallel orientation, as determined by NMR, paramagnetic relaxation enhancement would be observed for resonances at the N-terminal region (i.e., those close in sequence to residue 14). On the other hand, if the HR2 complex is in an anti-parallel orientation, as determined by X-ray crystallography, paramagnetic relaxation enhancement would be observed for resonances at the C-terminal region, as well as the N-terminal region.
In a first step, we measured the 15N-edited HSQC spectrum of S14C-MTSL in the presence of 30% TFE to test the validity of the previously determined NMR structure.14 In Figure 2(a), the relative intensities of the 1H–15N correlations are plotted as a function of residue number. The resonances near position 14 in sequence, the site of the MTSL, exhibit greatly reduced intensities with respect to HR2 not containing the spin label under identical experimental conditions. This observation implies that in the presence of TFE HR2 is a trimer of parallel helices, as previously determined.14
Figure 2.
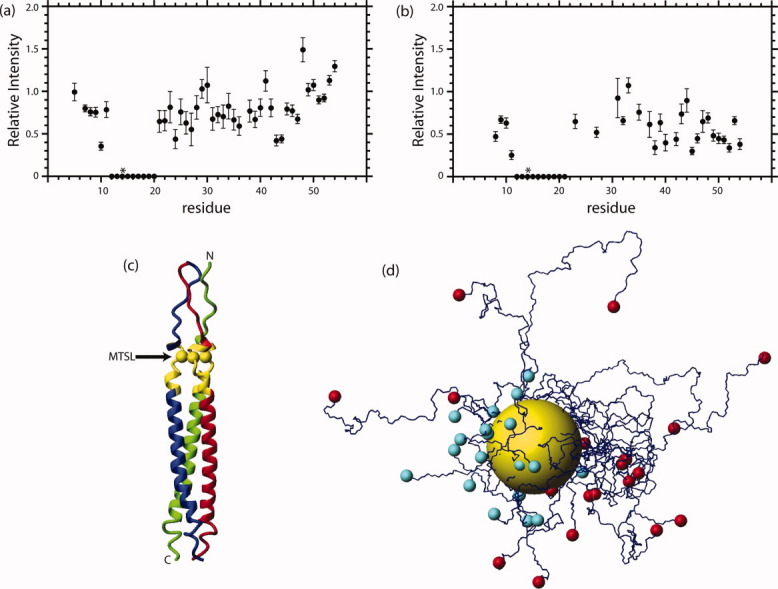
(a) Paramagnetic relaxation enhancement of S14C-MTSL as a function of residue number in the presence of TFE (the asterisk denotes the site of the MTSL). Experimental conditions were 1 mM S14C in 10 mM NaPO4, pH 7.0, 30% TFE-d3 and 10% 2H2O at 25°C. (b) Paramagnetic relaxation enhancement of S14C-MTSL as a function of residue number in the absence of TFE (the asterisk denotes the site of the MTSL). Experimental conditions were 1 mM S14C in 10 mM NaPO4, pH 7.0 and 10% 2H2O at 25°C. (c) SARS-CoV HR2 residues affected by paramagnetic relaxation enhancement. In this representation, individual monomers are colored red, green, and blue, respectively. 1H–15N correlations that exhibit less than 20% of the unlabeled intensity are depicted in yellow. The structure corresponds to that determined by NMR in the presence of 30% TFE.14 (d) Model of SARS-CoV monomers in the extended coil conformation. Structures were fit to the backbone atoms of residue 14 with an RMSD of 0.003 Å. The yellow sphere corresponds to a 15 Å radius from the residue 14 Cα. The cyan and red spheres depict the N- and C-terminal residues, respectively. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
As mentioned in the introduction, it is possible that the co-solvent TFE significantly affects the orientation and oligomerization state of HR2. Consequently, we measured the 15N-edited HSQC spectrum of S14C-MTSL under aqueous conditions (i.e., in the absence of TFE). In Figure 2(b), the relative intensities of the 1H–15N correlations are plotted as a function of residue number. Again the resonances near position 14 in sequence, the site of the MTSL, exhibit greatly reduced intensities with respect to HR2 not containing the spin label under identical experimental conditions. This observation is again consistent with the notion of a trimer of parallel helices, which was previously based on NMR and analytical ultracentrifugation studies in the presence and absence of TFE.14,15 Interestingly, under the aqueous conditions the intensity of other resonances is also affected to an intermediate degree. For example, the average intensity ratio for correlations corresponding to residues 30–55 is 0.85 and 0.53 for the TFE and aqueous conditions, respectively, suggesting long-range effects under aqueous conditions.
In Figure 2(c), the residues most affected by the spin label, defined by <20% relative intensity, are shown in yellow using the NMR structure determined in the presence of 30% TFE. Based on Cα–Cα distances, the residues most affected are <15 Å from the spin label site. Importantly, a similar effect is observed under aqueous conditions, which strongly supports the parallel orientation of SARS-CoV HR2. The differences between the NMR and X-ray structures may therefore be attributed to differences in the constructs. For example, the NMR construct contains nine additional native residues at the N-terminus with respect to the X-ray construct and it would seem that the additional residues push the equilibrium to the trimer of parallel helices. The propensity of heptad repeat regions to self-associate in different orientations and oligomerization states indicates that caution should be exercised in the interpretation of isolated domains. On the other hand, the ability of HR2 to assume different structures may have functional ramifications for the entry mechanism.13,15,16,20
As noted above, intermediate paramagnetic relaxation enhancement was observed for HR2 residues that are sequentially distant from the spin label under aqueous conditions. Previously, we have shown by CD, NMR, and analytical ultracentrifugation that HR2 exhibits a monomer–trimer equilibrium and that the monomer is unstructured.15,16,20 As a consequence, the intermediate effects shown in Figure 2(b) may be attributed to the approach of distant residues to the spin label attached to residue 14. To test the validity of this notion, we determined a family of HR2 structures without using experimentally determined NOE, H-bond, and dihedral restraints. A family of 20 monomer structures is shown in Figure 2(d). In this figure, the site of the spin label is depicted by a sphere of 15 Å, the approximate radius of correlations that are completely abolished in the 15N-edited HSQC spectra. The respective locations of the N- and C-terminal residues are depicted by cyan and red spheres, respectively. From this figure it is clear that the C-terminal residues that exhibit intermediate relaxation effects may often approach the local region of the spin label. For example, from a family of 100 HR2 structures the range of Cα–Cα distances observed for residues 20, 30, 40, and 50 to residue 14 is 8–20, 16–51, 15–68, and 11–80 Å, respectively. Based on the molecular dynamics simulations, it is predicted that the closest approach of residues 30, 40, and 50 is similar and thus the relatively uniform effects on relaxation may not be surprising. Finally, we note that we have previously suggested that the flexibility of the unstructured state facilitates conformational changes that are necessary for envelope-mediated membrane fusion.15,16,20
Materials and Methods
Protein preparation
Cysteine was introduced at residue 14 of SARS-CoV HR2 using the site-directed mutagenesis kit from Strategene and subsequently confirmed by DNA sequencing. The mutated version of HR2, termed S14C, was subcloned into the BamH1/HindIII restriction sites of a modified pQE30-expression vector (Qiagen, Valencia, CA). The resulting construct consists of an N-terminal polyhistidine tag, followed by PG (the IgG binding domain of streptococcus protein G),21 a TEV (tobacco etch virus) cleavage site for removal of the expression tag, and S14C. 15N-labeled S14C was prepared as previously described.14,15 Briefly, protein expression was achieved by growing E. coli strain SG13009 in the presence of the appropriate plasmid in 1 L of LB media supplemented with 100 μg/mL ampicillin and 50 μg/mL of kanamycin at 37°C until they reached an OD600 of 0.8. The cells were then pelleted, washed once with M9 salts, re-suspended in 250 mL of M9 minimal media supplemented with 1 g/L 15NH4Cl (Martek Biosciences, Columbia, MD), and set to recover in the absence of antibiotic selection. Protein expression was induced after 1 h by the addition of 0.8 mM IPTG and grown for an additional 4–5 h at 37°C. The HIS-PG-S14C fusion protein was purified from the soluble fraction using a Ni2+ fast-flow Sepharose column (Qiagen, Valencia, CA). The protein was then cleaved using TEV protease and run once more over the Ni2+ column to remove His-PG and TEV protease, which also contains a polyhistidine tag, as well as uncut HIS-PG-S14C. The flow-through fraction containing S14C was then dialyzed extensively against 10 mM NaPO4, pH 7.0, and concentrated by ultrafiltration (YM3, Amicon, Billerica, MA). The purity and identity of S14C was confirmed using SDS-PAGE and MALDI-TOF mass spectrometry. The newly introduced disulfide bond was reduced using a 5× molar excess of DTT. Excess reductant was removed using a Micro-Bio Spin chromotography column (Bio-Rad, Hercules, CA), then the reduced protein was incubated with a 7× molar excess of MTSL (S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl) methyl methanesulfonothioate) overnight at room temperature in 10 mM TRIS-HCl, pH 7.4, 0.02% sodium azide, to form S14C-MTSL. Finally, unbound MTSL was removed by dialysis against buffer containing 10 mM NaPO4, pH 7.0.
NMR spectroscopy
The experimental conditions for the NMR experiments were 1 mM S14C-MTSL in 10 mM NaPO4, pH 7.0, 30% TFE-d3 (Cambridge Isotope Laboratories, Andover, MA) and 10% 2H2O or alternatively in 10 mM NaPO4, pH 7.0, 10% 2H2O. Spectra were recorded at 25°C on a Bruker AVANCE 800 MHz spectrometer equipped with a 5 mm TCI cryogenic triple resonance probe. Processing of the spectra was done with NMRPipe and visualized with NMRDraw.22 The relative intensity was determined by comparing the intensity of S14C-MTSL correlations (Isl) with those of the wild-type HR2 (I) under the identical experimental conditions. Errors in the relative intensity were estimated as Isl/I ((NIsl/Isl)2 + (NI/I)2)0.5, where NIsl and NI are the noise calculated by NMRDraw in the appropriate spectrum.23
Molecular modeling
Structures were calculated by simulated annealing in torsion angle space starting from the HR2 in an extended conformation, followed by conventional simulated annealing, using the program CNS.24–26 A conformational database term was employed during the simulated annealing phase to favor energetically relevant dihedral angles.27 No NOE, H-bond, or other dihedral restraints were used in the simulated annealing phase. Figures were generated using the program MOLMOL.28
References
- 1.Simmons G, Reeves J, Rennekamp A, Amberg S, Piefer A, Bates P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS CoV) spike glycoprotein-mediated viral entry. Proc Natl Acad Sci USA. 2004;101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu S, Xiao G, Chen Y, He Y, Niu J, Escalente C, Xiong H, Farmar J, Debnath A, Tien P, Jiang S. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification effusion inhibitors. Lancet. 2004;363:938–947. doi: 10.1016/S0140-6736(04)15788-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tripet B, Howard M, Jobling M, Holmes R, Holmes K, Hodges R. Structural characterization of the SARS-coronavirus spike S fusion protein core. J Biol Chem. 2004;279:20836–20849. doi: 10.1074/jbc.M400759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simmons G, Gosalia D, Rennekamp A, Reeves J, Diamond S, Bates P. Inhibitors of cathespin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci USA. 2005;102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caffrey M. HIV envelope: challenges and opportunities for the discovery of entry inhibitors. Trends Microbiol. 2011;9:191–197. doi: 10.1016/j.tim.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matthews T, Salgo M, Greenberg M, Chung J, DeMasi R, Bolognesi D. Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat Rev Drug Discov. 2004;3:215–225. doi: 10.1038/nrd1331. [DOI] [PubMed] [Google Scholar]
- 8.Bosch B, Martina B, Van Der Zee R, Lepault J, Haijema B, Versluis C, Heck A, De Groot R, Osterhaus A, Rottier P. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc Natl Acad Sci USA. 2004;101:8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo Y, Tisoncik J, McReynolds S, Lou G, Martinez O, Farzan M, Prabhakar B, Gallagher T, Rong L, Caffrey M. Identification of a small region of SARS-CoV S protein critical for viral entry. J Mol Biol. 2009;394:600–605. doi: 10.1016/j.jmb.2009.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Supekar V, Bruckmann C, Ingallinella P, Bianchi E, Pessi A, Carfi A. Structure of a proteolytically resistant core from the severe acute respiratory syndrome coronavirus S2 fusion protein. Proc Natl Acad Sci USA. 2004;101:17958–17963. doi: 10.1073/pnas.0406128102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Y, Lou Z, Liu Y, Pang H, Tien P, Gao G, Rao Z. Crystal structure of the severe acute syndrome coronavirus spike protein fusion core. J Biol Chem. 2004;279:49414–49419. doi: 10.1074/jbc.M408782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duquerroy S, Vigouroux A, Rottier P, Rey F, Bosch B. Central ions an lateral asparagine/glutamine zippers stabilize the post-fusion hairpin conformation of the SARS coronavirus spike glycoprotein. Virology. 2005;335:276–285. doi: 10.1016/j.virol.2005.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng Y, Liu J, Zheng Q, Yong W, Lu M. Structures and polymorhic interactions of two heptad-repeat regions of the SARS virus S2 protein. Structure. 2006;14:889–899. doi: 10.1016/j.str.2006.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hakansson-McReynolds S, Jiang S, Rong L, Caffrey M. The solution structure of the SARS coronavirus heptad repeat 2 in the prefusion state. J Biol Chem. 2006;281:11965–11971. doi: 10.1074/jbc.M601174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McReynolds S, Jiang S, Ying G, Celigoy J, Schar C, Rong L, Caffrey M. Characterization of the prefusion and transition states of Severe Acute Respiratory Syndrome Coronavirus S2-HR2. Biochemistry. 2008;47:6802–6808. doi: 10.1021/bi800622t. [DOI] [PubMed] [Google Scholar]
- 16.McReynolds S, Jiang S, Rong L, Caffrey M. Dynamics of the SARS coronavirus prefusion and transition states. J Magn Reson. 2009;201:218–221. doi: 10.1016/j.jmr.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buck M. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q Rev Biophys. 1998;31:297–355. doi: 10.1017/s003358359800345x. [DOI] [PubMed] [Google Scholar]
- 18.Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 19.Park S, Caffrey M, Johnson M, Fung L. Solution structural studies on human erythrocyte alpha spectrin N-terminal tetramerization domain. J Biol Chem. 2003;278:21837–21844. doi: 10.1074/jbc.M300617200. [DOI] [PubMed] [Google Scholar]
- 20.Celigoy J, McReynolds S, Caffrey M. The SARS-CoV heptad repeat 2 exhibits pH-induced helix formation. Biochem Biophys Res Commun. 2011;412:483–486. doi: 10.1016/j.bbrc.2011.07.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huth J, Bewley C, Jackson B, Hinnebusch A, Clore G, Gronenborn A. Design of an expression system for detecting folded protein domains and mapping macromolecular interactions by NMR. Protein Sci. 1997;6:2359–2364. doi: 10.1002/pro.5560061109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 23.Jiang S, Jacobs A, Laue T, Caffrey M. Solution structure of the Coxsackievirus and Adenovirus receptor domain 1. Biochemistry. 2004;43:1847–1853. doi: 10.1021/bi035490x. [DOI] [PubMed] [Google Scholar]
- 24.Stein EG, Rice LM, Brunger AT. Torsion-angle molecular dynamics as a new efficient tool for NMR structure calculation. J Magn Reson. 1997;124:154–164. doi: 10.1006/jmre.1996.1027. [DOI] [PubMed] [Google Scholar]
- 25.Nilges M, Clore GM, Gronenborn AM. Determination of three-dimensional structures of proteins from interproton distance data by dynamical simulated annealing from a random array of atoms. Circumventing problems associated with folding. FEBS Lett. 1988;229:317–324. doi: 10.1016/0014-5793(88)80559-3. [DOI] [PubMed] [Google Scholar]
- 26.Brunger A, Adams P, Clore G, DeLano W, Gros P, Grosse-Kunstleve R, Jiang J-S, Kuszewski J, Nilges N, Pannu N, Read R, Rice L, Simonson T, Warren G. Crystallography and NMR system CNS: a new software system for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 27.Kuszewski J, Gronenborn AM, Clore GM. Improving the quality of NMR and crystallographic protein structures by means of a conformational database potential derived from structure databases. Protein Sci. 1996;5:1067–1080. doi: 10.1002/pro.5560050609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]